
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Revisiting cobaloxime(II) chemistry and clearing misconceptions of cobaloxime(II) in diamagnetic Ni(II) and Pd(II) matrixes using comprehensive magnetic measurements†
Yukina
Suzuki
 *a,
Mirosław
Arczyński
bi,
Masanori
Wakizaka
c,
Hisaaki
Tanaka
c,
Ryuta
Ishikawa
d,
Takefumi
Yoshida
ef,
Takeshi
Yamane
g,
Kazunobu
Sato
g,
Ryota
Sakamoto
a and
Masahiro
Yamashita
*ha
*a,
Mirosław
Arczyński
bi,
Masanori
Wakizaka
c,
Hisaaki
Tanaka
c,
Ryuta
Ishikawa
d,
Takefumi
Yoshida
ef,
Takeshi
Yamane
g,
Kazunobu
Sato
g,
Ryota
Sakamoto
a and
Masahiro
Yamashita
*ha
aDepartment of Chemistry, Graduate School of Science, Tohoku University, 6-3 Aramaki-Aza-Aoba, Aoba-Ku, Sendai 980-8578, Japan. E-mail: suzuki.yukina.s2@dc.tohoku.ac.jp; masahiro.yamashita.c5@tohoku.ac.jp
bFaculty of Chemistry, Jagiellonian University, Gronostajowa 2, 30-387 Kraków, Poland
cDepartment of Applied Chemistry and Bioscience, Faculty of Science and Technology, Chitose Institute of Science and Technology, 758-65 Bibi, Chitose 066-8655, Japan
dDepartment of Chemistry, Faculty of Science, Fukuoka University, Nanakuma 8-19-1, Fukuoka 814-0180, Japan
eCluster of Nanomaterials, Graduate School of Systems Engineering, Wakayama University, 930 Sakae-Dani, Wakayama, Japan
fPhysical and Chemical Research Infrastructure Group, RIKEN SPring-8 Center, RIKEN, Japan
gDepartment of Chemistry, Graduate School of Science, Osaka Metropolitan University, 3-3-138 Sugimoto, Sumiyoshi-Ku, Osaka 558-8585, Japan
hSchool of Chemical Science and Engineering, Tongji University, Siping Road 1239, Shanghai 200092, P. R. China
iDepartment of Chemistry, Graduate School of Advanced Science and Engineering, Hiroshima University, Higashihiroshima, Hiroshima 739-8526, Japan
First published on 15th April 2025
Abstract
Co(II) was doped into the diamagnetic one-dimensional framework of bis(dimethylglyoximato) Ni(II) and Pd(II) complexes [Ni/PdII(Hdmg)2] (Hdmg = dimethylglyoximate anion) (hereafter referred to as Co@Ni and Co@Pd samples) to study magnetic properties as a potential spin qubit. In a previous report of electron paramagnetic resonance (EPR) spectroscopy of this compound prepared using the same doping strategy in the Ni matrix, a spectrum assigned to S = 1/2 Co(II) with gx = 2.58, gy = 2.26 gz = 1.98 was observed. The relatively large gx value, compared to those typically observed in [Co(Hdmg)2Bx] complexes (B = Lewis base ligand, x = 1 or 2), which fall within the ranges gx = 2.1–2.4, gy = 2.0–2.2 and gz ≈ 2.01, led to the interpretation of this species to be Co(Hdmg)2 with an extremely axial interaction. However, our EPR analysis, combined with SQUID (superconducting quantum interference device) and XANES (X-ray absorption near-edge structure) analyses, revealed that the previously identified species are μ-O bridged dimers: [Co(Hdmg)(μ-Hdmg)]2 and [Co(Hdmg)(μ-Hdmg)][Ni/Pd(Hdmg)(μ-Hdmg)]. Furthermore, our liquid-helium temperature EPR spectra revealed a Co species with much greater axial g anisotropy (gx = 4.75 gy,z ≈ 0.75 for Co@Ni and gx = 4.2 gy,z ≈ 1.33 for Co@Pd). We assign this species to the truly planar Co(Hdmg)2 with negligibly weak axial interaction, which might have been overlooked in previous EPR studies.
Introduction
Molecular magnetism has been an active research area for decades with potential applications such as data storage devices and spintronics technologies. Furthermore, recent advancements in quantum information science have opened up an emerging field of molecular spin qubits. Among various types of qubit platforms, electronic spin-based molecular qubits can be relatively easily modified to tune their magnetic properties with precise spatial arrangement.1 Despite these advantages, molecular spin qubits tend to suffer from short spin–spin relaxation times (T2), with only a handful of systems that show coherence at room temperature.2–6 One technique to overcome such short T2 is to use clock transitions.This approach was first showcased on HoW10 ((Ho(W5O18)2)9−) complexes7 and a clear explanation of the role of hyperfine interactions was given.8 In brief, this method exploits the weak field dependence of the transition frequency near avoided crossings between two states, which can arise due to crystal field splitting or the hyperfine coupling of electronic and nuclear spins. For this reason, transition metals like 51V and 59Co with large nuclear spin (I = 7/2) and strong hyperfine coupling are good candidates for molecular S = 1/2 qubits.8 Freedman et al. previously reported a qubit exhibiting a clock transition based on a square planar Co(II) porphyrin in a Zr-based MOF.9 In this system, the MOF provided protection against additional axial ligand interactions with Co(II), which could otherwise weaken the hyperfine coupling required for achieving clock transition that can be accessed using a commercial EPR spectrometer.
Achieving longer coherence is crucial for qubit applications such as quantum computing10 and quantum sensing.11 Toward this goal, we decided to revisit the magnetic properties of a seemingly well-known low-spin Co(II) system, Co(Hdmg)2 (Hdmg = dimethylglyoximate anion), also known as cobaloxime(II).
