
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExperimental studies of high-temperature thermal dissociation of iso-propanol†
John H.
Kim
 *a,
Keunsoo
Kim
b,
Qinghui
Meng
a,
Ashish
Sutar
c,
Margaret S.
Wooldridge
ad and
Robert S.
Tranter
b
*a,
Keunsoo
Kim
b,
Qinghui
Meng
a,
Ashish
Sutar
c,
Margaret S.
Wooldridge
ad and
Robert S.
Tranter
b
aDepartment of Mechanical Engineering, University of Michigan, Ann Arbor, MI, USA. E-mail: johnhw@umich.edu
bChemical Sciences and Engineering Division, Argonne National Laboratory, Lemont, IL, USA
cDepartment of Mechanical Engineering, University of Illinois, Chicago, IL, USA
dDepartment of Aerospace Engineering, University of Michigan, Ann Arbor, MI, USA
First published on 19th March 2025
Abstract
iso-Propanol is an important biofuel for transportation and industrial applications, as well as a canonical compound for understanding alcohol reaction chemistry. There are few studies of the thermal decomposition of iso-propanol, hence the objective of this work was to investigate the mechanism of iso-propanol pyrolysis at high-temperature conditions. Shock tube studies were used to investigate the thermal decomposition of iso-propanol and identify the dominant reaction pathways and the intermediate species present in the temperature range of 1395–2053 K and at pressures of 0.3–4 bar. Time-resolved mass spectra were obtained from pyrolysis experiments in shock tube/mass spectrometer apparatuses using electron impact ionization or synchrotron vacuum ultraviolet photoionization. The relative concentrations of the intermediate species were used to identify the dominant reaction channels. iso-Propanol dissociated by water loss to propene and by elimination of a methyl radical yielding a 1-hydroxyethyl radical at the conditions studied. The time-dependent mass spectra allowed secondary products to be identified along with the orders of appearance. Notably, peaks at m/z = 50 and 52 were observed providing insight into secondary reactions. These features have not been previously reported in propanol-pyrolysis literature.
1. Introduction
Biofuels have attracted considerable attention from the energy sector. The U.S. Department of Energy's Co-Optimization of Fuels and Engines program identified four alcohols (ethanol, methanol, iso-propanol, and n-propanol) in a list of the ten most promising biofuels with the potential to deliver high engine efficiency and other benefits.1 Methanol and ethanol have been blended with gasoline for many years. However, longer chain alcohols have many desirable characteristics as alternative biofuels including larger lower heating values, favorable boiling points, better miscibility with conventional fuels, and lower corrosivity in engines.2Branched alcohols have some advantages over linear alcohols as fuels and additives, including low pour point, biodegradability, and greater solubility in hydrocarbon fluids.3 iso-Propanol, the smallest branched alcohol, can be produced commercially from bio-feedstocks or petrochemical ones providing flexibility in supply4 and is a promising low carbon fuel or fuel additive.5 Blends of iso-propanol and gasoline have been studied in engines6–9 and showed reductions in pollutants including CO, unburned hydrocarbons (HC), and NOx. The studies also showed that addition of iso-propanol reduced knocking compared with n-butanol when advanced combustion strategies were used.8 However, there are some shortcomings to using iso-propanol including increased carbonyl emissions.10,11
In contrast with other alcohols, and despite its industrial scale potential, there are few chemical kinetic studies of the pyrolysis12–19 or oxidation17–23 of iso-propanol. Bui et al. indicate the primary thermal decomposition reactions of iso-propanol are H2O elimination, reaction (R1),
| iC3H7OH ↔ C3H6 + H2O ΔHr,298K = 12.1 kcal mol−1 | (R1) |
and C–C bond scission, (R2),
| iC3H7OH ↔ sC2H4OH + CH3 ΔHr,298K = 87 kcal mol−1 | (R2) |
Three prior experimental studies have focused on iso-propanol pyrolysis at conditions relevant to the current work. Heyne et al.16 used a variable pressure flow reactor to study iso-propanol dissociation at 12.5 atm and 976–999 K. Heyne et al. measured k1 and k2. While k2 compared favorably with the calculated value from Bui et al., the Heyne et al. values for k1 were approximately four times faster than the theoretical predictions of Bui et al.15 Li et al.18 studied the pyrolysis of iso-propanol in a flow tube over 900–1350 K and 0.04–1 atm with photoionization mass spectrometry. The species concentration profiles obtained were simulated with a reaction mechanism the authors created based on analogies with butanol pyrolysis chemistry. Burnett et al.12 measured stable products formed from iso-propanol pyrolysis using a fast sampling method to extract gases from a rapid compression facility (T = 965 K to 1193 K, P = 4.4 to 10 atm). The results showed good agreement with the reaction mechanism for C3–C4 alcohols developed by Saggese et al.2
Unlike pyrolysis, the oxidation of iso-propanol has been studied experimentally over a broad range of temperatures and pressures. Ignition delay time (IDT) measurements were made by several groups. Johnson et al.20 used CH* emission (1 atm and 1400–1900 K). Man et al.21 used OH* emission (1.2–16 atm and 1100–1500 K) and also developed a reaction mechanism. CO absorption (3.5–11 atm and 1150–1500 K) was used by Jouzdani et al.17 who compared their results to simulations based on mechanisms from Johnson et al.,20 Man et al.21 and Sarathy et al.23 Jouzdani et al. found that predictions of IDTs with the Man et al. mechanism showed negative sensitivity to CH3 and HO2 radicals in contrast with the other mechanisms. Cooper et al.19 conducted shock tube/laser absorption experiments (1127–2162 K, 1.42 atm) to investigate the pyrolysis and oxidation of iso-propanol by monitoring water and CO formation. The measurements were in good agreement with simulations using reaction mechanisms from Li et al.18 and Saggese et al.2 Cheng et al.24 used pressure measurements in a rapid compression machine to measure IDTs (780–950 K, 20 and 40 bar). iso-Propanol was also studied in a counter flow burner by Frassoldati et al.22 (1000–1100 K, 1 atm) who extended chemical kinetics studies from methanol and ethanol to n-propanol and iso-propanol. Saggese et al.2 compared predictions of IDTs using their reaction mechanism for C3–C4 alcohols with experimental datasets from Man et al.21 and Cheng et al.24 with good agreement.
These works collectively provide important insight into iso-propanol chemistry, connecting theory with experimental results. However, the experimental data to date, cover limited temperatures and pressures. In particular there are no time-resolved speciation data at elevated temperatures and pressures that can provide insight into the reaction mechanisms. To address this lack, the current study used two shock tubes coupled via molecular beam sampling interfaces to time-of-flight mass spectrometers to obtain time-dependent species concentrations over a broad range of temperature and pressure. The results are used to identify intermediate species produced during iso-propanol pyrolysis and hence the main reaction paths. The data provide insights into the pyrolysis mechanism of iso-propanol and the relative importance of key primary and secondary reactions.
