
Auto-dissociation of atmospheric water on TiO2: insights from sum-frequency spectroscopy of Ti–O vibrations†
Hui
Li‡
a,
Wenqi
Zheng‡
a,
Xinyi
Liu
a,
Jiashi
Li
a,
Lianbing
Wen
a,
Fujie
Tang
 b and
Wei-Tao
Liu
*a
b and
Wei-Tao
Liu
*a
aPhysics Department, State Key Laboratory of Surface Physics, Key Laboratory of Micro and Nano Photonic Structures [Ministry of Education (MOE)], Fudan University, Shanghai 200433, China. E-mail: wtliu@fudan.edu.cn
bPen-Tung Sah Institute of Micro-Nano Science and Technology, Institute of Artificial Intelligence, Laboratory of AI for Electrochemistry (AI4EC), Tan Kah Kee Innovation Laboratory (IKKEM), Xiamen University, Xiamen, 361005, China
First published on 10th April 2025
Abstract
The dissociation of water on TiO2 surfaces, marked by the presence of TiOH groups, is pivotal for environmental and energy applications involving TiO2. Yet characterizing these surface groups has remained a challenge. Here, we employ in situ sum-frequency vibrational spectroscopy (SFVS) to unveil the vibrational signatures of surface TiOH and undercoordinated Ti–O groups in the Ti–O vibrational frequency range, offering a clear structural indicator of TiO2 hydroxylation. Our findings confirm the spontaneous dissociation of water molecules on TiO2 surfaces, a process significantly enhanced by structural defects such as oxygen vacancies. Through methanol titration experiments, we gain molecular-level insights into the adsorption/desorption dynamics, estimating a ∼70% TiOH coverage on amorphous TiO2 under ambient conditions. This work not only deepens our understanding of TiO2/water interactions but also lays the groundwork for future SFVS investigations into these interfaces.
1. Introduction
Since the seminal work by Fujishima and Honda in 1972 reporting its photocatalytic capabilities,1 titanium dioxide (TiO2) has found wide applications in photocatalysis, environmental remediation, energy production, etc.,2–4 owing to its natural abundance, chemical stability, and environmental compatibility. Despite the broad applications, many fundamental aspects related to reactions at TiO2 interfaces remain controversial. In particular, the information about water dissociation on TiO2 surfaces is still limited, despite it being the most relevant system to applications.5,6 Most experimental studies under ultra-high-vacuum (UHV), such as scanning tunneling microscopy (STM), temperature-programmed desorption (TPD), and X-ray photoelectron spectroscopy (XPS), suggested that water adsorbs primarily in the molecular form on defect-free TiO2 (anatase) surfaces as well as on reduced surfaces with subsurface defects.2,7–10 Yet UHV is very different from the ambient conditions under which most applications involving TiO2 surfaces take place. Surface X-ray diffraction (SXRD) experiments showed the dominance of dissociated water on reduced TiO2 (anatase) surfaces under ambient conditions.11 Still, analysis of diffraction results required single crystals, while in practical applications, amorphous or polycrystalline TiO2 surfaces are usually involved due to improved catalytic performance and easy production.12–14 Surface specific sum-frequency vibrational spectroscopy (SFVS) is a nonlinear optical technique that allows probing of surfaces under ambient conditions, whether single crystalline or amorphous. It has long been employed for investigating TiO2/water interfaces,14–17 with a focus on the OH (or OD) stretching vibrations of interfacial water molecules and/or dissociated hydroxyl (OH) groups. Nevertheless, due to the overlap in frequency, the molecular and dissociated water species are difficult to distinguish via OH vibrational spectra. More importantly, the dissociation processes involve two participants: water molecules and TiO2 surfaces. Information from the water side alone is insufficient, the complementary knowledge of the TiO2 surface would be essential for a complete view of the system.18Previously, we have implemented SFVS to explore lattice vibrations of various oxides such as crystalline/amorphous silicon oxides and crystalline titanium oxides,19–21 which provided unique insights into interfacial chemistry from the oxide perspective. In this work, we investigated lattice vibrations from amorphous and polycrystalline anatase TiO2 surfaces in the ambient atmosphere, in correlation with water adsorption and dissociation processes. Besides undercoordinated surface Ti–O bonds, we further identified resonances from Ti–O–H vibrations, which are well separated from water OH stretching and bending vibrations, and can serve as an unambiguous signature of chemisorbed OH groups on TiO2. In conjunction with the titration study between methanol and water, we monitored the adsorption and dissociation of atmospheric water on TiO2 and deduced a typical coverage of TiOH being ∼70% under ambient conditions. With ultraviolet (UV) irradiation and annealing treatments that converted the amorphous TiO2 to polycrystalline phase, the results again underscored the key role of structural defects in water dissociation, as previously shown for single crystalline TiO2 surfaces.20,21 These vibrational signatures can serve as direct indicators of the TiO2 surface hydroxylation status to be monitored in situ, paving the way for a wholistic understanding of the TiO2/water interaction as combined with complementary spectroscopic information.