Cobaloxime derivatives have been well studied specifically for their catalytic properties and as a model of the coenzyme vitamin B12.12–14 Despite the extensive investigation of those cobalt complexes, knowledge of unmodified Co(Hdmg)2 is very limited.15 Its first synthesis was reported by Schrauzer, describing it as a “long-sought compound” due to challenging preparation.15 The difficulties in preparing Co(Hdmg)2 arise from a high propensity to form axial coordination, which makes it impossible to crystallize cobaloxime(II) from polar solvents without the presence of axially ligating solvent molecules. The strong tendency for axial interactions is also evidenced by a library of more than 600 entries in the Cambridge Structural Database (CSD) found for cobaloxime(II) derivatives and other axially ligated cobaloxime(II) compounds like dimers/polymers.13,16–19
Schrauzer's synthesis was later adopted by other researchers to investigate the electronic structure of “square planar” Co(Hdmg)2 using EPR spectroscopy.20–22 In order to do this, the Co(II) ions were typically doped in square planar diamagnetic Ni(Hdmg)2 crystals (Fig. 1) by simply starting the preparation procedure with a mixture of Co and Ni acetates instead of Co alone. In these studies, Co@Ni was often used as a model for the square planar cobaloxime(II) complex with negligibly weak axial interactions.20,22–24 Indeed, EPR data for all Co@Ni samples consistently showed relatively strong axiality with gx = 2.58, gy = 2.26, gz = 1.98 as compared to the range of g1 = 2.1–2.4, g2 = 2.0–2.2, and g3 ≈ 2.01 for axially coordinated cobaloximes(II).19
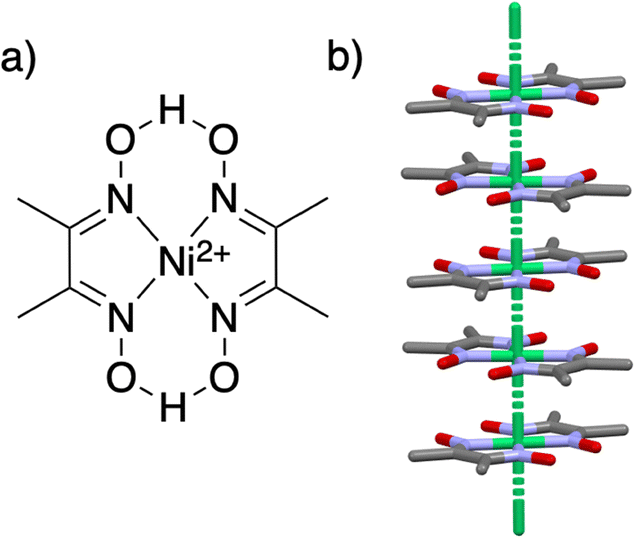 | ||
| Fig. 1 (a) The schematic structure of Ni(Hdmg)2. (b) The crystal structure of Ni(Hdmg)2 (Ibam space group) forms a one-dimensional chain. Color code Ni – green, N – blue, O – red, C – grey, H – omitted for clarity. | ||
In this study, however, we made two main observations that challenge the previous understanding. First, EPR peaks that were not observed in previous studies20–22 were detected below 80 K for Co@Ni and below 120 K for Co(II) in diamagnetic Pd(Hdmg)2 (Co@Pd). These species, in both the Ni(II) and Pd(II) matrices, exhibit extremely high axiality, with their EPR peaks splitting into approximately 150–250 mT and 800–1000 mT sets for Co@Ni, and 150–250 mT and 400–600 mT for Co@Pd. We attribute this low-temperature component of the EPR spectra to a pseudo-square planar Co(Hdmg)2 that retains the original planar geometry of Ni(Hdmg)2 or Pd(Hdmg)2 (1) (Fig. 2). Hereafter, we will refer to them as “species (1)”. As discussed in detail later, this “pseudo-square planar Co(Hdmg)2” shows different EPR parameters depending on whether it is in the Ni or Pd matrix. Therefore, strictly speaking, these species in Ni(II) and Pd(II) have slightly different axial interactions of Co(II) with Pd(II) or Ni(II). However, due to the similar coordination environments around Co(II) and magnetic behaviors, we use the same designation for them. For clarity, when discussing species (1), we indicate the host metal after the number as species (1@Ni) and species (1@Pd). The clear differences in g factors between species (1@Ni) and species (1@Pd) (Table 1) are consistent with the assigned pseudo-square planar structure, where Co(II) ions are situated in an environment where axial positions are easily accessible within the host matrices. As a result, the EPR parameters of these species (1@Ni/Pd) are readily perturbed by the different metal ions in the axial direction in the host matrices.
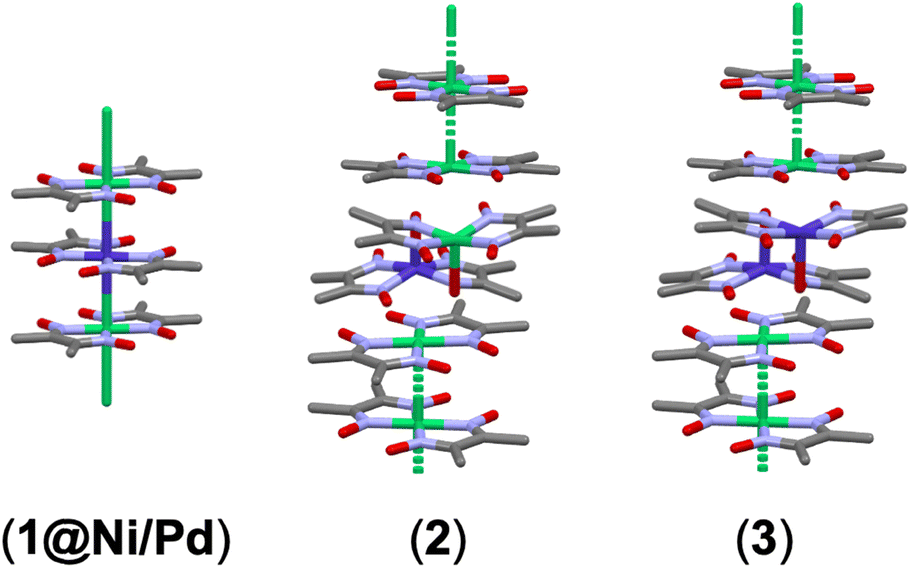 | ||
| Fig. 2 The three Co(II) species found in the Ni(Hdmg)2 and Pd(Hdmg)2 matrices in this study: Co(Hdmg)2 dispersed in Ni(Hdmg)2 or Pd(Hdmg)2 matrix (1@Ni/Pd), heterometallic dimer [Co(Hdmg)(μ-Hdmg)][M(Hdmg)(μ-Hdmg)] (M = Ni or Pd) (2), and its homometallic variations [Co(Hdmg)(μ-Hdmg)]2 (M = Ni or Pd) dimer (3). | ||
| Planar CoLn (low-temperature species) | Dimeric [CoLn][MLn] (M = Ni, Pd, Zn) (RT species) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| g x | g y | g z | A x | A y | A z | g x | g y | g z | A x | A y | A z | |
| Co@Ni | 4.75 | 0.65–0.85 (0.76) | 0.65–0.85 (0.75) | 1700 | −(100) | −(100) | 2.58 | 2.26 | 1.98 | 349 | 148 | 373 |
| Co@Pd | 4.22 | 1.2–1.7 (1.55) | 1.2–1.7 (1.34) | 1630 | −(145) | −(140) | 2.57 | 2.23 | 1.98 | 363 | 163 | 373 |
| Co(Hdmg)2 by NEVPT2 | 4.40 | 1.96 | 1.57 | — | — | — | ||||||
| Co(salen)25,26 | 3.805 | 1.660 | 1.740 | 872 | 155 | 90 | 2.63 | 2.31 | 2.01 | 340 | 138 | 357 |
| Co(a2phen)@Ni27 | 5.147 | 0.7 | 0.7 | — | — | — | ||||||
| Co(a2phen)@Pd27 | 4.546 | 1.33 | 1.33 | — | — | — | ||||||
Second, there is virtually no difference between the EPR parameters of Co@Ni and Co@Pd at RT (Table 1). This lack of matrix influence on the magnetic properties suggests that axial interactions of Co(II) remain barely changed in different host materials. Consequently, the corresponding Co(II) species is not, as opposed to the previous interpretation, a planar complex that would have different Co–M (M = Ni or Pd) interactions inside the Ni(Hdmg)2 or Pd(Hdmg)2 crystals, but a μ-O bridged heterometallic dimer [Co(Hdmg)(μ-Hdmg)][Ni/Pd(Hdmg)(μ-Hdmg)] (2) (Fig. 2). Additionally, an EPR silent component was suggested by SQUID (superconducting quantum interference device) magnetic measurements. This species is tentatively assigned as a μ-O bridged [Co(Hdmg)(μ-Hdmg)]2 dimer (3) due to remarkable similarities with analogous [Co(μ-salen)]2 dimers.