2. Experimental methods, experimental
2.1. Shock-tube facilities
Pyrolysis of iso-propanol was conducted behind reflected shock waves in a diaphragmless shock tube, DFST, and miniature high repetition rate shock tube, HRRST. Both shock tubes are connected to time-of-flight mass spectrometers (TOF-MS) using differentially pumped molecular beam sampling (MBS) systems. The HRRST and DFST generate very reproducible reaction conditions which allow signal averaging over many experiments. Both apparatuses have been fully described previously and brief descriptions are given here.The DFST25,26 consists of two main parts: a driver section and a driven section. The driver section is a stainless-steel tube with a length of 58 cm and an inner diameter of 22 cm. The driven section is a ∼7.5 m long stainless-steel tube with an inner diameter of 6.35 cm. A custom built pneumatically-actuated valve separates the driver and driven sections at the start of an experiment. The driven section is filled to pressure P1 with the reagent mixture and the driver section to pressure P4 with helium, where P4 ≫ P1. (Standard shock wave notation is used throughout the manuscript.) The conditions generated by the reflected shock wave, temperature T5 and pressure P5, are determined by the ratio P4/P1. In the driven section, three pressure transducers (PCB 132A35) are positioned at intervals of 76.2 mm, with the last transducer located 25.4 mm from the endwall of the shock tube to monitor the passage of the shock wave. The known distances and time intervals between the triggering of each pressure transducer are used to calculate the incident shock velocity which is extrapolated to obtain the shock velocity at the end wall. The temperature and pressure behind the reflected shock waves are calculated using the shock velocity at the end wall, the initial temperature, pressure and mixture composition of the driven gas, the specific heat capacity of the driven gas, and ideal normal-shock relations.
The HRRST27–29 is a miniature fully automated shock tube which is typically fired once every second. The driver and driven sections of the HRRST are separated by a solenoid-actuated valve.28 The driver section has an internal volume of approximately 100 cm3, and is linked to a 1 L reservoir, ensuring an almost constant P4 over many cycles of the HRRST. The driven section has an internal diameter of 6.35 mm and a length of 879 mm. It is built in a modular fashion and contains pneumatic valves for filling and venting. The firing sequence is described in ref. 27. The reagent mixture is supplied at P1via one of the pneumatic valves and the mixture is prepared automatically on-demand in a mixing rig.30 The driven section is equipped with six evenly-spaced piezoelectric pressure transducers (Dynasen CA1135) at 50 mm intervals, with the last pressure transducer positioned 75 mm from the nozzle. The methodology used to calculate T5 and P5 is similar to that used with the DFST.
2.2. Time-of-flight mass spectrometry
The DFST and HRRST are coupled to TOF-MSs (Kasedorf CB-5) by differentially-pumped molecular-beam sampling interfaces (MBS). Although the shock tubes are different, the MBSs are very similar, see Tranter et al.27,31 and Banyon et al.32 The driven section end-wall of each shock tube is equipped with a small nozzle (300 μm orifice in the DFST, 200–300 μm in the HRRST) through which gases continuously elute generating a supersonic jet that quenches the reactions initiated in the shock heated gases. A skimmer (Beam Dynamics Model 2, orifice: 0.5 mm DFST; 0.2 mm HRRST) samples gases from the core of the jet creating a molecular beam that is directed into the ion source of the TOF-MS. In this study, electron-impact ionization (EI) in the laboratory (DFST) and photoionization (HRRST) at the advanced light source (ALS), Lawrence Berkeley National Laboratory (LBNL), were used to create cations in the molecular beam. The cations were extracted into the mass spectrometer by pulsing the voltages applied to the repeller and extractor plates. The extraction period was 1 μs and the frequency was 100 kHz providing a time resolution of 10 μs. For each experiment, mass spectra were obtained for 3 ms (providing a total of 300 mass spectra) that spanned the pre- and post-shock regions. The ionized species were detected by a multichannel plate (MCP) detector. The analog signals from the MCP detector passed through a preamplifier (ORTEC VT120A) and were digitized at 1 GS per s with a 12-bit data acquisition board (GageApplied PCIe EON series).| P 5 (Torr) | T 5 (K) | Composition |
|---|---|---|
| 236 | 1673 | 1% iso-propanol, 1% Kr, 98% Ne |
| 176 | 1395 |
| Set | N | P 5 (bar) | σ P 5 (bar) | T 5 (K) | σ T 5 (K) | hν (eV) | Composition |
|---|---|---|---|---|---|---|---|
| N = number of experiments; hν = range of photon energies. | |||||||
| 1 | 7500 | 4.4 | 0.31 | 2053 | 90.4 | 9.45–12.8 | 0.25% iso-propanol, 99.75% Argon |
| 2 | 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 500 |
3.9 | 0.14 | 1660 | 33.2 | 9.3–12.8 | |
| 3 | 17500 |
3.7 | 0.18 | 1481 | 35.8 | 9.5–12.8 | |
Each dataset contains a series of experimental results all at the same conditions and mixture compositions, but with different values of photon energy, PE. At each PE, data from 500 shocks were acquired. The mass spectra for each photon energy were averaged to improve signal-to-noise ratios. Similarly, the reaction conditions were averaged over all photon energies for a nominal set of temperatures and pressures. The information for each dataset along with uncertainty bounds is given in the ESI;† from hereon only the nominal T5 and P5 are given. A baseline correction code34 was applied to the averaged mass spectra to eliminate background features that may interfere with peak identification.
The measured signal (S) is a function of m/z, PE and reaction time, t. The data obtained at each T5 and P5 form a multidimensional dataset as described in Banyon et al.32 The data set can be processed in various ways yielding different insights into the reaction chemistry. The most useful subsets are two-dimensional (2D) plots of t vs. m/z, referred to as XT, and PE vs. m/z, referred to as EX. The XT data can be obtained for individual PE or a range of PE. Similarly, the EX datasets can represent a range of time or an individual time, t. From the XT plots, time-dependent signal intensities for each m/z can be extracted. These are equivalent to concentration versus time data. Additionally, at each t, S versus m/z data can be obtained which are equivalent to classical mass spectra. From the EX datasets, photoionization spectra (PES), S vs. PE, are obtained for individual masses.
The measured signal is directly proportional to the concentration (x) of a species, the photoionization cross section of the species at the PE (σhν), and the photon flux (Φ) at the PE. A photodiode (OSI Optoelectronics, XUV-100C) connected to a picoammeter (Keithley 6485), was used to monitor the photon flux for T5 = 1481 K. The current values (I) measured by the picoammeter are proportional to Φ and are used to scale the signal intensity to account for variations in Φ. This approach provides an estimate of the effective concentration of a species viaeqn (1).
| x ∝ S/Iσhν | (1) |
Determining the absolute concentrations of species is more challenging and requires the mass discrimination factor and instrument-specific constants as discussed in ref. 32. Additionally, the photon flux was not measured for all experiments, although for a given PE it is reasonably stable for the duration of the experiments.