2. Methods
2.1. Laser system and the experimental setup of SFVS
The principles of SFG were described in detail elsewhere.20–23 In our experimental configuration, two incident beams, an 800 nm near-infrared (NIR) and a broadband infrared (IR) beam, were overlapped on the TiO2 surface to generate the SF signal in the reflection direction (refer to Fig. 1a). The basic theory of SFG can be briefly described as follows: when the IR frequency (ωIR) is near vibrational resonances, the SF signal (SSF) generated by the two incident beams is:where χNR and χR are the non-resonance and resonance contributions, with Aq, ωq, and Γq being the amplitude, frequency, and damping coefficient of the qth resonance mode, respectively. Throughout the paper, the polarization combination is SSP, where S, S, and P denote the polarization of SF, NIR, and IR beams, respectively, unless otherwise specified. The spectra in the Ti–O vibrational frequency range were normalized to those of Au (or GaAs), while the spectra in the C–H range were normalized to those of quartz.20,22

 | ||
| Fig. 1 (a) Schematic of the SFG experimental geometry. (b) Calculated local field intensity at the air/TiO2 (red curve) and TiO2/Si (black curve) interfaces of the thin film sample at various film thicknesses. (c) SF spectra in the C–H stretching vibrational range from the as-grown sample (red curve) and the sample after cleaning (blue curve). (d) The AFM topography of the pristine sample. (e) Raman and (f) XRD spectra of the as-grown sample (black curves) and annealed sample (red curves). | ||
The SFVS experiments were carried out with a femtosecond broadband laser system. Briefly, a Ti:sapphire amplifier (Spitfire ACE, Spectra Physics) produces ∼7 W of 800 nm, 35 fs pulses at a 2 kHz repetition rate. 35% of the 800 nm beam passed through a beam-splitter to pump an optical parameter amplifier, followed by a difference frequency generation stage (TOPAS-C, Spectra Physics). The rest of the light was either used to generate a narrowband beam of approximately 3 nm bandwidth by passing through an interference filter (LL01-808-25, Semrock) or reflected from a Bragg filter (N013-14-A2, OptiGrate) to generate a narrowband beam of approximately 0.5 nm bandwidth. The broadband IR and narrowband 800 nm pulses overlapped at the sample surface with incident angles of 57° and 30° (or 45°), respectively. The SF signal was collected along the reflected direction using a spectrograph (Acton SP300i, Princeton Instruments) and a CCD camera (PyLoN: 400BR eXcelon, Princeton Instruments). All SFVS measurements were conducted under ambient conditions at room temperature. The relative humidity (RH) of the environment was maintained at 40%.