Experimental
Synthesis
Detailed descriptions of the synthesis and discussions of the associated issues are presented in the ESI.† Other experimental methods are also described in the ESI.† Essentially, we followed Schrauzer's original procedure15 and its variations modified for cobalt doping.20–22 We mixed Co(II) acetate with Ni(II) acetate in MeOH or EtOH under an inert atmosphere and then added dimethylglyoxime to obtain quickly precipitating solid solutions. This product was then washed thoroughly with MeOH to remove unwanted byproducts like [CoxNi1−x(Hdmg)2B2] (B = MeOH or EtOH), which are more soluble than [CoxNi1−x(Hdmg)2]. The resulting precipitate was then dried under a vacuum. Using this method, we have prepared 5% Co@Ni and 5% Co@Pd samples. To increase the Co(II) concentration, we modified this procedure by adding triethylamine, which is expected to deprotonate dimethylglyoxime and facilitate its coordination. This resulted in 11% Co@Pd samples. Elemental analysis confirmed the composition, and powder X-ray diffraction showed no crystalline impurities (Fig. S1–S4, ESI†). Inductively coupled plasma optical emission spectroscopy (ICP-OES) and X-ray fluorescence (XRF) were used to determine Co concentrations (Table S1, ESI†).Compared to the previous EPR studies,20,22–24 where the initial Co(II) to Ni(II) acetate ratios changed from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100 to 1:1000, our samples were prepared with a relatively high 1:1 ratio. This was necessary to achieve sufficiently high Co concentrations (at least above 1%) to further characterize the magnetic properties by SQUID measurements (ESI†) by outweighing the diamagnetic contribution.
:100 to 1:1000, our samples were prepared with a relatively high 1:1 ratio. This was necessary to achieve sufficiently high Co concentrations (at least above 1%) to further characterize the magnetic properties by SQUID measurements (ESI†) by outweighing the diamagnetic contribution.
Results
ESR spectroscopy
To investigate the spin state of cobalt doped in the diamagnetic Ni(Hdmg)2 and Pd(Hdmg)2 matrices, EPR spectra were measured at different temperatures from 3.8 K up to room temperature (RT) for 5% Co@Ni and 11% Co@Pd. The spectra at RT and a selected low temperature are shown in Fig. 3 and 4, respectively. The collective temperature dependency down to 3.8 K is shown in Fig. S5 and S8 (ESI†). | ||
| Fig. 3 The EPR spectrum of (a) 5% Co@Ni (b) 11% Co@Pd at room temperature (blue) along with a simulated spectrum (red). | ||
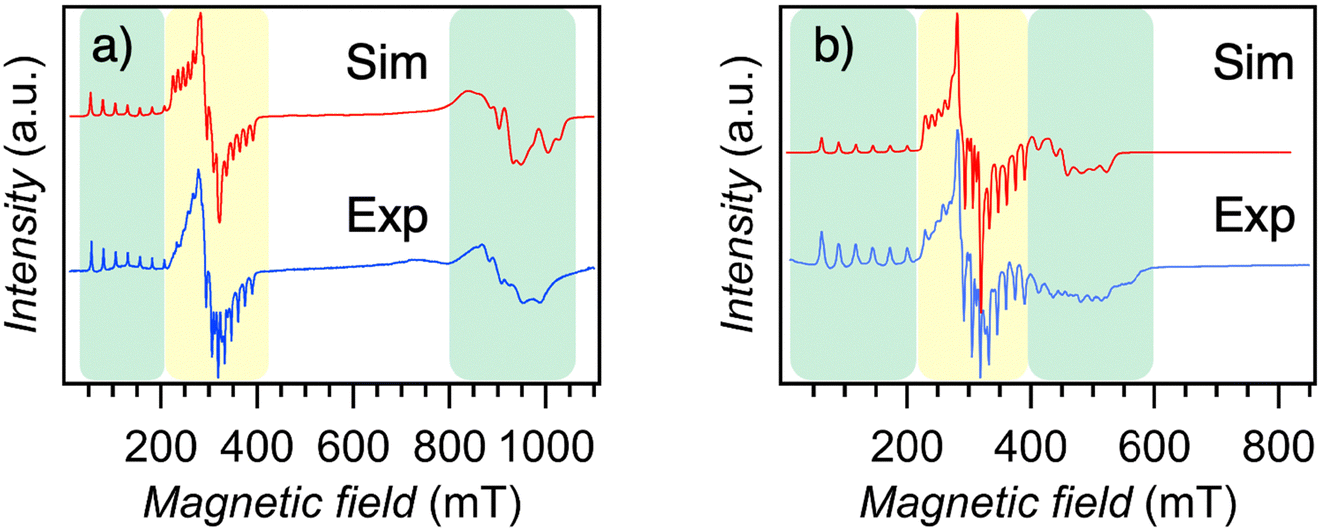 | ||
| Fig. 4 (a) The EPR spectra of 5% Co@Ni at 40 K (blue) and the simulated spectrum (red). The components in green and yellow originate from different species (1@Ni/Pd) and species (2), respectively. (b) The EPR spectra of 11% Co@Pd at 3.9 K (blue) and the simulated spectrum (red). | ||
Experimental spectra were fit to simulations based on a following spin Hamiltonian (eqn (1)) using the Easyspin program,28 where g and ACo are g and hyperfine tensors, respectively (assuming that both tensors are coaxial), B magnetic field, μB Bohr magnetons, S and I electron and nuclear spins of Co(II). In the following analysis, we use the same custom cartesian coordinate system as established by Hitchman and von Zelewsky,23,27 in which the x and y axes lie in the plane of the molecule while the z axis lies perpendicular to it.
| Ĥ = μBŜ·g·B + ηACo·Ŝ | (1) |
The spectra of Co@Ni and Co@Pd at room temperature can be simulated well with very similar parameters: gx = 2.58, gy = 2.26, gz = 1.98 and hyperfine splitting parameters ACo,x = 349 MHz, ACo,y = 148 MHz, ACo,z = 373 MHz for Co@Ni and gx = 2.57, gy = 2.23, gz = 1.98 and ACo,x = 363 MHz, ACo,y = 163 MHz, ACo,z = 373 MHz for Co@Pd (Table 1). The orientation of g and A is given based on the previous EPR studies20,23 and these g factors are essentially the same as those of the previous EPR studies of S = 1/2 “square planar” Co(Hdmg)2.20,22–24 However, the negligibly small changes of EPR parameters between Co@Ni and Co@Pd are inconsistent with the previous structural assignment, where Co(II) can easily be accessed by the axial metal ions (Ni2+ and Pd2+) and the EPR parameters can be readily perturbed by the difference between the metal ion (Ni or Pd) and the metal–metal distance (3.168Å for Ni(Hdmg)2 and 3.195 for Pd(Hdmg)2 at 123 K29). Based on the negligibly small difference between EPR parameters of Co@Ni and Co@Pd at RT (Table 1), here, we tentatively assign it to the μ-O bridged dimers [Co(Hdmg)(μ-Hdmg)][M(Hdmg)(μ-Hdmg)] (M = Ni(II) or Pd(II)) (2) (Fig. 2). The structural assignment will be further elaborated on in the Discussion section.