2.3. Reagents
For the DFST/TOF-MS studies, iso-propanol was mixed with krypton and neon in a 50 L glass mixing tank using a partial pressure method. The small amount of krypton, Table 1, acted as an internal standard to account for pressure variation in the ion source of the mass spectrometer.31 The mixing tank was evacuated to ∼10−5 Torr prior to preparing a mixture. iso-Propanol was degassed through repeated freeze–pump–thaw cycles using liquid nitrogen. The reagent mixture consisted of 1% of iso-propanol (99% purity, Sigma Aldrich), krypton (99.999%, Airgas), and neon (99.999%, Airgas). Helium (99.996%, Airgas) was the driver gas.For the HRRST/TOF-MS experiments, helium (99.999%, Airgas) was the driver gas, and the reagent mixture consisted of 0.25% iso-propanol (99% purity, Sigma-Aldrich) diluted in argon (99.999%, Airgas). iso-Propanol was placed in a high pressure bubbler and the mixture was prepared on demand in a mixing rig using the method described by Dalmiya et al.30
3. Results and discussion
3.1. Pre-shock region
Mass spectra of iso-propanol from the pre-shock region (i.e., prior to reaction) at PE = 10.7 eV and 12.8 eV are shown in Fig. 1. The mass spectra have been normalized to the largest peak in the displayed range. At both PE, the dominant peak is m/z = 45 (C2H5O+) and there is only a minor peak at m/z = 60, the parent mass. The ionization energy (IE) of iso-propanol is 10.17 eV,33 and the appearance energy (AE) of m/z = 45 is 10.20 eV. The ratio of m/z = 45 to m/z = 60 is reduced at 10.7 eV indicating dissociative ionization (DI) is less significant. The mass spectra are consistent with the literature 70 eV EI spectrum,33 although the photoionization spectra have fewer DI products as expected. At PE = 12.8 eV, a peak is observed at m/z = 18. Potentially, this could be water from dissociative ionization. However, DI creates a neutral water molecule and a C3H6+ cation (AE ∼ 12.0 eV33). Thus m/z = 18 is attributed to background water vapor in the chamber (H2O, IE = 12.6 eV35). Similarly, the peak at m/z = 32 is from background O2 (IE = 12.07 eV33) in the chamber. In Fig. 1b, peaks at m/z = 43, 44, and 59 are also observed. The AE of m/z = 43 and m/z = 59 are close to or greater than 10.7 eV and hence these fragments are not seen in Fig. 1a. Conversely, m/z = 44 is stronger relative to m/z = 45 in Fig. 1a than Fig. 1b suggesting the process forming it, CH4 elimination, becomes less favorable at higher PE. | ||
| Fig. 1 Mass spectra in the pre-shock region with photon energy of (a) 10.7 eV and (b) 12.8 eV. The signal levels at 10.7 eV are weaker than those at 12.8 eV and noise from pulsing the repeller and extractor plates is seen at <20 amu in panel (a). M denotes the ion of iso-propanol, M–X denotes the fragments generated from dissociative ionization of iso-propanol, and X denotes non-ionized species. The mass spectra are normalized to the largest peak in each plot. | ||
In Fig. 2 the photoionization spectra for iso-propanol, m/z = 60, and the products of dissociative ionization are shown. Each spectrum was normalized to its maximum signal. Spectra with the original signal intensities are provided in the ESI† to allow direct comparison of the relative fragment intensities. The sharp increase in the m/z = 60 signal between 10.1 and 10.2 eV is in good agreement with the IE of Refaey et al.36 of 10.12 eV for iso-propanol. The appearance energies of the DI products at m/z = 43, 44 and 45 are also in good agreement with literature values.33
 | ||
| Fig. 2 Photoionization spectrum of non-reacting iso-propanol and iso-propanol DI products. | ||
3.2. Dissociation of iso-propanol
Pyrolysis of iso-propanol produced several species which varied in concentration and identity depending on the reaction conditions. The following discussion focuses on the higher-pressure PI experiments and a comparison with the low pressure EI data is made in Section 3.5. Mass spectra at PE = 10.7 eV and 12.8 eV from the post-shock region for iso-propanol at 1481 K and 3.7 bar and 2053 K and 4.0 bar are shown in Fig. 3 and 4, respectively. Mass spectra at other photon energies and reaction conditions are provided in the ESI.† In addition, a table summarizing all observed m/z values, their species identifications, chemical formulae, and ionization energies is provided in the ESI† for reference. The mass spectra were obtained by integrating over the first 500 μs after the arrival of the shock wave, i.e., the start of reaction. | ||
| Fig. 3 Mass spectra at T5 = 1481 K integrated over the first 500 μs of reaction in the post-shock region with photon energy of (a) 10.7 eV and (b) 12.8 eV. The mass spectra are normalized to the largest peak in each plot. | ||
 | ||
| Fig. 4 Mass spectra at T5 = 2053 K integrated over the first 500 μs of reaction in the post-shock region with photon energy of (a) 10.7 eV and (b) 12.8 eV. The mass spectra are normalized to the largest peak in each plot. | ||
The dehydration reaction, R1, and the Cα–Cβ bond scission reaction eliminating CH3, R2, were identified by Bui et al.15 as the initial dissociation channels for iso-propanol and are discussed in the following sections. For reference, values of the rate coefficients for R1, R2 and other important reactions during iso-propanol pyrolysis are provided in Table 3 for T = 1400 K and 2000 K. For pressure-dependent reactions, the rate coefficients were calculated at 4 atm and 200 Torr, corresponding to the conditions studied here, using the expressions in the cited sources.