2.2. Sample preparation and Raman and XRD characterization
TiO2 film samples were prepared via vacuum evaporation coating technology commissioned by the Shanghai Institute of Technical Physics, and the substrate was 0.5 mm N-type silicon. Before coating, the silicon substrate underwent sequential cleaning in an ultrasonic bath with acetone, ethanol, and deionized water for 30 minutes, followed by irradiation in an ultraviolet ozone cleaner (BZS250GF-TC) for 1 hour. While evaporating titanium dioxide, the air pressure in the chamber is maintained at 2 × 10−4 Pa, with a working gas of 30 sccm argon and 40 sccm oxygen, at a temperature of 180 °C, and an evaporation rate of 0.3 nm s−1. The typical sample thickness measuring about 90 nm was determined by X-ray reflectometry (XRR) (Fig. S1, ESI†). Here, this particular thickness was chosen to specifically enhance the total local field strength at the air/TiO2 interface (see Fig. 1b and details in the ESI†). The TiO2 films were thoroughly cleaned before the measurement, showing no detectable contaminant signal in the C–H stretching vibrational range (Fig. 1c). Fig. 1d shows a typical atomic force microscopy (AFM, MultiMode 8, Bruke) image of the film surface, with the root-mean-square (RMS) roughness being ∼0.9 nm. The ultraviolet (UV) treatment was performed using the apparatus (BZS250GF-TC, hwotech) for 12 hours. The annealing treatment was carried out using a box furnace (KSL-1400X, Ke Jing) at 400 °C for 4 hours.We further used Raman spectroscopy and XRD to characterize the TiO2 films.13,24 Both Raman and XRD spectra of the as-grown sample showed no characteristic peaks of crystalline TiO2, indicating amorphous nature (Fig. 1e and f, black curves). (The Raman signals observed at 300 cm−1 and 517 cm−1, as well as the diffraction peak at 33°, were all due to the Si substrate.) After annealing at 400 °C, the sample turned into the anatase phase, characterized by prominent Raman peaks at 140 and 632 cm−1 (Fig. 1e, red curve).13 Meanwhile, the XRD analysis of the annealed sample showed four distinct peaks at 25.4°, 33°, 38°, and 48.2°, corresponding to anatase (101), (004), and (200), and the peak at 33° was from Si (200) (Fig. 1f, red curve).24
3. Results and discussion
3.1. SFVS from the TiO2 film surface in the Ti–O vibrational frequency range
We then recorded SFG spectra at the air/TiO2 interface in the Ti–O vibrational frequency range following the sample preparation procedure described in Section 2.2. As shown in Fig. 2a, within 750–1200 cm−1, the spectrum exhibited two prominent resonance features at ∼900 cm−1 and ∼1060 cm−1. Spectra of different TiO2 film samples exhibited the same two major bands, with frequency variations within ten wavenumbers (Fig. S2, ESI†). Previously, we have identified a surface phonon mode at 860 cm−1 on the single crystalline anatase (101) surface,20,21 arising from the stretching vibration of the Ti–O bonds between surface under-coordinated titanium and subsurface oxygen ions. Considering that the surface structure of the amorphous film would be different from that of the single crystal sample, we tentatively assigned the 900 cm−1 mode to the stretching of surface under-coordinated Ti–O bonds, which would be discussed in more detail later. Meanwhile, the high-frequency mode at 1060 cm−1 is close to the Ti–O–H bending vibrational frequency that falls within the range of ∼1050 to 1100 cm−1.25–27 We therefore attributed the 1060 cm−1 mode to the surface TiOH groups of the TiO2 film. | ||
| Fig. 2 (a) A typical SF spectrum of the air/TiO2 film interface in the Ti–O vibrational frequency region. (b) Typical SF spectra from air/TiO2 film surfaces in the Ti–O vibrational frequency region: as-grown sample (black), UV irradiated as-grown sample (purple), and sample annealed at 400 °C for 4 hours (red). The spectrum from the single crystalline anatase (101) surface is shown for comparison (dashed grey curve). (c) Ti–O–H vibrational spectra from the as-grown sample (black), annealed sample (red), and annealed sample treated by UV irradiation again (purple). (d) Schematics of the TiO2 film surface structural change upon UV irradiation (left) and annealing (right) treatments. | ||
To further verify our assignment of both surface modes, we conducted UV and annealing treatments of the TiO2 surface. First, we irradiated an as-grown sample surface with UV light, which is known to promote the water dissociation on TiO2 surfaces under ambient conditions.28–30 The contrasting spectra before (black) and after (purple) UV irradiation of the as-grown TiO2 film are shown in Fig. 2b. The UV irradiation caused a notable increase in the intensity of the 1060 cm−1 mode, in accordance with its assignment to the TiOH species. Besides, the low-frequency intensity grew after the treatment, possibly contributed from UV-generated free carriers.31 Next, we annealed the as-grown samples at 400 °C for 4 hours, which would remove adsorbed water molecules and hydroxyl groups from the TiO2 surfaces.32,33 As shown in Fig. 2b (red), the 1060 cm−1 peak intensity of the annealed sample decreased drastically, which again is consistent with our assignment. This 1060 cm−1 band can therefore characterize the dissociated TiOH species on TiO2.