Low-temperature X-band CW-EPR spectra of 11% Co@Pd and 5% Co@Ni (Fig. 4 and Fig. S5–S8, ESI†) revealed the presence of additional fast-relaxing species that might have been overlooked by previous EPR measurements at 103 K.20 As the temperature decreases, new peaks of g > 4 appear in the range of 50–200 mT at ∼70 K for Co@Ni and at ∼120 K for Co@Pd (Fig. 4 and Fig. S5, S8, ESI†). These low-temperature peaks show 8 hyperfine splitting lines of electron spin localized on Co(II) with nuclear spins I = 7/2, confirming that the signals originate from doped Co(II). In addition, complex broad peaks were observed in the high field around 800–1000 mT (g ∼ 0.65–0.85) for Co@Ni and 400–600 mT (g ∼ 1.2–1.7) for Co@Pd. These low field peaks g > 4 disappeared along with the high field peaks g < 2, suggesting that both sets of peaks originate from the same species.
The fitting results of the spectra at a selected low temperature are plotted in Fig. 4. The obtained g and A parameters are compiled in Table 1. Unfortunately, we were unable to find an unambiguous set of all parameters. Only gx and Ax values for peaks at the low field could be well-fitted and established unequivocally due to a good separation from other peaks in the spectra (Table 1). For the y and z components (the high field peaks), the complex structure with small peaks on the broad feature expanding over 200 mT makes the optimization difficult as they are convoluted and mostly hidden under the broad feature. Therefore, only the possible ranges of g factors are shown in Table 1 based on the magnetic field of the peaks (Fig. 4 and Fig. S9, ESI†). In the simulation spectra in Fig. 4, a set of selected simulation parameters (Table 1) that rather well reflect the main features of the experimental spectra were used. Reproducing the small peaks in the y and z components of Co@Pd will probably require additional parameters due to superhyperfine coupling with 105Pd (I = 5/2 22.33% natural abundance) nuclear spins (Fig. S10, ESI†).
Due to the fast relaxation of these low-temperature species and a relatively large g factor as high as 4.75, we suspected a high spin S = 3/2 Co(II) impurity at first. However, it is unlikely because such high spin Co(II) complexes are rarely observable above the liquid nitrogen temperature.30–33 Moreover, obtained average g factors are much closer to the free electron value (gav = 2.0–2.2 for Co@Ni and gav = 2.2–2.5 for Co@Pd) (Table 1), compared to the typical average effective g value of high spin Co(II) (∼4.3).9,30,31,34,35 Therefore, we assign the spin of these low-temperature species to S = 1/2. As further discussed in detail in Discussion, we attribute these species to planar Co(Hdmg)2 dispersed in Ni(Hdmg)2 or Pd(Hdmg)2 (1) (Fig. 2). This assignment was further supported by the theoretical calculations of g factors by CASSCF (complete active space self-consistent field) with the active space of seven electrons on the five 3d-orbitals (CAS(7,5) space). The theoretically calculated g factors for S = 1/2 square planar Co(Hdmg)2 (Fig. S13, ESI†) gave a qualitatively consistent result with the experimentally obtained g factors for the low-temperature species (Exp.: g = 4.75, 0.65–0.85, 0.65–0.85 for Co@Ni and g = 4.22, 1.2–1.7, 1.2–1.7 for Co@Pd, Theory: gx = 4.40, gy = 1.96, gz = 1.57). The theoretical prediction of g factors for 3d metals is known to be notoriously difficult and including only metal-based orbitals in the active space tends to overestimate the g values even after dynamic electron correlation is included using the NEVPT2 method.36 Our results largely overestimated gy and gz but may be improved in the future by the extension of the active space with the second d orbital shell.
SQUID magnetic measurements
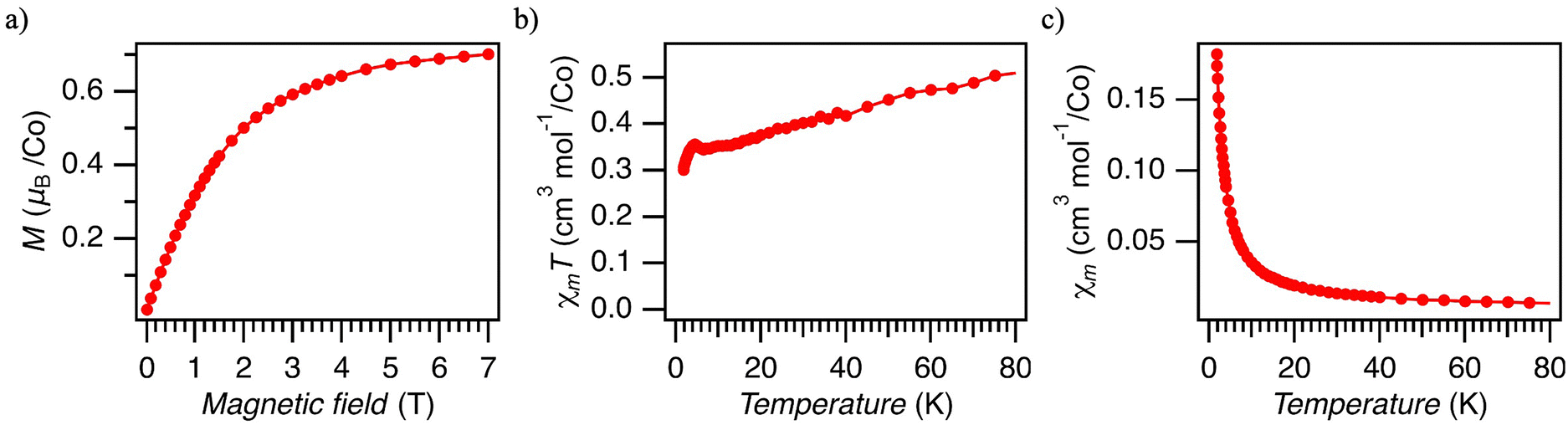 | ||
| Fig. 5 DC magnetic properties of 5% Co@Ni. (a) The magnetization at 1.8 K. The red circles are measured points, and the solid line is a visual guide. (b) Temperature dependence of the DC susceptibility (χmT) at 1000 Oe. (c) Temperature dependence of the DC susceptibility (χm) at 1000 Oe. | ||
The magnetization of 5% Co@Ni measured at 1.8 K approaches saturation at 7 T, reaching ∼0.7μB per Co atom (Fig. 5a), as calculated by using the Co concentration estimated by XRF and ICP-OES (Table S1, ESI†). This value is significantly smaller than the expected value of 1.05–1.15 for S = 1/2 with gav = 2.1–2.3 (the range expected for species (1@Ni) and dimer (2) based on EPR). We attribute the reduced magnetization to the presence of antiferromagnetic interaction, which we identify as originating from antiferromagnetically coupled [Co(Hdmg)(μ-Hdmg)]2 dimers (3). This conclusion is based on the magnetic studies on the pristine Co(Hdmg)2 (Fig. S18–S22, ESI†). Pristine Co(Hdmg)2 shows a very similar antiferromagnetic exchange coupling interaction (J = −21.8 cm−1) with analogous Co(II) salen dimer [Co(salen)]2 (salen = N,N′-bis(salicylaldehyde)ethylenediimine) (J = −21 cm−1).37,38 The similarity of this complex to the Co(Hdmg)2 was also noted by the previous EPR study of Co@Ni.20 Though the precise geometry of the dimerized Co(Hdmg)2 could not be conclusively determined due to the lack of crystallinity, the similarity in their magnetic properties suggests that the pristine Co(Hdmg)2 forms a dimeric species [Co(Hdmg)(μ-Hdmg)]2 similar to [Co(salen)]2. Furthermore, the presence of the heterometallic dimer (2), as revealed by EPR, reinforces the presence of analogous [Co(Hdmg)(μ-Hdmg)]2. The possibility of diamagnetic oxidized Co(III) impurities is ruled out by control measurements under an Ar atmosphere in a sealed NMR tube (Fig. S23, ESI†).