| Reaction | k 1400K | k 2000K | Ref. | |
|---|---|---|---|---|
| a sC2H4OH = H3CCH(OH), tC3H6OH = H3CC(OH)CH3, C3H6OH-2-1 = H3CCH(OH)CH2. | ||||
| R1 | iso-Propanol ↔ C3H6 + H2O | 4.84 × 10+3 (2.45 × 103) | 1.10 × 106 (3.18 × 105) | 16 |
| R2 | iso-Propanol ↔ sC2H4OHa + CH3 | 2.06 × 103 (4.51 × 102) | 9.61 × 105 (9.90 × 104) | 15 |
| R3 | C 3 H 6 + H ↔ C2H4 + CH3 | 9.67 × 1012 (1.18 × 1013) | 1.75 × 1013 (2.44 × 1013) | 37 |
| R4 | C 3 H 6 + H ↔ C3H5 + H2 | 4.03 × 1012 | 1.53 × 1013 | 37 |
| R5 | C3H5 ↔ C3H4 + H | 1.74 × 109 (4.21 × 108) | 2.47 × 1011 (3.34 × 1010) | 38 |
| R6 | sC 2 H 4 OH ↔ CH3CHO + H | 1.77 × 108 (9.11 × 107) | 5.48 × 109 (1.38 × 109) | 39 |
| R7 | sC 2 H 4 OH ↔ C2H3OH + H | 5.12 × 107 (9.56 × 106) | 9.58 × 108 (8.37 × 107) | 39 |
| R8 | 2CH3 ↔ C2H6 | 1.44 × 1013 (1.44 × 1013) | 1.15 × 1013 (1.14 × 1013) | 40 |
| R9 | iso-Propanol + H ↔ tC3H6OHa + H2 | 7.05 × 1012 | 1.95 × 1013 | 41 |
| R10 | iso-Propanol + CH3 ↔ tC3H6OHa + CH4 | 4.81 × 1010 | 2.93 × 1011 | 41 |
| R11 | iso-Propanol + H ↔ C3H6OH-2-1a + H2 | 5.41 × 1012 | 2.95 × 1013 | 41 |
| R12 | iso-Propanol + CH3 ↔ C3H6OH-2-1a + CH4 | 4.76 × 1010 | 4.75 × 1011 | 41 |
| R13 | tC3H6OHa ↔ CH3COCH3 + H | 7.14 × 107 (1.26 × 107) | 4.20 × 108 (4.72 × 107) | 42 |
| R14 | C3H6OH-2-1a ↔ C2H3OH + CH3 | 1.09 × 109 | 3.40 × 1010 | 43 |
| R15 | C 2 H 3 OH ↔ CH3CHO | 1.98 × 104 (7.85 × 103) | 1.53 × 106 (3.76 × 105) | 44 |
 | ||
| Fig. 5 Photoionization spectrum of m/z = 42 obtained from current study (solid line) at T5 = 1481 K in the post-shock region and the reference data for photoionization spectrum of propene (dash line) reported by Cool et al.46 | ||
 | ||
| Fig. 6 Time histories of the peak intensities of m/z = 18 (12.8 eV) and m/z = 42 (12.8 eV) at 1481 K and 3.7 bar based on PI data. Time t = 0 μs corresponds to the start of reaction, i.e., the arrival of the reflected shock. The as-recorded signals have been normalized, and the lines are moving 5-point averages for clarity. | ||
The products of R1 are stable over most of the temperature range of this study. However, at the highest temperatures studied in the current work propene can decompose by two channels yielding allyl + H, and vinyl + methyl.49 Propene also reacts readily with H-atoms at elevated temperatures to yield ethylene (C2H4) R3 or allyl radicals (C3H5) R4. From Table 3k3/k4 varies from 2.9 to 1.6 over 1400–2000 K. Potentially, propene can also be removed by reaction with a CH3 but, k is ∼4.0 × 109 1.9 × 1011 cm3 mol−1 s−1 for propene + CH3 over 1000–2000 K50 or at least two orders of magnitude lower than k3.
From reactions R3 to R5, ethylene (IE = 10.5 eV, m/z = 2851) and allene (IE = 9.7 eV, m/z = 4048) and/or propyne (IE = 10.36 eV, m/z = 4051) will be formed. Fig. 7 shows the PES for m/z = 28 and 40 at 1481 K and 3.7 bar in the post-shock region. The observed IE for m/z = 28 is consistent with ethylene. For m/z = 40 there is a noticeable increase in signal starting at 9.9 eV. This is 0.2 eV higher than the IE of allene and 0.46 eV below the IE of propyne. However, the photoionization cross-section of allene is very small (<1 Mb) below 9.8 eV.48 Consequently, the increase at 9.9 eV is likely due to allene, and the second increase observed at 10.4 eV due to propyne. Thus, the m/z = 40 peak is attributed to both isomers of C3H4 and from here on C3H4 will be used to represent a mixture of allene and propyne.
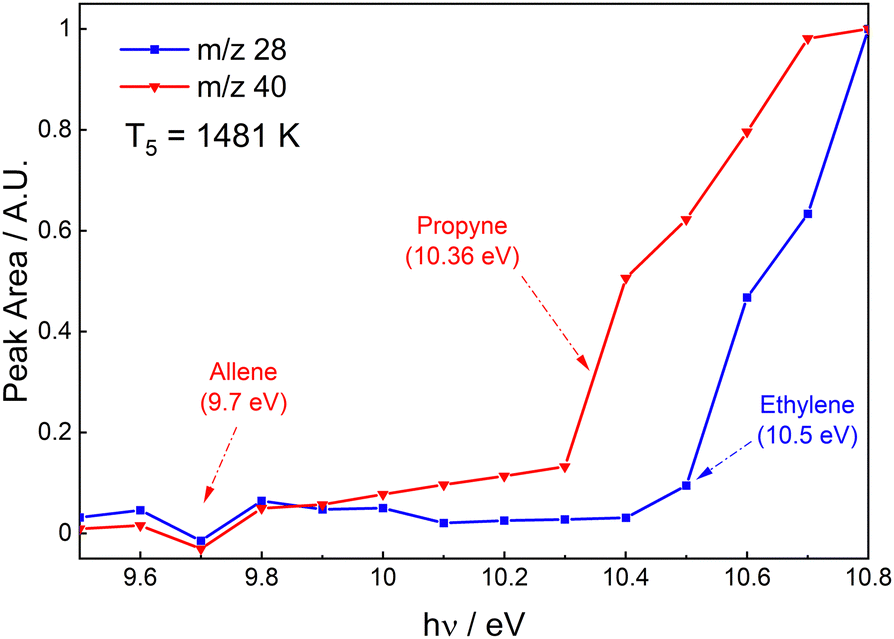 | ||
| Fig. 7 Photoionization spectra of m/z = 28 and = 40 at T = 1481 K in the post-shock region. | ||
In Fig. 8, relative concentrations for ethylene and C3H4 at 1481 K and 3.7 bar are shown as a function of reaction time. Ionization energies of 11.7 eV and 11.0 eV were selected for m/z = 28 (σ11.7eV = 8.42 Mb51) and m/z = 40 respectively. The C3H4 data include both allene and propyne. For propyne, σ11.0eV = 43.84 Mb51 and is considerably larger than that of allene. Consequently, the m/z = 40 signal in Fig. 8 was calculated assuming only propyne contributes to the signal and is thus an upper limit. The data show ethylene and C3H4 are produced simultaneously about 70 μs after reflection of the shock wave. Production of both species ceases after around 210 μs with around six times more C2H4 being formed than C3H4.
