
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVacuum ultraviolet spectroscopy of pyrimidine derivatives: the effect of halogenation†
Fábris
Kossoski
 *a,
Mónica
Mendes
*b,
Ana I.
Lozano
c,
Rodrigo
Rodrigues
b,
Nykola C.
Jones
d,
Søren V.
Hoffmann
d and
Filipe Ferreira
da Silva
b
*a,
Mónica
Mendes
*b,
Ana I.
Lozano
c,
Rodrigo
Rodrigues
b,
Nykola C.
Jones
d,
Søren V.
Hoffmann
d and
Filipe Ferreira
da Silva
b
aLaboratoire de Chimie et Physique Quantiques (UMR 5626), Université de Toulouse, CNRS, UPS, France. E-mail: fkossoski@irsamc.ups-tlse.fr
bCEFITEC, Department of Physics, NOVA School of Science and Technology, NOVA University Lisbon, Caparica, 2829-516, Portugal. E-mail: mf.mendes@fct.unl.pt
cInstitut de Recherche en Astrophysique et Planétologie (IRAP), Université de Toulouse, CNRS, CNES, 9 Avenue du Colonel Roche, Toulouse, F-31028, France
dISA, Department of Physics and Astronomy, Aarhus University, Ny Munkegade 120, Aarhus C, DK-8000, Denmark
First published on 7th April 2025
Abstract
As a prototypical molecule in the important class of halopyrimidines, 2-chloropyrimidine has been the subject of numerous spectroscopic studies. However, its absorption spectrum under vacuum ultraviolet (VUV) radiation has not yet been reported. Here, we close this gap by presenting high-resolution VUV photoabsorption cross-sections in the 3.7–10.8 eV range. Based on time-dependent density functional theory (TDDFT) calculations performed within the nuclear ensemble approach, we are able to characterize the main features of the measured spectrum. By comparing the present results for 2-chloropyrimidine with those of 2-bromopyrimidine and pyrimidine, we find that the effect of the halogen atom increases remarkably with the photon energy. The two lowest-lying absorption bands are overall similar for the three molecules, apart from some differences in the vibrational progressions in band I (3.7–4.6 eV) and minor energy shifts in band II (4.6–5.7 eV). Larger shifts appear in band III (5.7–6.7 eV), especially when comparing pyrimidine with the two halogenated species. The three molecules absorb more strongly in the region of band IV (6.7–8.2 eV), where the bands look qualitatively different because the mixing of excited configurations is strongly dependent on the species. At higher energies (8.2–10.8 eV) the three spectra no longer resemble each other. An important finding of this study is the very satisfactory comparison between experiment and theory, as the combination of TDDFT calculations with the nuclear ensemble approach yields cross-sections much closer to experiments than the simpler vertical approximation, in shape and magnitude, and across the whole spectral range surveyed here.
1. Introduction
The halogenation of organic molecules is one of the most significant chemical modifications in molecular synthesis. The halogen atom acts as an electrophilic species, leading to alterations in the functional properties of the molecule, as well as in its geometry, polarizability and electron affinity.1,2 It is well-established that the halogen bond strength is tuneable by modifying the halogen atom according to its polarizability scale (F < Cl < Br < I). This represents a very useful tool to control intermolecular recognition and assembly, with impacts in a variety of research areas.1,3 Indeed, halogenated compounds are found in 20% of medical drugs4–6 and in 30% of agrochemicals.7 Moreover, they can be used as dyes and flame retardants, and in other areas such as medical diagnosis and materials science.2,7,8Pyrimidine and its derivatives are part of the diazine group and are found in natural and synthetic compounds. In pyrimidines, nitrogen atoms replace the CH groups at positions 1 and 3 of the benzene ring, which gives rise to interesting properties relevant to applications in industry, chemistry, and medicine.9,10 Therefore, the physicochemical properties of pyrimidine have been attracting considerable attention in recent decades, and several experimental and theoretical methodologies have been employed in order to deepen the knowledge on its spectroscopy and dynamics.11–20
In particular, halogenated pyrimidines are found in various therapeutic applications in medicinal chemistry, since pyrimidines are building blocks of nucleic acids (DNA and RNA) and, therefore, they are good candidates in pharmacology.10 The replacement of thymine in DNA by a halogenated pyrimidine enhances radiation damage due to its increased radiosensitivity.21,22 Bromodeoxyuridine, iododeoxyuridine, 5-fluorouracil and gemcitabine are examples of tumour-specific radiosensitizers used in cancer therapy.23–30 The irradiation of halopyrimidines by UV, X-ray or ion beams can trigger a variety of chemical alterations, as induced by photochemical hydration, dimerization, excitation, ionization, and bond dissociation. Therefore, a detailed description of the electronic excitation and ionization of pyrimidine and its derivatives is essential to understand the underlying radiation-induced and photo-induced reactions. In light of this motivation, many spectroscopic studies on halopyrimidines31–35 and halobenzenes36,37 have been reported.