Using the magnetization value at 1.8 K (∼0.7μB) and the average g factors of the dimer (2) (gav = 2.27), the amount of the antiferromagnetically coupled dimer (3) can be estimated to be roughly 38%. Although the g factors of species (1@Ni) are not precisely determined based on EPR data, gav values of species (1@Ni) are similar to species (2), making it difficult to determine the composition of species (1@Ni) and dimer (2) based on this approach. Similar magnetization values were obtained for 5% Co@Pd and 11% CoPd (Fig. S16 and S17, ESI†) as well, indicating that the relative amount of homometallic dimer (2) remains the same regardless of the Co(II) doping concentration.
The temperature dependence of the magnetic susceptibility for 5% Co@Ni is presented in Fig. 5b and c. The temperature is limited only up to 80 K due to a large diamagnetic contribution from the host material and sample holder. The χmT value decreases as the temperature drops down to 6 K, and then shortly rises, reaching a small maximum at 4 K (Fig. 5b). Below 4 K, χmT decreases again, reaching the χmT value of ∼0.3 cm3 mol−1 K at 1.8 K. The decrease of the χmT signal is in line with antiferromagnetic interactions. Similar behavior could be also explained by zero-field splitting; however, S > 1/2 is required and we did not find any indication of high spin (HS) Co(II) impurities by using any other method. HS Co(II) complexes are also chemically very unlikely for cobaloximes(II) prepared in alcohol solutions. Cobaloximes(II) axially coordinated by solvent show small g factors and low spin states,19,23 also supported by the pure Co(Hdmg)2 experiment (ESI†).
The presence of antiferromagnetic exchange interactions in a material can, in general, be inferred from the existence of maximum in χm(T) vs. T curves. The absence of such a feature in χm(T) in our materials (Fig. 5c) can be explained by the presence of other magnetic species like (1@Ni) and (2) which effectively mask the maximum. Indeed, the simulated χm(T) vs. T with a mixture of S = 1/2 Co(II) and antiferromagnetically coupled Co(II) shows no maximum when the dimer composition is less than 60% (Fig. S25, ESI†). As discussed, the dimer (3) consists of roughly 40% based on the magnetization value at 7 T at 1.8 K. Therefore, the absence of maximum in χm does not contradict the presence of antiferromagnetic interaction.
The small maximum of χmT at 4 K suggests another intermolecular magnetic interaction. The more concentrated 11% Co(II)@Pd shows a significantly larger peak than 5% Co(II)@Ni/Pd (Fig. S16, ESI†). This observation is reasonable because a more concentrated sample has a higher probability of having intermolecular interactions with neighboring Co(II). Though the exact origin of this interaction remains elusive, it indicates an inhomogeneous distribution of Co(II) in the Ni and Pd matrices, likely with structural disorders inferred from the absence of crystalline impurities in PXRD. The inhomogeneous nature of our samples is further supported by the presence of homometallic Co(II) dimer (3) and AC susceptibility (vide infra). In such a highly defected amorphous phase with high Co concentration, various types of exchange coupling between different species (1@Ni/Pd) – (3) in different environments and distances from each other are possible.
AC susceptibility
The field-dependent AC susceptibility of 5% Co@Ni, 5% Co@Pd, and 11% Co@Pd at 1.8 K is shown in Fig. 6. The field dependence of the relaxation times was obtained by fitting the experimental data to the generalized Debye model by using the Relacs program.39 The Cole–Cole plots are found in the ESI† (Fig. S26–S30, ESI†). The two species with essentially the same Co concentration, 5% Co@Ni and 5%@Pd, show a very similar field dependence of relaxation times (Fig. S31, ESI†). A maximum at around 10000 Hz at 0 T was observed in the out-of-phase  component. Upon field increase, the maximum shifts to lower frequencies and it becomes mostly field-independent above 3000 Oe at around 400 Hz. Similar behavior was observed for 5% Co@Pd. This indicates that the metal of the diamagnetic matrix (Ni(II) or Pd(II)) has a minimum impact on the behavior of this relaxation pathway of Co(II).
component. Upon field increase, the maximum shifts to lower frequencies and it becomes mostly field-independent above 3000 Oe at around 400 Hz. Similar behavior was observed for 5% Co@Pd. This indicates that the metal of the diamagnetic matrix (Ni(II) or Pd(II)) has a minimum impact on the behavior of this relaxation pathway of Co(II).
 | ||
| Fig. 6 AC susceptibility of 5%Co@Ni (a) and (b), 11% Co@Pd (c) and (d), 5% Co@Pd (e) and (f) at 2 K. The circles mark the experimental data points. The solid lines are the fits to the generalized Debye model. | ||
For 11% Co@Pd, the maximum of 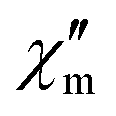 occurs at frequencies generally smaller than those of 5% Co@Ni or Co@Pd at around 10–1000 Hz across different fields, indicating that a more concentrated sample has longer relaxation times. This is counterintuitive as the spin–spin interaction typically shortens the relaxation time of paramagnetic ions. One possible explanation for this behavior is the spin-glass state and a similar concentration dependence was observed previously.40 Furthermore, evaluation of the so-called Mydosh parameter and additional results of magnetic experiments discussed in ESI† (Fig. S32–S38, ESI†) suggest that slow spin relaxation originates from the spin glass phase. While our sample exhibits properties like a spin-glass state, its manifestation occurs only at a very low temperature (∼4 K) under a small magnetic field (<1000 Oe). Therefore, our DC magnetic measurements under a high field can mainly be interpreted only by the S = 1/2 model. Additionally, the spin-glass phase has a negligible effect on the EPR spectra, as the measurement temperature is high enough to overcome its magnetic interactions.
occurs at frequencies generally smaller than those of 5% Co@Ni or Co@Pd at around 10–1000 Hz across different fields, indicating that a more concentrated sample has longer relaxation times. This is counterintuitive as the spin–spin interaction typically shortens the relaxation time of paramagnetic ions. One possible explanation for this behavior is the spin-glass state and a similar concentration dependence was observed previously.40 Furthermore, evaluation of the so-called Mydosh parameter and additional results of magnetic experiments discussed in ESI† (Fig. S32–S38, ESI†) suggest that slow spin relaxation originates from the spin glass phase. While our sample exhibits properties like a spin-glass state, its manifestation occurs only at a very low temperature (∼4 K) under a small magnetic field (<1000 Oe). Therefore, our DC magnetic measurements under a high field can mainly be interpreted only by the S = 1/2 model. Additionally, the spin-glass phase has a negligible effect on the EPR spectra, as the measurement temperature is high enough to overcome its magnetic interactions.