 | ||
| Fig. 8 Time histories of relative concentrations (i.e., concentrations corrected for photon flux and photoionization cross section) of ethylene (11.7 eV) and C3H4 (11 eV) at T = 1481 K (based on PI data). The points and lines represent the as-recorded experimental data and a five-point moving average of the as-recorded data, respectively. Time t = 0 s corresponds to the passage of the shock wave and the start of reaction. | ||
 | ||
| Fig. 9 Photoionization spectra of m/z = 44 and m/z = 30 at T = 1481 K in the post-shock region. | ||
3.3. H-abstraction from iso-propanol
Reaction R2 and subsequent reactions R5 to R7 release H and CH3 atoms during the dissociation of iso-propanol. H-abstraction reactions by these early-generated radicals play a significant role in the consumption of iso-propanol, particularly at high pressures that favor bimolecular reactions. The radical H3C-C(OH)-CH3 is produced by the abstraction of a secondary hydrogen via reactions R9 and R10, while reactions R11 and R12 involve the abstraction of a primary hydrogen to form the radical H3C-CH(OH)-CH2. The radicals generated from the H abstraction reactions rapidly dissociate by the β-scission reactions R13 and R14, Table 3, yielding acetone (m/z = 58) + H and ethenol (m/z = 44) + CH3, respectively and are chain-propagating reactions.Burnett et al.12 identified acetone (m/z = 58) as a major stable species from iso-propanol pyrolysis at low temperatures (T = 965 K to 1193 K, P = 4.4 to 10 atm). However, in this work the m/z = 58 signal at 1481 K and 3.7 bar is low, Fig. 3, and at 2053 K and 4.0 bar it was almost negligible, Fig. 4. The experiments by Burnett et al. were at long residence times and much lower temperatures than the present work, which probably accounts for the differences in m/z = 58. At the highest temperatures of the current work, acetone dissociates readily to acetyl (CH3CO) and methyl radicals (Saxena et al.52k ≈ 2.11 × 103–4.91 × 106 s−1 for 1400–2000 K and 4 bar). Followed by the subsequent rapid dissociation of CH3CO to CO and CH3 (Senosiain et al.53k > 1010 s−1).
The β-scission reaction of C3H6OH-2-1, R14, generates ethenol and methyl radicals. As previously discussed, ethenol isomerizes to acetaldehyde and the m/z = 44 signal includes acetaldehyde generated from H-abstraction reactions as well as the dissociation of iso-propanol via R2 followed by R6.
3.4. Secondary reactions
In addition to the primary stable species and radicals previously discussed, secondary reactions also yield distinct species. The mass spectra in Fig. 3 and 4 show peaks at m/z = 26 and m/z = 50 for 1481 K and 2053 K, with an additional m/z = 52 peak observed solely at 1481 K. Photoionization spectra of m/z = 26, 50, and 52 at 1481 K are shown in Fig. 10. These indicate that m/z = 26, 50 and 52 correspond to acetylene (C2H2, IE = 11.4 eV), diacetylene (C4H2, IE = 10.17 eV), and vinylacetylene (C4H4, IE = 9.58 eV), respectively.51 A peak at m/z 74 was also observed, see Fig. S1–S3 (ESI†). This feature is barely discernable in 1481 K data but becomes more significant as T5 increases. The peak is assigned to 1,3,5-hexatriyne (C6H2, IE = 9.5 eV54). Photoionization spectra of m/z = 74 are provided in Fig. S6 and S7 in the ESI† to support this assignment. Additional PES of m/z = 26, 50, 52 and 74 at 1660 K and 2053 K are provided in the ESI† and are similar to Fig. 10. | ||
| Fig. 10 Photoionization spectra of m/z = 26, m/z = 50, and m/z = 52 at T5 = 1481 K in the post-shock region. | ||
The time histories of vinylacetylene and diacetylene at 1481 K and 3.7 bar are shown in Fig. 11. Vinylacetylene does not appear until about 100 μs and is clearly not a primary product. Diacetylene lags in time relative to the production of vinylacetylene. Li et al.18 provide an explanation for the relative orders of appearance via the sequence C3H4 → C3H3 → C3H2 followed by C3H2 + CH3 → C4H4 + H where C4H4 is vinylacetylene. Vinylacetylene undergoes H-abstraction ultimately yielding diacetylene. In the 1660 K and 2053 K experiments, the m/z = 52 peak becomes diminishingly small suggesting that under these conditions C4H4 is either consumed much more rapidly than at 1481 K or the sequence of reactions leading to C4H4 is interrupted at higher temperatures. As noted earlier, Saggese et al.2 recently proposed a C3–C4 alcohol reaction mechanism. However, the mechanism does not account for the chemistry of C4H2 and C4H4 and therefore fails to explain the appearance of m/z = 50 and m/z = 52 observed in this study.
 | ||
| Fig. 11 Species time histories of m/z = 50 and 52 at T = 1481 K based on PI data. The plots represent the relative order of appearance of each species rather than relative concentrations. The points and lines represent the as-recorded experimental data and a five-point moving average of the as-recorded data, respectively. Time t = 0 μs corresponds to the passage of the reflected shockwave and the start of reaction. | ||
3.5. Comparison of EI and PI spectra
The DFST/EI experiments covered much lower pressures, ∼200 Torr, than the HRRST/PI experiments, ∼4 bar, although similar temperatures were used. In Fig. 12, EI mass spectra are compared with PI mass spectra. The temperatures are similar, but the pressures are significantly different and the iso-propanol concentrations differ by a factor of four. | ||
| Fig. 12 Mass spectra from photoionization (PI) and electron-impact ionization (EI) experiments in the (a) pre-shock and (b) post-shock regions. PI mass spectra integrated over the first 500 μs of reaction were taken at T = 1660 ± 40 K, P = 3.9 ± 0.1 bar, 0.25% iso-propanol/99.75% Ar, and 12.8 eV. EI mass spectra integrated over the first 1000 μs of reaction were taken at T = 1673 K, P = 0.31 bar (236 Torr), 1% iso-propanol/1% Kr/98% Ne, and 24 eV. | ||
As seen in Fig. 12, the PI and EI spectra are quite similar, although there are some key differences. The EI pre-shock mass spectra include a signal at m/z = 29; that is absent from the PI mass spectra. The discrepancy can be attributed to the appearance energy of m/z = 29, which is ∼12.5 eV and σ12.8eV = 0.54 Mb.47 Additionally, m/z = 18 and 32 are present in the PI spectra due to background water and oxygen. While the base pressures in the EI and PI TOF-MS chambers are similar background gases are rarely observed in the DFST/EI experiments. This difference is mainly due to the PI mass spectra being averaged over hundreds of experiments whereas the EI spectra are from single experiments. In Fig. 12b, neon, the bath gas used in EI experiments, forms the peak at m/z = 20, and a small peak due to the 22Ne isotope is seen. The features at m/z = 30 and 40–44 are absent from the EI spectra, while the m/z = 50 peak is strong in the EI mass spectrum. The stable species represented by m/z = 40–44 have IEs that are considerably lower than the electron energy used and thus should be observable in the EI experiments if they are formed. While fragmentation may decrease the intensity of the parent ion peaks, the literature 70 eV mass spectra for these species still show a strong parent ion peak. Thus, the absence of m/z = 40–44 peaks in the post-shock EI mass spectra suggests the species if formed are below the detection limit. Similarly, m/z = 30 and 29 which are characteristic of, ethane33 were not observed in the EI mass spectra whereas m/z 30 was assigned to ethane in the PI data. As discussed above, diacetylene serves as an indication of the water elimination reaction and the differences in the m/z = 50 peak between the DFST and HRRST experiments suggest that the water elimination reaction, R1, is more dominant at lower pressures, which is consistent with the values of the rate coefficients in Table 3.