Meanwhile, the SF signal near ∼900 cm−1 grew appreciably upon annealing (Fig. 2b, red), showing a strong peak at 870 cm−1, closely resembling the surface phonon mode on single crystalline anatase (101) (Fig. 2b, grey dashed curve, adopted from ref. 20). This is consistent with the Raman and XRD results shown above (Fig. 1e and f), that the thermal annealing treatment converted the amorphous sample to the (poly)crystalline anatase phase. Such conversion underscored our assignment of the low-frequency mode to the vibration of surface Ti–O bonds. Overall, the TiO2 samples exhibited multiple peaks near ∼900 cm−1, with the single crystalline sample dominated by the peak at ∼870 cm−1, and amorphous samples by the peak at ∼900 cm−1. The higher frequency on the amorphous surface possibly arose from the structural variation in Ti–O bond length, Ti–O–Ti bonding angle, and/or the localization of the vibrational mode. Collectively, the UV and thermal annealing treatments agreed with our assignment of the spectral features. Fig. 2d schematically summarizes the structural changes of the amorphous TiO2 surface upon UV and annealing treatments.
It is noted that, in contrast to the amorphous TiO2 surface, our previous study on the single crystalline anatase (101) surface did not observe TiOH resonance under ambient conditions.20 Even for the annealed (poly)crystalline sample, the UV irradiation afterwards could not restore the TiOH signal (Fig. 2c). Overall, since annealing (UV irradiation) would reduce (increase) the amount of structural defects, the corresponding drop (increase) in the TiOH signal intensity upon the above treatments emphasizes that surface structural defects, including oxygen vacancies (VO) and others, are crucial in promoting the auto-dissociation of water in the ambient atmosphere.
3.2. Methanol titration of surface TiOH species
To gain more insights into the interaction between amorphous TiO2 and adsorbates, we investigated the titration behaviour between methanol and water on the surface. Previous studies on the competitive adsorption between water and methanol showed that the adsorption of methanol was more favourable than that of water from the vapor phase.16,34 In our measurement, we first exposed the TiO2 film to saturated methanol vapor at t = 0 for ∼2 minutes, then the sample was re-exposed to the ambient atmosphere. During the process, we monitored the adsorption and desorption of methanol using SFVS in the C–H vibrational frequency range, in correlation with the change in the Ti–O–H band. Fig. 3a presents the series of C–H spectra recorded over elapsed time upon methanol exposure. The black dotted curve is the spectrum of the original pristine surface. Subsequent colored spectra are those that were acquired at one-minute intervals.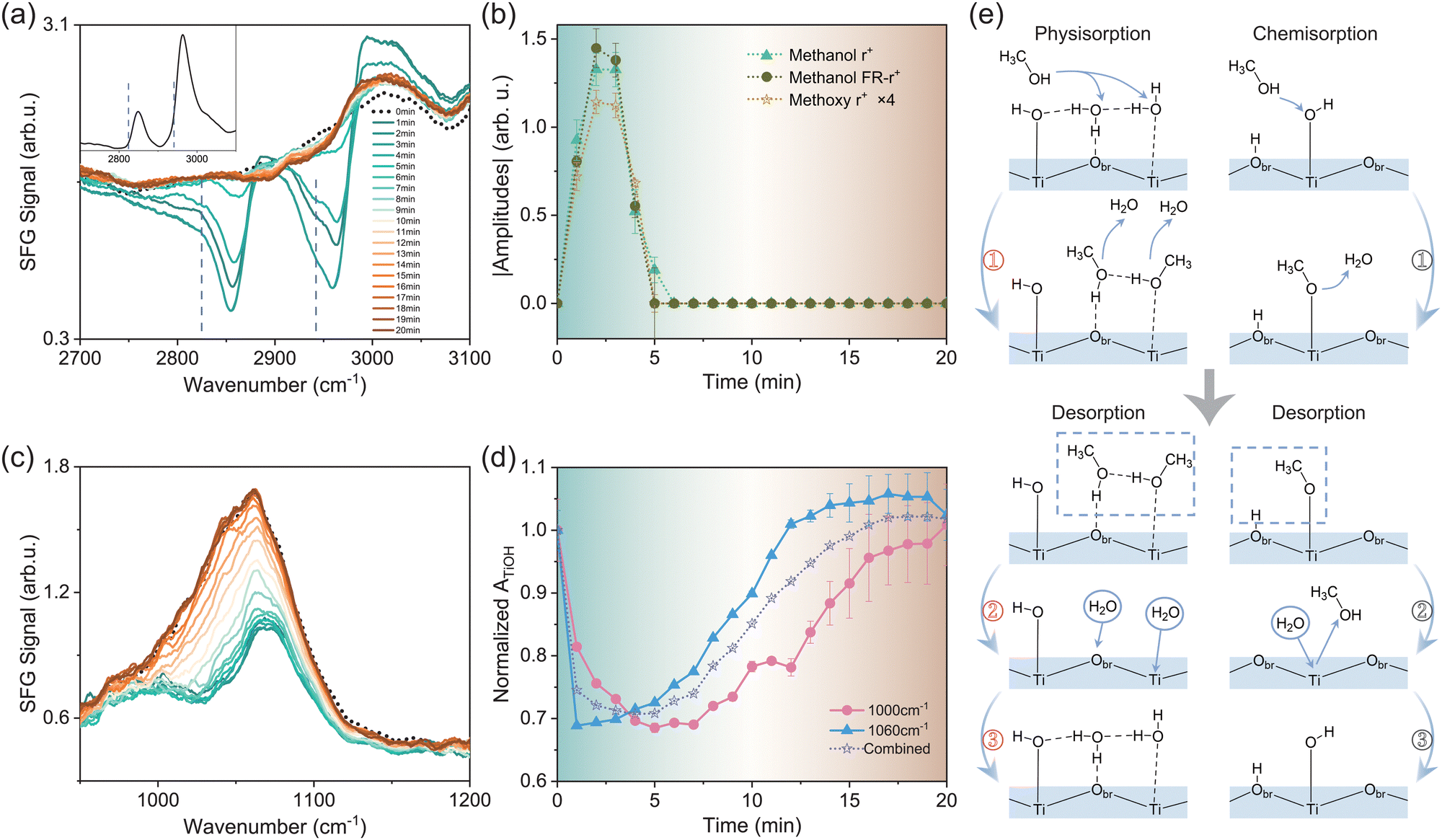 | ||