XANES (X-ray absorption near edge structure) spectroscopy
In addition to the insights into the electronic structure and the coordination geometry obtained from EPR spectroscopy and SQUID, XANES (X-ray absorption near edge structure) spectroscopy can further elucidate the local environment of Co(II) in the Ni/Pd(Hdmg)2 matrix, providing complementary confirmation of the conclusions drawn from the EPR analysis. The XANES spectra of Co(II) in Ni/Pd(Hdmg)2 are shown in Fig. S39 (ESI†).Square planar Co(II) complexes typically exhibit a strong peak around 7716 eV, which is attributed to either 1s–4p + shakedown17,41 or a direct 1s–4p transition.41 Notably, the intensity of this feature significantly decreases as the Co(II) geometry shifts from square planar to square pyramidal or octahedral. This characteristic feature for square planar complexes is well-documented in cobalt(II) phthalocyanine with/without pyridine,41 and the absence of this peak is also observed in the pentacoordinate dimeric [Co(salen)]2 complex,42 a compound analogous to our (2) and (3).
The weak intensity of this peak in spectra of Co@Ni and Co@Pd samples indicates that the majority of Co(Hdmg)2 in our sample adopts a non-planar geometry, consistent with a square pyramidal geometry of Co(II) in species (2) and (3) as inferred using EPR analysis and magnetic measurements. This does not exclude the presence of Co(Hdmg)2 (1@Ni/Pd) in a planar geometry suggested by EPR. Rather, the small intensity indicates that this planar Co(Hdmg)2 (1@Ni/Pd) is minor compared to the species (2) and (3). This conclusion is also consistent with the magnetic data that show that roughly 40% of the sample is species (3).
Discussion
EPR spectroscopy – RT components
As already mentioned, the species observed by CW-EPR at RT was previously considered as Co(Hdmg)2. However, the striking similarity of EPR parameters between Co(II) doped in Pd(Hdmg)2 and Ni(Hdmg)2 matrices is at first puzzling; the metal–metal interactions along vertical stacks of M(Hdmg)2 complexes (M = Ni, Pd or Co) should certainly be different due to changes in metal–metal distances between Ni(Hdmg)2 and Pd(Hdmg)2, and much more diffuse character of the heavier Pd metal orbitals. Raynor et al. encountered the same issue in their study of Co(II) doped in Ni(Hdmg)2.20 The similarity of hyperfine A and g parameters of Co(II) in Ni(Hdmg)2 with those of [Co(μ-salen)]2 spectra (Table 1) led them to conclude that Co(Hdmg)2 is strongly influenced by metal–metal interactions with Ni(II). However, as both Co@Ni and Co@Pd exhibit nearly identical EPR spectra, (Fig. 3, Fig. S9, ESI†), the Co–M interaction is unlikely. Other possibilities of axial interaction are nitrogen or oxygen on the glyoxime ligand. Since the interaction with N atom (I = 3/2) will result in the superhypefine splittings, as observed in the pyridine adduct of Co(salen),26 the lack of superhyperfine splittings in the EPR spectra suggests the interaction with O. Such metal interaction with the glyoxime oxygen is reported in the dimeric form of Cu(Hdmg)2 and Ni(Hdmg)2.43 In addition to the possible dimers formation suggested by Cu(Hdmg)2 and Ni(Hdmg)2,43 the similarity of EPR parameters of this RT species to those of μ-O bridged [Co(μ-salen)]2 dimers further suggests a dimeric form of [Co(Hdmg)(μ-Hdmg)][M(Hdmg)(μ-Hdmg)] (M = Ni2+ or Pd2+) (3) for the RT species, where Co(II) ions are axially coordinated by bridging oxygen atoms.In Co(Hdmg)2B (B = monodentate ligand) complexes, the donor atom pulls the Co(II) ion above the molecular plane, simultaneously tilting both glyoxime ligands in the opposite direction of the donor ligand.20 The coordination of the oxygen atom to the glyoxime ligand in the dimers (2) and (3) is likely to induce a structural distortion similarly. This geometry of the glyoxime ligands outward from the complex introduces additional steric hindrance, further hampering interactions along the axis of cobaloxime(II).
In addition to the minimal interaction with the metal ions in the diamagnetic matrix (Ni(II) or Pd(II)), the invariance of EPR parameters of the dimer between Co@Ni and Co@Pd compared to the low-temperature species is also likely to be a result of its electronic structure. The theoretical analysis by Zelewsky and Hitchman27,44 provides diagrams of calculated g and hyperfine parameter values in relation to the separation between 2A2 (dyz) and 2A1 (dz2) states. In these diagrams, one can easily see that the EPR parameter change with respect to the energy separation change is much smaller around the g factors (gx ∼2.6) found for this dimer than the g factors (gx > 4).
EPR spectroscopy – low-temperature component
The negligibly small change of the g factor of the RT species (3) in the Ni and Pd matrices sharply contrasts with the behavior of the low-temperature species (1@Ni/Pd), which shows very high sensitivity of the EPR parameters to metal ions in the host matrix. A similar high sensitivity of EPR parameters to the diluting host matrix is known for some planar Schiff base Co(II) complexes, which are structurally very similar to Co(Hdmg)2 systems.27 A good illustration of that is Co(II)(a2phen) ((E,E)-diethyl 2,2′-[1,2-phenylenebis-(iminomethylidyne)]bis[3-oxobutanoato](2-)-N,N′,O3,O3′]cobalt(II)) in the Ni(II) based matrix g1 = 5.147, g2 = 0.7, g3 = 0.7 and g1 = 4.546, g2 = 1.33, g3 = 1.33 in the Pd(II) based matrix.27 The large g factors of 4.75 and 4.22 for the low-temperature species of Co@Ni and Co@Pd as S = 1/2 Co(II) are justified by the theoretical analysis by Zelewsky and Hitchman,27,44 which we introduced above, and are useful for intuitive understanding of the range of possible parameters and the high sensitivity to perturbations in those complexes. They predict the maximum possible gx to be 5.46 when gy, gz approach zero. Moreover, our EPR parameters align well with their predicated plot of EPR parameters. Therefore, based on the g factors and the EPR parameter change observed in the different matrixes, we assign the low-temperature species to a planar Co(Hdmg)2 (1@Ni/Pd).The high sensitivity of EPR parameters originates from the electronic structure of Co(II) in the square planar ligand field. The electronic structure of Schiff base Co(II) complexes in a tetradentate planar ligand field, such as Co(salen) (C2v symmetry), is well documented27 and their analysis also applies to cobaloximes(II) (approximate D2h point group), as both C2v and D2h point groups are described by the same rhombic crystal field parameters.45 To briefly summarize their findings, those complexes are characterized by a set of four 3d molecular orbitals lying close to each other in energy because the fifth orbital (3dxy) lies much higher in energy because it points directly toward the equatorial donor atoms. This orbital splitting gives rise to a large energy separation and consequently weak coupling between ground and excited states of 3d8 and 3d9 complexes (Cu(II) and Ni(II)). For 3d7 complexes like Co(II); however, the electronic structure results in one unpaired electron in one of the four closely spaced orbitals. This leads to multiple low-lying doublet and quartet states that are strongly coupled via spin–orbit interaction, producing g values that deviate significantly from the free-electron value (ge = 2.0023) and making them highly sensitive to structural perturbations.