Fig. 13 shows the time histories and sequential appearance of several species from the PI and EI experiments. From the PI experiments acetylene, ethylene, ethane, and diacetylene appear in that order.
 | ||
| Fig. 13 Time histories of acetylene, ethylene, ethane, and diacetylene from PI and EI. The PI data were taken at T = 1660 ± 40 K, P = 3.9 ± 0.1 bar, 0.25% iso-propanol/99.75% Ar, and 11.7 eV. The EI data were taken at T = 1673 K, P = 236 Torr, 1% iso-propanol/1% Kr/98% Ne, and 24 eV. Ethane was absent from the EI datasets. The plots represent the relative order of appearance of each species. The lines are the moving averages of the species measurements (symbols). | ||
The reaction pathways active during thermal decomposition of iso-propanol were further considered using reaction path analysis based on the reaction mechanism by Li et al.18 A reaction path diagram is presented in Fig. 14 for 2% iso-propanol at 1600 K and 4 bar, 20 μs after the start of reaction. The analysis indicates iso-propanol is primarily consumed through H abstraction reactions by H and CH3 radicals and by the water elimination reaction, R1. Recombination of methyl radicals produces ethane, followed by production of ethylene from the dissociation of ethane and also propene. Although the reaction mechanism includes pressure-dependent reactions, the predicted reaction pathways remain largely unchanged between 4 atm and 200 Torr. At higher temperatures, the reaction pathway also remains largely unchanged, except the water elimination and C–C bond scission reactions become more significant, and the recombination of methyl radicals is moved to later times. Consequently, based on the simulation results one would expect to observe ethane in the 200 Torr experiments contrary to the experimental results. This suggests that there may be inadequacies in the reaction mechanism.
 | ||
| Fig. 14 Reaction path diagram 20 μs after the start of reaction based on the reaction mechanism by Li et al.18 for 2% iso-propanol at 1600 K, and 4 bar. The percentage values are the relative molar fluxes (greater than 10%) of the species consumption pathways. The species highlighted in bold text were observed in the experimental data. The values in blue (e.g., 1, 2, 3) indicate the relative order of appearance of the species observed in the experiments. | ||
While the reaction mechanism by Li et al.18 includes thermodynamic data and reactions for the formation of vinylacetylene and diacetylene, the reaction channels are negligibly small compared with the overall reaction pathways for iso-propanol and their pathway fluxes are below the 10% cutoff limit used to highlight the primary reaction paths in Fig. 14. Using photoionization cross sections at 10.7 eV for propene (11.9 Mb46), diacetylene (24.14 Mb51), and vinylacetylene (38.97 Mb51), effective concentrations of 0.045, 0.02, and 0.005, respectively, are obtained. While vinylacetylene is a relatively minor species diacetylene is much more abundant.
4. Conclusions
The thermal decomposition of iso-propanol was systematically investigated using time-of-flight mass spectrometry in diaphragmless and high-repetition-rate shock-tube studies. Electron impact ionization and photoionization techniques were applied to obtain high-fidelity spectra that provided direct measurements of intermediate species produced during iso-propanol pyrolysis. In addition to the identification of key species, the dominant reaction pathways were determined by evaluating the relative concentration of intermediate species from the mass spectra. The main observation is that at high pressures iso-propanol dissociates by both R1 and R2. Whereas at low pressures, the lack of m/z = 30 indicates formation of methyl radicals is suppressed. As R2 is the primary source of methyl this indicates that the C–C scission channel is minor at low pressures. These observations are consistent with those of Bui et al.15 However, the literature chemical mechanisms for iso-propanol pyrolysis indicate that significant amounts of ethane should be formed at 200 Torr on the experimental timescale.Reaction R1 produced propene and water, both of which were observed. The data indicate that propene dissociated into ethylene and methyl radicals, or underwent hydrogen elimination to yield C3H4, with the latter being favored at high temperatures. Reaction R2, a C–C bond scission reaction, produced the 1-hydroxyethyl radical, which was expected to rapidly dissociate to acetaldehyde through β-scission reactions. However, the m/z = 44 peak in the mass spectra, associated with acetaldehyde, also contained dissociative ionization products from iso-propanol rendering it difficult to assess the acetaldehyde concentration. While H-abstraction reactions of iso-propanol were expected to yield acetone and ethenol, there was little indication of these species. For ethenol, this is likely due to rapid isomerization to acetaldehyde. The lack of an m/z = 58 peak suggests acetone was not formed, contrary to a lower temperature study by Burnett et al.12 Finally, peaks were observed in both PI and EI experiments at m/z = 50 and 52. Based on the photoionization spectra, these were assigned to diacetylene and vinylacetylene, respectively. Prior studies of iso-propanol have not observed these species, and they are likely produced from secondary reactions initiated from propene. Comparison of the experimental results with model predictions indicates discrepancies with the ethane, diacetylene and vinylacetylene reaction pathways in the available reaction mechanisms and highlight areas for future focus. The observation of 1,3,5-hexatriyne is consistent with molecular growth processes initiated by polyacetylenes rather than through formation single ring aromatics. The novel experimental observations presented in this work can significantly enhance the development of the reaction theory and elementary chemistry of iso-propanol pyrolysis, enabling more accurate predictive understanding of iso-propanol decomposition at high-temperature conditions.
Author contributions
J. H. Kim: investigation, data curation, visualization, writing – original draft. K. Kim: investigation. Q. Meng: investigation A. Sutar: investigation M. S. Wooldridge: conceptualization, investigation, supervision, writing – review & editing. R. S. Tranter: conceptualization, investigation, data curation, supervision, writing – review & editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This material is based on work supported by the U.S. Department of Energy, Office of Basic Energy Sciences, Division of Chemical Sciences, Geosciences, and Biosciences through Argonne National Laboratory. Argonne is a U.S. Department of Energy laboratory managed by UChicago Argonne, LLC, under contract DE-AC02-06CH11357. JHW was supported by the U.S. Department of Energy, Office of Science, Office of Workforce Development for Teachers and Scientists, Office of Science Graduate Student Research (SCGSR) program. The SCGSR program is administered by the Oak Ridge Institute for Science and Education (ORISE) for the DOE. ORISE is managed by ORAU under contract number DE-SC0014664. All opinions expressed in this paper are the authors’ and do not necessarily reflect the policies and views of DOE, ORAU, or ORISE. MSW and QM acknowledge support through The College of Engineering at the University of Michigan. This research used resources of the Advanced Light Source, a U.S. DOE Office of Science User Facility under contract no. DE-AC02-05CH11231. The work was conducted using the T4 terminal at beamline 9.0.2. We gratefully acknowledge the assistance of Oleg Kostko and Bruce Rude.References
- D. J. Gaspar, Top Ten Blendstocks For Turbocharged Gasoline Engines: Bioblendstocks With Potential to Deliver the for Highest Engine Efficiency. PNNL-28713, Pacific Northwest National Laboratory, 2019.