Among the class of halopyrimidines, 2-chloropyrimidine (2ClPyr) in particular has been extensively investigated. Thorough analyses by X-ray photoemission spectroscopy,38 near edge X-ray absorption fine structure spectroscopy,39 and Auger spectroscopy40 have been reported by Bolognesi and co-workers. Its valence photoelectron spectrum was investigated by O’Keeffe et al.41 The photoionization mass spectrum of 2ClPyr was studied by Castrovilli et al.42,43 at 14 eV. He et al.44 explored the first electronically excited state of 2ClPyr and its cationic ground state using two-colour resonance-enhanced multiphoton ionization (REMPI) and zero kinetic energy photoelectron spectroscopy. The UV absorption spectrum of 2ClPyr in solution was reported in a previous publication.45 Additionally, the photochemistry of 2ClPyr in solution was investigated by Lindquist et al.46 at 254 nm (4.88 eV). We further mention studies on its vibrational frequencies, via infrared47 and Raman spectroscopy,31,34 and on its resonant anions, through electron transmission and dissociative electron attachment experiments,48 as well as theoretically.49,50 Moreover, the total cross-sections for the single electron-impact ionization of pyrimidine and halogenated molecules have been calculated using the binary-encounter-Bethe model, from the ionization threshold up to 5 keV.51 Despite numerous studies on 2ClPyr, it has not yet been probed throughout the vacuum ultraviolet (VUV) spectral range, which is a great spectroscopic technique to better understand electronically excited states, photochemistry, and radiation chemistry.
Here, we present a combined experimental–theoretical investigation on the VUV photoabsorption of gas-phase 2ClPyr, in the 3.7–10.8 eV energy range. The present goals are threefold. First, to present and discuss the VUV photoabsorption cross-sections of 2ClPyr, which are the first ever measured for this molecule, to the best of our knowledge. The interpretation of its many absorption bands is done with the support of time-dependent density functional theory (TDDFT) calculations. Our second goal is to carry out a comparison among the present results for 2ClPyr and the available results for pyrimidine12 and 2-bromopyrimidine (2BrPyr),52 thus addressing the effect of halogenation on the VUV spectra. Here, we only compare with the latest and better resolved spectrum of pyrimidine,12 but we refer to older experiments in ref. 53 and 54. The third goal of this contribution is to assess three different treatments for the nuclei in the calculation of photoabsorption cross-sections, namely, the vertical approximation, the nuclear ensemble approach (NEA), and the Franck–Condon Herzberg–Teller (FCHT) approach. The same electronic structure level of theory is employed for the three models. We are particularly interested in how going beyond the vertical approximation through the NEA changes the shape and magnitude of the cross-section across the wide (3.7–10.8 eV) energy range covered here.