This high sensitivity to structural changes may be responsible for strongly broadened peaks of species (1@Ni/Pd) (Fig. 4). Our samples were precipitated very quickly, and one can expect high structural defects and disorder. We believe that Co(Hdmg)2 in our Co@Ni and Co@Pd samples exist in at least a few environments that differ slightly, making it difficult to reproduce the EPR spectra well via simulation.
It is well known that the electronic spectra of M(Hdmg)2 (M = Ni, Pd, Pt) crystals are highly sensitive to external pressure, owing to the modulation of metal–metal interactions. For example, the absorption of Pd(Hdmg)2 at 6.2 GPa is at ∼ 460 nm (yellow) while under 0.1 GPa ∼ it is 700 nm (green).46 Here, by the comparison of species (1@Ni) and (1@Pd), we have shown that magnetic parameters are also prone to changes in metal–metal interactions. In this context, our finding paves a way to controlling the magnetic properties of Co(Hdmg)2 through g factor changes by external pressure. This can be utilized in sensing applications and warrants further investigation. Furthermore, the sensitivity of EPR parameters to external factors, such as the diamagnetic matrix and pressure combined with the versatility of glyoxime-based ligands, could enable systematic investigations into the dependence of T2 on EPR parameters at clock transitions.
Magnetic data
Our magnetic measurements showed the presence of homometallic dimeric [Co(Hdmg)(μ-Hdmg)]2 (3) and various intermolecular interactions in our sample. For example, we have shown that slow spin relaxation at low temperatures in 5% Co@Ni, as well as 5% and 11% Co@Pd samples comes from the spin glass phase. This behavior is most likely due to the non-uniform dispersion of Co within the sample, as evidenced by a large amount (ca. 38%) of dimers (3).The formation of species (2) and (3) in the Ni/Pd matrix can be rationalized by the strong tendency to form axial coordination of Co(Hdmg)2 and the presence of a dimeric phase of Ni(Hdmg)2. The crystal structure of Ni(Hdmg)2, though is predominantly reported as a pseudo-1D chain structure as in Fig. 1 (space group: Ibam), a dimeric phase characterized by μ-O bridges through the oxime oxygen has also been observed (space group: P21/c).43 This P21/c crystal was prepared by refluxing Ni(Hdmg)2 in DMF and then slowly cooling the filtrate solution. Since the formation of this phase requires slow crystallization at elevated temperatures, it likely represents the thermodynamically favored structure, in contrast to the bulk Ibam phase, which forms via rapid precipitation.
Given that Co(Hdmg)2 exhibits a strong tendency to dimerize, supported by both pristine experiments and previous reports,12,13,17 the presence of Co(II) in the reaction mixture during the synthesis of Co@Ni/Pd may induce partial conversion of Ni(Hdmg)2 into the dimer form. The absence of PXRD diffraction peaks for this phase suggests that species (2) and (3) are likely occluded in a highly defective manner. The inhomogeneous distribution of Co(II), driven by its strong preference for axial coordination and the structural disorder supported by the absence of a crystalline phase other than the Ibam phase in PXRD, also provides an explanation for the observed spin glass behavior, which often appear in doped systems of moderate concentration of magnetic species due to competing exchange interactions, spin frustration or structural disorder.
Conclusions
Motivated by quantum technology development, we have prepared and studied Co(II) doped into Pd(Hdmg)2 and Ni(Hdmg)2. A comprehensive study of the magnetic properties of Co@Ni/Pd revealed a mixture of at least three different Co(II) species: Co(Hdmg)2 dispersed in Ni/Pd(Hdmg)2 (1@Ni/Pd), [Co(Hdmg)(μ-Hdmg)][M(Hdmg)(μ-Hdmg)] (M = Ni or Pd) species (2) and its bimetallic variations of antiferromagnetically coupled [Co(Hdmg)(μ-Hdmg)]2 species (3).Our findings challenge the previous studies on Co@Ni, which identified only species (2) and often misattributed it to monomeric planar cobaloxime(II) (1@Ni).20,22,23 The unexpected mixture of three different Co(II) complexes with more dominance of axially coordinated complexes species (2) and (3) in the planar Ni/Pd(Hdmg)2 matrix is due to the strong propensity of Co(II) to form axial coordination. This is evidenced by the lack of a crystal structure of pristine Co(Hdmg)2 despite over 600 structures with some axial coordination in the CSD database and the reported ease of forming dimers13,17 or trimers.18
Author contributions
The manuscript was written by Y. S., M. A., and edited by M. W., and M. Y. Experiments were performed by Y. S. and then analyzed by Y. S. and M. A. The project was conceptualized by M. W. and M. Y. XANES measurements were performed by T. Y. Variable temperature EPR spectroscopy was performed by H. T, T. Y., and K. S. Theoretical calculations were performed by R. I. All authors have given approval to the final version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The X-ray absorption fine structure (XAFS) measurements were carried out at the BL36XU beamline on SPring-8. M. Y. is thankful for the 111 project (B18039) from China. This work was partially supported by the JSPS KAKENHI (No. JP23K23203, JP20H05870, JP23K26618, JP23KJ0169, and JP23K13761, JP25K01656). R. I. gratefully acknowledges funding from the Japan Research Institute of Industrial Science (Fukuyama) and the Central Research Institute of Fukuoka University (Grant Number GR2303). The OMU group acknowledges financial support from the office of Naval Research - Global (Grant No. N62909-23-1-2079).References
- M. J. Graham, J. M. Zadrozny, M. S. Fataftah and D. E. Freedman, Chem. Mater., 2017, 29, 1885–1897 CrossRef CAS.
- M. J. Amdur, K. R. Mullin, M. J. Waters, D. Puggioni, M. K. Wojnar, M. Gu, L. Sun, P. H. Oyala, J. M. Rondinelli and D. E. Freedman, Chem. Sci., 2022, 13, 7034–7045 RSC.
- M. Atzori, E. Morra, L. Tesi, A. Albino, M. Chiesa, L. Sorace and R. Sessoli, J. Am. Chem. Soc., 2016, 138, 11234–11244 CrossRef CAS PubMed.
- M. Atzori, L. Tesi, E. Morra, M. Chiesa, L. Sorace and R. Sessoli, J. Am. Chem. Soc., 2016, 138, 2154–2157 CrossRef CAS PubMed.
- M. S. Fataftah, M. D. Krzyaniak, B. Vlaisavljevich, M. R. Wasielewski, J. M. Zadrozny and D. E. Freedman, Chem. Sci., 2019, 10, 6707–6714 RSC.