- C. Saggese, C. M. Thomas, S. W. Wagnon, G. Kukkadapu, S. Cheng, D. Kang, S. S. Goldsborough and W. J. Pitz, An improved detailed chemical kinetic model for C3–C4 linear and iso-alcohols and their blends with gasoline at engine-relevant conditions, Proc. Combust. Inst., 2021, 38, 415–423 CAS.
- S. Atsumi, T. Hanai and J. C. Liao, Non-fermentative pathways for synthesis of branched-chain higher alcohols as biofuels, Nature, 2008, 451, 86–89 CAS.
- C. R. Shen and J. C. Liao, Metabolic engineering of escherichia coli for 1-butanol and 1-propanol production via the keto-acid pathways, Metab. Eng., 2008, 10(6), 312–320 CAS.
- S. P. Singh, S. Bansal and A. Pandey, Basics and Roots of Synthetic Biology, Current Developments in Biotechnology and Bioengineering, 2019, ch. 1, pp. 3–22 Search PubMed.
- A. Keskin and M. Gürü, The effects of ethanol and propanol additions into un- leaded gasoline on exhaust and noise emissions of a spark ignition engine, Energy Sources, Part A, 2011, 33(23), 2194–2205 CrossRef CAS.
- B. Masum, H. Masjuki, M. A. Kalam, S. Palash and M. Habibullah, Effect of alcohol—gasoline blends optimization on fuel properties, performance and emissions of a si engine, J. Cleaner Prod., 2015, 86, 230–237 CrossRef CAS.
- A. Uyumaz, An experimental investigation into combustion and performance characteristics of an hcci gasoline engine fueled with n-heptane, iso-propanol and n-butanol fuel blends at different inlet air temperatures, Energy Convers. Manage., 2015, 98, 199–207 CrossRef CAS.
- L. Xingcai, H. Yuchun, J. Libin, Z. Linlin and H. Zhen, Heat release analysis on combustion and parametric study on emissions of hcci engines fueled with 2-propanol/n-heptane blend fuels, Energy Fuels, 2006, 20(5), 1870–1878 CrossRef.
- S. Smith and A. Gordon, Studies of diffusion flames. ii. diffusion flames of some simple alcohols, J. Phys. Chem., 1956, 60(8), 1059–1062 CrossRef CAS.
- K. Kohse-Höinghaus, P. Oßwald, T. Cool, T. Kasper, N. Hansen, F. Qi, C. West-brook and P. Westmoreland, Biofuel combustion chemistry: from ethanol to biodiesel, Angew. Chem., Int. Ed., 2010, 49(21), 3572–3597 CrossRef PubMed.
- M. A. Burnett, J. Kim, S. W. Wagnon, A. B. Mansfield and M. S. Wooldridge, An Experimental Study of 2-Propanol Pyrolysis Chemistry, J. Phys. Chem. A, 2022, 126, 9097–9107 CrossRef CAS PubMed.
- J. A. Barnard, The pyrolysis of iso-propanol, Trans. Faraday Soc., 1960, 56, 72–79 RSC.
- A. B. Trenwith, Thermal decomposition of iso-propanol, J. Chem. Soc., Faraday Trans. 1, 1975, 71, 2405–2412 RSC.
- B. H. Bui, R. S. Zhu and M. C. Lin, Thermal decomposition of iso-Propanol:First-principles prediction of total and product-branching rate constants, J. Chem. Phys., 2002, 117, 11188 CrossRef CAS.
- J. S. Heyne, S. Dooley, Z. Serinyel, F. L. Dryer and H. Curran, Decomposition Studies of Iso-propanol in a Variable Pressure Flow Reactor, Z. Phys. Chem., 2015, 229(6), 881–907 CrossRef CAS.
- S. Jouzdani, A. Zhou and B. Akih-Kumgeh, Propanol isomers: Investigation of ignition and pyrolysis time scales, Combust. Flame, 2017, 176, 229–244 CrossRef CAS.
- W. Li, Y. Zhang, B. Mei, Y. Li, C. Cao, J. Zou, J. Yang and Z. Cheng, Experimental and kinetic modeling study of n -propanol and i- propanol combustion: Flow reactor pyrolysis and laminar flame propagation, Combust. Flame, 2019, 207, 171–185 CrossRef CAS.
- S. P. Cooper, C. M. Grégoire, D. J. Mohr, O. Mathieu, S. A. Alturaifi and E. L. Petersen, An Experimental Kinetics Study of Iso-propanol Pyrolysis and Oxidation behind Reflected Shock Waves, Energies, 2021, 14(20), 6808 CAS.
- M. V. Johnson, S. S. Goldsborough, Z. Serinyel, P. O’Toole, E. Larkin, G. O’Malley and H. J. Curran, A Shock Tube Study of n- and iso-Propanol Ignition, Energy Fuels, 2009, 23, 5886–5898 CAS.
- X. Man, C. Tang, J. Zhang, Y. Zhang, L. Pan, Z. Huang and C. K. Law, An experimental and kinetic modeling study of n-Propanol and i-propanol ignition at high temperatures, Combust. Flame, 2014, 161(3), 644–656 CAS.
- A. Frassoldati, A. Cuoci, T. Faravelli, U. Niemann, E. Ranzi, R. Seiser and K. Seshadri, An experimental and kinetic modeling study of n-Propanol and iso-Propanol combustion, Combust. Flame, 2010, 157(1), 2–16 CAS.
- S. M. Sarathy, P. Oßwald, N. Hansen and K. Kohse-Höinghaus, Alcohol combustion chemistry, Prog. Energy Combust. Sci., 2014, 44, 40–102 Search PubMed.
- S. Cheng, D. Kang, S. S. Goldsborough, C. Saggese, S. W. Wagnon and W. J. Pitz, Experimental and modeling study of C2–C4 alcohol autoignition at intermediate temperature conditions, Proc. Combust. Inst., 2021, 709–717 CAS.
- J. B. Randazzo and R. S. Tranter, Note: An improved driver section for a diaphragmless shock tube, Rev. Sci. Instrum., 2015, 86, 016117 CAS.
- R. S. Tranter and B. R. Giri, A diaphragmless shock tube for high temperature kinetic studies, Rev. Sci. Instrum., 2008, 79, 94103 Search PubMed.
- R. S. Tranter and P. T. Lynch, A Miniature High Repetition Rate Shock Tube, Rev. Sci. Instrum., 2013, 84, 094102 CAS.
- P. T. Lynch, Note: An Improved Solenoid Driver Valve for Miniature Shock Tubes, Rev. Sci. Instrum., 2016, 87, 056110 CAS.
- P. T. Lynch, T. P. Troy, M. Ahmed and R. S. Tranter, Probing Combustion Chemistry in a Miniature Shock Tube with Synchrotron VUV Photo Ionization Mass Spectrometry, Anal. Chem., 2015, 87, 2345–2352 CAS.