| Fig. 3 (a) SF spectra of the TiO2 film surface in the C–H vibrational region upon methanol adsorption/desorption. The inset shows the methanol adsorption spectrum on the anatase (101) surface for reference. Dashed lines mark the position of resonance modes from methoxy groups. (b) Time-dependent absolute values of the fitted amplitudes of C–H vibrational signals in (a). (c) SF spectra of the TiO2 film surface in the Ti–O–H vibrational frequency region upon methanol adsorption/desorption. The colour scheme is the same as that in (a). (d) Time dependence of the fitted amplitudes of Ti–O–H vibrational signals in (c). (e) Illustrations of the adsorption/desorption processes of methanol and water in the vapor phase. The left and right panels depict the physisorption and chemisorption processes, respectively. | ||
Upon exposure to methanol, spectra in the C–H stretching vibrational frequency range showed major resonance bands centered at 2850–2870 cm−1, 2960–2980 cm−1, and 2990–3050 cm−1 (Fig. 3a, see details in the ESI†). Some of the modes appeared as dips due to their interference with the non-resonant background of the TiO2 surface.20,35 These modes can be assigned to the CH3 symmetric stretching mode (r+), its Fermi resonance mode with the bending overtone (FR-r+), antisymmetric stretching and/or OH stretching mode from physisorbed methanol molecules,22 respectively. Meanwhile, we observed notable SF responses at the lower frequency sides of the methanol r+ and FR-r+ peaks, marked by dashed lines in Fig. 3a, which was absent on single crystalline TiO2 surfaces including anatase (101) (presented in the inset of Fig. 3a) and rutile (110), (001), and (100).22 According to the literature,16,36 these modes arise from the CH3 symmetric stretching of methoxy species (CH3O) dissociated from methanol. Observation of these modes indicated the auto-dissociation of methanol on the amorphous TiO2 film. Again, this showcases the essential role played by TiO2 structural defects in methanol dissociation. Fig. 3b illustrates the time-dependent magnitudes of the fitted amplitudes of methanol r+ and FR-r+ modes, as well as the methoxy r+ mode (magnified by 4 times for clarity). The background colour of Fig. 3b was matched that of the corresponding SF spectrum in Fig. 3a to guide the eye. It was seen that all three amplitudes rose simultaneously and reached the maxima at t = 2–3 minutes, then diminished at t ≈ 5 minutes. The same adsorption/desorption processes were repeatable for multiple cycles.