2. Methods
2.1. Experimental methods
The high-resolution VUV photoabsorption spectrum of 2ClPyr was measured using the AU-UV beam line of the ASTRID2 storage ring facility at Aarhus University in Denmark, which is described in detail elsewhere.36,55 Briefly, the apparatus comprises a gas cell coupled to a monochromator with a MgF2 window which plays the role of separating it from the ultra-high vacuum (UHV) of the beam line. After the interaction of synchrotron radiation with a static gas sample, the transmitted light intensity is detected by a photomultiplier tube (PMT). By making use of a capacitance manometer (Chell CDG100D), it is possible to record the absolute pressure of the effusive molecular gas. The wavelength is selected using a toroidal dispersion grating with 2000 lines per mm providing a resolution of 0.075 nm, corresponding to 3 meV at the midpoint of the present energy range. As described in detail in ref. 55, the full wavelength range of the monochromator is calibrated through the measurement of fine peaks for nitrogen and sulphur dioxide, with an accuracy of ±0.05 nm. Below 200 nm (energies above 6.20 eV) it is necessary to avoid any absorption of O2 from the air, and for that, there is a small gap between the PMT and the MgF2 gas cell exit window which is evacuated using a scroll pump. For higher wavelength measurements, air is admitted into the gap at atmospheric pressure to allow O2 to absorb any higher orders of light produced by the monochromator.Absolute photoabsorption cross-sections (σ) were obtained at room temperature (∼25 °C) by using the Beer–Lambert attenuation law It = I0 exp(−Nσl), where It is the light intensity transmitted through the gas sample, I0 is the intensity transmitted through the evacuated cell, N is the molecular density, and l is the absorption path length (15.5 cm). The range of pressures used for the present measurements was 0.03 to 0.25 mbar. The spectrum was measured in the energy range from 3.757 eV (330 nm) to 10.781 eV (115 nm) using data points separated by 0.05 nm (see Fig. 1). The accuracy of the cross-sections is estimated to be ∼10%, which is ensured by recording the VUV spectra in small (5 or 10 nm) sections to allow optimization of pressure according to the local cross-sections, with at least 10 points overlapping between the adjoining ranges.55,56 The vapour pressure of the sample at room temperature was sufficient for measurement, so no heating was required. The reproducibility of the spectrum was confirmed through repeated measurement of each region, ensuring a good overlap between adjoining ranges and also with a broad scan taken in larger step sizes. An in-depth description of the experimental procedure used to measure gas phase photoabsorption spectra is given in ref. 55.
 | ||
| Fig. 1 High-resolution VUV photoabsorption cross-section of 2-chloropyrimidine (present work, green), pyrimidine12 (grey), and 2-bromopyrimidine52 (purple). | ||
The 2ClPyr sample (CAS: 1722-12-9) was purchased from Alfa Aesar with a stated purity of ∼98%. The sample was used with no further purification and degassed before measurement through repeated freeze–pump–thaw cycles.
2.2. Theoretical methods
Because we are interested in comparing the VUV spectra of 2ClPyr with those of 2BrPyr52 and pyrimidine,12 here we employed the same methods as in our previous study on 2BrPyr.52 The excited states of 2ClPyr were described with time-dependent density functional theory (TDDFT) calculations. We adopted the CAM-B3LYP57 functional with the aug-cc-pVDZ basis set supplemented with 2s2p2d even-tempered diffuse functions placed at the carbon atom at position 2 of the ring (bonded to the chlorine atom). Their exponents were generated from the smallest exponents of the aug-cc-pVDZ set by successive divisions by 4. The choice of functional (CAM-B3LYP) is motivated by its favourable performance for excitation energies58 and oscillator strengths59 when compared with alternative functionals. In addition, range-separated functions like CAM-B3LYP are more advisable to treat Rydberg states. Similarly, an improved description of these states requires the inclusion of more diffuse functions, hence the augmented 2s2p2d set. Moving to the larger aug-cc-pVTZ+2s2p2d (augmented analogously) basis set produces an average absolute shift of 0.01 eV in the vertical excitation energies of the 40 lowest-lying excited states. This minor effect and the need to perform repeated excited-state calculations (see below) justify the adoption of the aug-cc-pVDZ+2s2p2d basis set. To have a fair comparison among the calculated excited states of 2ClPyr, 2BrPyr and pyrimidine, we performed additional excited-state calculations for pyrimidine with the same level of theory. The ground-state geometry and the vibrational normal modes and frequencies were obtained with density functional theory calculations and the same functional and basis set described above. The ESI† contains the Cartesian coordinates, vibrational modes, and harmonic and anharmonic frequencies, the latter computed with the generalized second-order vibrational perturbation method.60,61 We employed Gaussian 16 software62 to perform all the electronic structure calculations.Three different models were employed to calculate the photoabsorption cross-sections. The first and simplest one is the vertical approximation, which only considers the excited-state energies and oscillator strengths evaluated at the ground-state equilibrium geometry. It completely neglects vibrational effects of ground and excited states. The second model is the nuclear ensemble approach (NEA).63,64 It goes beyond the vertical approximation by emulating nuclear quantum effects of the electronic ground state (before photoexcitation). This is achieved by properly averaging excitation energies and oscillator strengths over a collection of configurations that sample the underlying vibrational wave function. These two models were used for the full spectral range investigated experimentally (between 3.7 and 10.8 eV). Finally, to understand the nature of the very rich vibrational structures of band I (between 3.7 and 4.6 eV), we employ the Franck–Condon Herzberg–Teller (FCHT) method. This third model describes the nuclei quantum-mechanically in both ground and excited states (before and after photoexcitation) but relies on harmonic approximations for energies and transition properties.