- T. Yamabayashi, M. Atzori, L. Tesi, G. Cosquer, F. Santanni, M.-E. Boulon, E. Morra, S. Benci, R. Torre, M. Chiesa, L. Sorace, R. Sessoli and M. Yamashita, J. Am. Chem. Soc., 2018, 140, 12090–12101 CrossRef CAS PubMed.
- M. Shiddiq, D. Komijani, Y. Duan, A. Gaita-Ariño, E. Coronado and S. Hill, Nature, 2016, 531, 348–351 CrossRef CAS PubMed.
- S. Giménez-Santamarina, S. Cardona-Serra, J. M. Clemente-Juan, A. Gaita-Ariño and E. Coronado, Chem. Sci., 2020, 11, 10718–10728 RSC.
- J. M. Zadrozny, A. T. Gallagher, T. D. Harris and D. E. Freedman, J. Am. Chem. Soc., 2017, 139, 7089–7094 CrossRef CAS PubMed.
- M. Atzori and R. Sessoli, J. Am. Chem. Soc., 2019, 141, 11339–11352 CrossRef CAS PubMed.
- C.-J. Yu, S. von Kugelgen, D. W. Laorenza and D. E. Freedman, ACS Cent. Sci., 2021, 7, 712–723 CrossRef CAS PubMed.
- A. Rockenbauer, É. Budó-Záhonyi and L. I. Simándi, J. Chem. Soc., Dalton Trans., 1975, 1729–1737 RSC.
- G. N. Schrauzer and L.-P. Lee, J. Am. Chem. Soc., 1968, 90, 6541–6543 CrossRef CAS PubMed.
- M. Bacchi, G. Berggren, J. Niklas, E. Veinberg, M. W. Mara, M. L. Shelby, O. G. Poluektov, L. X. Chen, D. M. Tiede, C. Cavazza, M. J. Field, M. Fontecave and V. Artero, Inorg. Chem., 2014, 53, 8071–8082 CrossRef CAS PubMed.
- G. N. Schrauzer and R. J. Windgassen, Chem. Ber., 1966, 99, 602–610 CrossRef CAS PubMed.
- D. Dolui, S. Khandelwal, P. Majumder and A. Dutta, Chem. Commun., 2020, 56, 8166–8181 RSC.
- M. Rangel, A. Leite, A. Silva, B. de Castro, W. Schlindwein and L. Murphy, J. Organomet. Chem., 2014, 760, 11–18 CrossRef CAS.
- D. C. Lacy, G. M. Roberts and J. C. Peters, J. Am. Chem. Soc., 2015, 137, 4860–4864 CrossRef CAS PubMed.
- G. N. Schrauzer and L. P. Lee, J. Am. Chem. Soc., 1970, 92, 1551–1557 CrossRef CAS PubMed.
- B. de Castro, M. Rangel and J. B. Raynor, J. Chem. Soc., Dalton Trans., 1990, 3311–3318 RSC.
- C. J. Winscom, W. Lubitz, H. Diegruber and R. Möseler, Stud. Surf. Sci. Catal., 1982, 12, 15–21 CrossRef CAS.
- W. Lubitz, C. J. Winscom, H. Diegruber and R. Möseler, Z. Naturforsch., A:Phys. Sci., 1987, 42, 970–986 CrossRef CAS.
- M. Baumgarten, W. Lubitz and C. J. Winscom, Chem. Phys. Lett., 1987, 133, 102–108 CrossRef CAS.
- S. Deshpande, D. Srinivas and P. Ratnasamy, J. Catal., 1999, 188, 261–269 CrossRef CAS.
- A. von Zelewsky and H. Fierz, Helv. Chim. Acta, 1973, 56, 977–980 CrossRef CAS.
- C. Busetto, F. Cariati, P. Fantucci, D. Galizzioli and F. Morazzoni, J. Chem. Soc., Dalton Trans., 1973, 1712–1716 RSC.
- C. Daul, C. W. Schläpfer and A. Von Zelewsky, The electronic structure of cobalt(II) complexes with schiff bases, and related ligands, Springer Berlin Heidelberg, 1979, pp. 129–171 Search PubMed.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed.
- A. Sugimoto, S. Kusumoto, M. Nakaya, Y. Sekine, L. F. Lindoy and S. Hayami, CrystEngComm, 2022, 24, 4656–4660 RSC.
- J. R. Pilbrow, Transition ion electron paramagnetic resonance, Oxford University Press, 1990 Search PubMed.
- L. Banci, A. Bencini, C. Benelli, D. Gatteschi and C. Zanchini, Struct. Bond., 2007, 37–86 Search PubMed.
- R. Boca, H. Elias, W. Haase, M. Huber, R. Klement, L. Muller, H. Paulus, I. Svoboda and M. Valko, Inorg. Chim. Acta, 1998, 278, 127–135 CrossRef CAS.
- P. H. Haffner and J. E. Coleman, J. Biol. Chem., 1973, 248, 6630–6636 CrossRef CAS PubMed.
- G. Magri, A. Folli and D. M. Murphy, Eur. J. Inorg. Chem., 2022,(9), e202101071 CrossRef CAS.
- A. Ozarowski, H. M. Lee and A. L. Balch, J. Am. Chem. Soc., 2003, 125, 12606–12614 CrossRef CAS PubMed.
- S. K. Singh, M. Atanasov and F. Neese, J. Chem. Theory Comput., 2018, 14, 4662–4677 CrossRef CAS PubMed.
- K. S. Min, J. Arthur, W. W. Shum, M. Bharathy, H.-C. zur Loye and J. S. Miller, Inorg. Chem., 2009, 48, 4593–4594 CrossRef CAS PubMed.
- K. Murray, A. Vandenbergen, B. Kennedy and B. West, Aust. J. Chem., 1986, 39, 1479–1493 CrossRef CAS.
- M. Liberka, M. Zychowicz, W. Zychowicz and S. Chorazy, Chem. Commun., 2022, 58, 6381–6384 RSC.
- G. Eiselt, J. Kötzler, H. Maletta, D. Stauffer and K. Binder, Phys. Rev. B: Condens. Matter Mater. Phys., 1979, 19, 2664–2676 CrossRef CAS.
- Y. Liu, A. Deb, K. Y. Leung, W. Nie, W. S. Dean, J. E. Penner-Hahn and C. C. L. McCrory, Dalton Trans., 2020, 49, 16329–16339 RSC.
- C. Johnson, B. Long, J. G. Nguyen, V. W. Day, A. S. Borovik, B. Subramaniam and J. Guzman, J. Phys. Chem. C, 2008, 112, 12272–12281 CrossRef CAS.
- J. Wan, Y.-F. Miao, X.-M. Li and S.-S. Zhang, Asian J. Chem., 2010, 18, 1600–1606 Search PubMed.
- M. A. Hitchman, Inorg. Chem., 1977, 16, 1985–1993 CrossRef CAS.
- E. Belin-Ferré, E. Bauer and M. Rotter, Book Ser. Complex Met. Alloys, 2009, 183–248 Search PubMed.
- I. Shirotani, K. Suzuki and T. Yagi, Proc. Jpn. Acad., Ser. B, 2006, 68, 57 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cp00629e |
| This journal is © the Owner Societies 2025 |