We then acquired Ti–O spectra under the same conditions to track the changes of the TiO2 surface structure. The low-frequency mode near ∼900 cm−1 did not exhibit notable changes upon methanol adsorption/desorption (see Fig. S3 in the ESI†). In contrast, the high-frequency band from TiOH groups evolved in close correlation with the methanol response, as shown in Fig. 3c. The black dotted curve represents the spectrum before methanol adsorption. Within the first 2–3 minutes, the intensity of the TiOH band dropped rapidly in accordance with the methanol adsorption, showing the titration behaviour between methanol and water. Meanwhile, the band split into two bands at ∼1000 and 1070 cm−1, respectively (see details in the ESI†). After t ≈ 3 minutes, upon methanol desorption, the TiOH band started to increase but at a much slower pace, and eventually recovered to the initial profile in about 20 minutes. By analysing the time-dependent TiOH spectra, we found that all spectra could be fitted with two bands around 990–1010 cm−1 and 1060–1070 cm−1, respectively. According to ref. 37, the frequency of the R–O–H bending mode is affected by the hydrogen bonding (H-bonding) environment. The stronger the H-bonding it senses, the higher the R–O–H bending frequency becomes. We therefore attribute the bands near 1000 cm−1 and 1060 cm−1 to weak and strong H-bonded TiOH groups, respectively. Fig. 3d displays the time-dependent amplitudes of the two modes extracted from fitting (solid curves), as well as their combined amplitude for reference (dot curve). The amplitudes were all normalized to their respective values at t = 0 minute.
With both CH and TiOH spectra available, we now discuss the microscopic picture underlying the titration procedure. Before introducing methanol molecules, the TiO2 surface is covered by both molecular water17 and dissociated OH groups (Fig. 3e). Upon the introduction of methanol vapor (t = 0–2 minute), both physisorption and chemisorption of methanol would occur. Fig. 3e (left panel ①) presents the typical physisorption scheme of methanol molecules, which could replace physisorbed water molecules on both surface Ti and bridging oxygen (Obr) sites. Accordingly, we observed the increase of the r+ and FR-r+ amplitudes of methanol molecules. Although physisorbed methanol molecules could not directly replace TiOH groups, they would weaken the overall surface H-bonding network, as methanol cannot form as many H-bonds as water per molecule. This agreed with the trend observed in Fig. 3d, that when the intensities of both strong and weak H-bonded TiOH groups dropped within the first few minutes, the former dropped more rapidly than the latter due to the overall weakened surface H-bonding network.
For the chemisorption of methanol, it involves the dissociation of methanol molecules into methoxy groups via the reaction with surface terminal hydrogen groups, described by CH3OH (g) + Ti–OH → CH3O–Ti + H2O (g) (Fig. 3e, right panel ①).12 Meanwhile, as mentioned earlier, the strong H-bonded TiOH groups dropped more rapidly than weak H-bonded ones (Fig. 3d). This may also be contributed by the proton transfer process involved in the above reaction facilitated by the H-bonding network.38 In addition, as we previously observed on anatase (101), the configuration energy of surface VO could be lower upon the adsorption of methanol.20 This could further facilitate the methanol dissociation in a “self-catalysis” manner as demonstrated in ref. 39, that a methanol molecule would auto-dissociate on the surface VO site.
We could further estimate the coverage of TiOH groups based on the above results. By assuming a one-to-one conversion between methoxy and desorbed TiOH species, we derived that the coverage of TiOH is about 72.5% ± 14.5% under ambient conditions (1 atm, RH = 40%, see details in the ESI†). This value is greater than that predicted for pristine crystalline surfaces,5 for example, 20–30% on rutile (110),6 but is close to the 75% found by SXRD on heavily reduced anatase (101) in the ambient atmosphere.11
At 3–5 minutes, the methanol started to desorb. The physisorbed methanol molecules could simply leave the surface (Fig. 3e, left panel ② and ③). For chemisorbed methoxy species, they are usually assumed to be hydrolysed by water via CH3O–Ti + H2O (g) → CH3OH (g) + Ti–OH.40 However, this reaction would lead to TiOH formation, yet the TiOH intensity hardly increased during the same period of time. Such an observation suggested that, in our case, the methoxy desorption could also occur via processes other than hydrolysis. One possibility is that the chemisorbed methoxy could recombine with neighbouring protons on Obr sites and desorb from the surface (Fig. 3e, right panel ②). Meanwhile, the slow recovery of the TiOH signal in comparison to the rapid rise of the methoxy signal (upon adsorption) is in accordance with the fact that methoxy adsorbs more favorably than water.