- A. Dalmiya, J. M. Mehta, R. S. Tranter and P. T. Lynch, High pressure, high flow rate batch mixing apparatus for high throughput experiments, Rev. Sci. Instrum., 2021, 92, 114104 CAS.
- R. S. Tranter, B. R. Giri and J. H. Kiefer, Shock Tube/Time-of-Flight Mass Spectrometer for High Temperature Kinetic Studies, Rev. Sci. Instrum., 2007, 78, 034101 CrossRef.
- C. Banyon, T. Sikes and R. S. Tranter, Reactions of propyl radicals: A shock tube–VUV photoionization mass spectrometry study, Combust. Flame, 2021, 224, 14–23 CrossRef CAS.
- ed. P. J. Linstrom and W. G. Mallard, NIST chemistry WebBook, NIST Standard Reference Database Number 69, National Institutes of Standards and Technology, Gaithersburg, MD, 2023 Search PubMed.
- C. M. Galloway, E. C. Le Ru and P. G. Etchegoin, An iterative algorithm for background removal in spectroscopy by wavelet transforms, Appl. Spectrosc., 2009, 63, 1370–1376 CrossRef CAS PubMed.
- G. N. Haddad and J. A. R. Samson, Total absorption and photoionization cross sections of water vapor between 100 and 1000 Å, J. Chem. Phys., 1986, 84(12), 6623–6626 CrossRef CAS.
- K. M. A. Refaey and W. A. Chupka, Photoionization of the lower aliphatic alcohols with mass analysis, J. Chem. Phys., 1968, 48, 5205 CrossRef CAS.
- J. A. Miller and S. J. Klippenstein, Dissociation of Propyl Radicals and Other Reactions on a C3H7 Potential, J. Phys. Chem. A, 2013, 117, 2718–2727 CrossRef CAS PubMed.
- J. A. Miller, J. P. Senosiain, S. J. Klippenstein and Y. Georgievskii, Reactions over Multiple, Interconnected Potential Wells: Unimolecular and Bimolecular Reactions on a C3H5 Potential, J. Phys. Chem. A, 2008, 112, 9429–9438 CrossRef CAS PubMed.
- E. E. Dames, Master Equation Modeling of the Unimolecular Decompositions of α-Hydroxyethyl (CH3CHOH) and Ethoxy (CH3CH2O), Int. J. Chem. Kinet., 2014, 46(3), 176–188 CrossRef CAS.
- B. Wang, H. Hou, L. M. Yoder, J. T. Muckerman and C. Fockenberg, Experimental and Theoretical Investigations on the Methyl–Methyl Recombination Reaction, J. Phys. Chem. A, 2003, 107(51), 11414–11426 CrossRef CAS.
- S. N. Elliott, K. B. Moore III, A. V. Copan, M. Keçeli, C. Cavallotti, Y. Georgievskii, H. F. Schaefer III and S. J. Klippenstein, Automated theoretical chemical kinetics Predicting the kinetics for the initial stages of pyrolysis, Proc. Combust. Inst., 2021, 38, 375–384 CrossRef CAS.
- J. Zador and J. A. Miller, Unimolecular dissociation of hydroxypropyl and propoxy radicals, Proc. Combust. Inst., 2013, 34(1), 519–526 CrossRef CAS.
- J. M. Simmie and H. J. Curran, Energy Barriers for the Addition of H, ĊH3, and Ċ2H5 to CH2
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHX [X = H, CH3, OH] and for H-Atom Addition to RCHO [R = H, CH3, Ċ2H5, n-C3H7]: Implications for the Gas-Phase Chemistry of Enols, J. Phys. Chem. A, 2009, 113, 7834–7845 CrossRef CAS.
CHX [X = H, CH3, OH] and for H-Atom Addition to RCHO [R = H, CH3, Ċ2H5, n-C3H7]: Implications for the Gas-Phase Chemistry of Enols, J. Phys. Chem. A, 2009, 113, 7834–7845 CrossRef CAS. - R. Sivaramakrishnan, J. V. Michael, L. B. Harding and S. J. Klippenstein, Resolving Some Paradoxes in the Thermal Decomposition Mechanism of Acetaldehyde, J. Phys. Chem. A, 2015, 119(28), 7724–7733 CrossRef CAS.
- C. Banyon and R. S. Tranter, High-Temperature Dissociation of Neopentanol: Shock Tube/Photoionization Mass Spectrometry Studies, J. Phys. Chem. A, 2023, 127(5), 1293–1301 CrossRef CAS PubMed.
- T. A. Cool, K. Nakajima, T. A. Mostefaoui, F. Qi, A. McIlroy, P. R. Westmoreland, M. E. Law, L. Poisson, D. S. Peterka and M. Ahmed, Selective detection of isomers with photoionization mass spectrometry for studies of hydrocarbon flame chemistry, J. Chem. Phys., 2003, 119(16), 8356–8365 CrossRef CAS.
- J. Wang, B. Yang, T. A. Cool, N. Hansen and T. Kasper, Near-threshold absolute photoionization cross-sections of some reaction intermediates in combustion, Int. J. Mass Spectrom., 2008, 269(3), 210–220 CrossRef CAS.
- B. Yang, J. Wang, T. A. Cool, N. Hansen, S. Skeen and D. L. Osborn, Absolute photoionization cross-sections of some combustion intermediates, Int. J. Mass Spectrom., 2012, 309, 118–128 CrossRef CAS.
- L. Ye, Y. Georgievskii and S. J. Klippenstein, Pressure-dependent branching in the reaction of 1CH2 with C2H4 and other reactions on the C3H6 potential energy surface, Proc. Combust. Inst., 2015, 35, 223–230 CAS.
- W. Tsang, Chemical kinetic data base for combustion chemistry. Part V. Propene, J. Phys. Chem. Ref. Data, 1991, 20, 221–273 CAS.
- T. A. Cool, J. Wang, K. Nakajima, C. A. Taatjes and A. Mcllroy, Photoionization cross sections for reaction intermediates in hydrocarbon combustion, Int. J. Mass Spectrom., 2005, 247, 18–27 CAS.
- S. Saxena, J. H. Kiefer and S. J. Klippenstein, A shock-tube and theory study of the dissociation of acetone and subsequent recombination of methyl radicals, Proc. Combust. Inst., 2009, 32, 123–130 CAS.
- J. P. Senosiain, S. J. Klippenstein and J. A. Miller, Pathways and Rate Coefficients for the Decomposition of Vinoxy and Acetyl Radicals, J. Phys. Chem. A, 2006, 110(17), 5772–5781 CAS.
- G. Bieri, F. Burger, E. Heilbronner and J. P. Maier, Valence ionization energies of hydrocarbons, Helv. Chim. Acta, 1977, 60, 2213–2233 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cp00562k |
| This journal is © the Owner Societies 2025 |