4. Conclusions
To conclude, we identified SF spectroscopic signatures corresponding to chemisorbed TiOH groups in the Ti–O vibrational frequency range, which can serve as a direct, unambiguous indicator of dissociated water on TiO2 surfaces. UV irradiation and thermal annealing treatments of the amorphous TiO2 surface underscore the key role played by structural defects in promoting the water dissociation, in line with our previous studies on single crystalline TiO2 surfaces. By monitoring the competitive adsorption between methanol and water with both C–H and Ti–O vibrational spectra, we gained insights into this process at the molecular level and estimated the TiOH coverage to be ∼70% in the ambient atmosphere. This study paves the way for future investigations of TiO2/water interfaces and reactions based on this structural indicator.Author contributions
H. Li and W. Zheng contributed equally to this work. H. Li: experimentation, characterization, data curation; W. Zheng: data curation, formal analysis, writing – original draft; X. Liu & F. Tang: calculations and discussion; J. Li & L. Wen: characterization; W.-T. Liu: conceptualization, methodology, final writing, funding acquisition, project administration. All authors have approved the final version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research and Development Program of China (2024YFA1409803 and 2024YFA1210804) and the National Natural Science Foundation of China (12250002), and the Science and Technology Commission of Shanghai Municipality (Grant No. 22XD1400200, 23JC1400400, and 23DZ2260100). F. T. acknowledges the start-up fund from Xiamen University. The sample fabrication was conducted at the Shanghai Institute of Technical Physics Chinese Academy of Sciences.References
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- A. Selloni, Annu. Rev. Phys. Chem., 2024, 75, 47–65 CrossRef CAS PubMed.
- A. H. Navidpour, S. Abbasi, D. Li, A. Mojiri and J. L. Zhou, Catalysts, 2023, 13, 232 CrossRef CAS.
- R. Li, T. Li and Q. Zhou, Catalysts, 2020, 10, 804 CrossRef CAS.
- Z. Zeng, F. Wodaczek, K. Liu, F. Stein, J. Hutter, J. Chen and B. Cheng, Nat. Commun., 2023, 14, 6131 CrossRef CAS PubMed.
- B. Wen, M. F. C. Andrade, L. Liu and A. Selloni, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2212250120 CrossRef CAS PubMed.
- L. Yang, M. Huang, N. Feng, M. Wang, J. Xu, Y. Jiang, D. Ma and F. Deng, Chem. Sci., 2024, 15, 11902–11911 RSC.
- F. Fasulo, G. Piccini, A. B. Muñoz-García, M. Pavone and M. Parrinello, J. Phys. Chem. C, 2022, 126, 15752–15758 CrossRef CAS.
- C. Dette, M. A. Perez-Osorio, S. Mange, F. Giustino, S. J. Jung and K. Kern, J. Phys. Chem. C, 2018, 122, 11954–11960 CrossRef CAS.
- J. Li, J. Hu and J. Cheng, Phys. Chem. Chem. Phys., 2023, 25, 29143–29154 RSC.
- I. M. Nadeem, J. P. W. Treacy, S. Selcuk, X. Torrelles, H. Hussain, A. Wilson, D. C. Grinter, G. Cabailh, O. Bikondoa, C. Nicklin, A. Selloni, J. Zegenhagen, R. Lindsay and G. Thornton, J. Phys. Chem. Lett., 2018, 9, 3131–3136 CrossRef CAS PubMed.
- Y. Wu, F. Gao, H. Wang, L. Kovarik, B. Sudduth and Y. Wang, J. Phys. Chem. C, 2021, 125, 3988–4000 CrossRef CAS.
- E. H. G. Backus, S. Hosseinpour, C. Ramanan, S. Sun, S. J. Schlegel, M. Zelenka, X. Jia, M. Gebhard, A. Devi, H. I. Wang and M. Bonn, Angew. Chem., Int. Ed., 2024, 63, e202312123 CrossRef CAS PubMed.
- S. Hosseinpour, F. Tang, F. Wang, R. A. Livingstone, S. J. Schlegel, T. Ohto, M. Bonn, Y. Nagata and E. H. G. Backus, J. Phys. Chem. Lett., 2017, 8, 2195–2199 CrossRef CAS PubMed.
- S. J. Schlegel, S. Hosseinpour, M. Gebhard, A. Devi, M. Bonn and E. H. G. Backus, Phys. Chem. Chem. Phys., 2019, 21, 8956–8964 RSC.
- C. Y. Wang, H. Groenzin and M. J. Shultz, J. Am. Chem. Soc., 2005, 127, 9736–9744 CrossRef CAS PubMed.
- M. Qu, G. Huang, X. Liu, X. Nie, C. Qi, H. Wang, J. Hu, H. Fang, Y. Gao, W. Liu, J. S. Francisco and C. Wang, Chem. Sci., 2022, 13, 10546–10554 RSC.
- M. F. C. Andrade, H. Ko, R. Car and A. Selloni, J. Phys. Chem. Lett., 2018, 9, 6716–6721 CrossRef PubMed.
- X. Li, F. S. Brigiano, S. Pezzotti, X. Liu, W. Chen, H. Chen, Y. Li, H. Li, X. Lin, W. Zheng, Y. Wang, Y. R. Shen, M. Gaigeot and W. Liu, Nat. Chem., 2025, 17, 198–203 CrossRef CAS PubMed.
- Y. Cao, S. Chen, Y. Li, Y. Gao, D. Yang, Y. R. Shen and W. Liu, Sci. Adv., 2016, 2, e1601162 CrossRef PubMed.
- X. Liu, T. Zhou and W. Liu, J. Chem. Phys., 2019, 150, 84701 CrossRef PubMed.
- D. Yang, Y. Li, X. Liu, Y. Cao, Y. Gao, Y. R. Shen and W. Liu, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E3888–E3894 CrossRef CAS PubMed.
- X. Liu, D. Yang, Y. Li, Y. Gao and W. Liu, J. Phys. Chem. C, 2019, 123, 29759–29764 CrossRef CAS.
- Y. Son, M. Lee and Y. Park, Langmuir, 2021, 37, 1850–1860 CrossRef CAS PubMed.
- G. Mattioli, F. Filippone and A. Amore Bonapasta, J. Am. Chem. Soc., 2006, 128, 13772–13780 CrossRef CAS PubMed.
- X. Y. Ling, R. Yan, S. Lo, D. T. Hoang, C. Liu, M. A. Fardy, S. B. Khan, A. M. Asiri, S. M. Bawaked and P. Yang, Nano Res., 2014, 7, 132–143 CrossRef CAS.
- J. Li, L. Zhu, Y. Wu, Y. Harima, A. Zhang and H. Tang, Polymer, 2006, 47, 7361–7367 CrossRef CAS.
- Y. Zhang, Z. Xu, G. Li, X. Huang, W. Hao and Y. Bi, Angew. Chem., Int. Ed., 2019, 58, 14229–14233 CrossRef CAS PubMed.
- I. M. Nadeem, G. T. Harrison, A. Wilson, C. L. Pang, J. Zegenhagen and G. Thornton, J. Phys. Chem. B, 2018, 122, 834–839 CrossRef CAS PubMed.
- Y. Li and Y. Gao, Phys. Rev. Lett., 2014, 112, 206101 CrossRef.
- X. Liu, T. Zhou, Z. Qin, C. Ma, F. Lu, T. Liu, J. Li, S. H. Wei, G. Cheng and W. Liu, Sci. Adv., 2023, 9, eadg7037 CrossRef CAS PubMed.
- C. E. Nanayakkara, J. Pettibone and V. H. Grassian, Phys. Chem. Chem. Phys., 2012, 14, 6957–6966 RSC.
- Y. Gao, Y. Masuda, W. Seo, H. Ohta and K. Koumoto, Ceram. Int., 2004, 30, 1365–1368 CrossRef CAS.
- C. Fu, Y. Zhang, X. Sun, F. Fang and W. Huang, J. Phys. Chem. C, 2022, 126, 8615–8626 CrossRef CAS.
- R. Feng, A. Liu, S. Liu, J. Shi, R. Zhang and Z. Ren, J. Phys. Chem. C, 2015, 119, 9798–9804 CrossRef CAS.
- Z. Ren, R. Zhang, T. Luo, W. Zeng, C. Zhou and X. Yang, J. Phys. Chem. C, 2023, 127, 1049–1056 CrossRef.
- T. Seki, K. Chiang, C. Yu, X. Yu, M. Okuno, J. Hunger, Y. Nagata and M. Bonn, J. Phys. Chem. Lett., 2020, 11, 8459–8469 CrossRef CAS PubMed.
- X. Ma, Y. Shi, J. Liu, X. Li, X. Cui, S. Tan, J. Zhao and B. Wang, J. Am. Chem. Soc., 2022, 144, 13565–13573 CrossRef CAS PubMed.
- A. Tilocca and A. Selloni, J. Phys. Chem. B, 2004, 108, 19314–19319 CrossRef CAS.
- C. Y. Wang, H. Groenzin and M. J. Shultz, J. Am. Chem. Soc., 2004, 126, 8094–8095 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cp00400d |
| ‡ These authors contributed equally to this work. |
| This journal is © the Owner Societies 2025 |