For the NEA calculations, the configurations were sampled from the Wigner distribution of independent harmonic potentials at 298 K (matching the experimental condition). Each harmonic potential corresponds to a vibrational normal mode of the electronic ground state. For each geometry qi, excitation energies ΔE0n(qi) and oscillator strengths f0n(qi) between the ground (0) and the n-th excited state are calculated. This allows us to obtain the photoabsorption cross-section (expressed in atomic units),

![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) is probably excessive if the goal is to reproduce the main trends at lower energies. Here, however, as in our previous study for 2BrPyr,52 we are equally interested in the higher energies of the VUV spectra, where a high density of overlapping states is found. To obtain properly converged cross-sections with the NEA at this regime, while keeping artificial undulations to a minimum, a large number of configurations and excited states must be considered. The Newton-X package65 was used to handle the NEA calculations.
000) is probably excessive if the goal is to reproduce the main trends at lower energies. Here, however, as in our previous study for 2BrPyr,52 we are equally interested in the higher energies of the VUV spectra, where a high density of overlapping states is found. To obtain properly converged cross-sections with the NEA at this regime, while keeping artificial undulations to a minimum, a large number of configurations and excited states must be considered. The Newton-X package65 was used to handle the NEA calculations.
The cross-sections calculated in the vertical approximation can be seen as a limiting case of the NEA where the equilibrium geometry is the single configuration taken. Here, we employed a Gaussian function with a linewidth of 0.5 eV to obtain the cross-sections in the vertical approximation. This value was chosen such that the calculated spectrum becomes overall similar in shape to the experimental one. The FCHT calculations were employed in a time-independent framework and with the adiabatic Hessian model, as implemented in Gaussian 16.62 Following the procedure described in ref. 66, we employed excited-state anharmonic frequencies, estimated through a mode-specific scaling of the calculated ground-state anharmonic frequencies. Temperature effects were considered in the FCHT calculations by including transitions from vibrationally excited levels. Here, we accounted for all the levels whose initial population is larger than 1% for a temperature of 298 K (which matches the experimental condition). Exploratory calculations with different thresholds showed minute changes in the computed spectrum. Finally, the intensity of each vibronic transition is weighted by the corresponding Boltzmann population of the initial vibrational level. A Gaussian linewidth of 2.5 meV was adopted to plot the FCHT spectrum. Throughout the discussion, we adopt the Mulliken convention (normal modes are first ordered by symmetry and then by decreasing frequencies). The character of the excited state normal modes and their vibrational frequencies can be found in the ESI.†
3. Results and discussion
Fig. 1 compares the presently measured high-resolution VUV photoabsorption spectrum of 2ClPyr with those previously reported for 2BrPyr52 and pyrimidine,12 in the 3.7–10.8 eV photon energy range. A key finding is that three spectra become increasingly more dissimilar as the photon energy increases. Moreover, the differences between the spectrum of pyrimidine and of its halogenated derivatives are more pronounced than when comparing the spectra of 2ClPyr and 2BrPyr. While this is clearly seen at lower energies, at higher energies the qualitative similarities between 2ClPyr and 2BrPyr also wane off.The measured and calculated photoabsorption cross-section curves of 2ClPyr are compared in Fig. 2. It is important to recall that the vertical approximation and the NEA make use of the same electronic structure level of theory. Whereas the vertical approximation yields an overall qualitatively correct spectrum, accounting for vibrational effects through the NEA substantially improves the agreement with experiment. This allowed us to confidently interpret the various absorption bands of 2ClPyr.
 | ||
| Fig. 2 Comparison between the experimental (green) and calculated photoabsorption cross-section of 2-chloropyrimidine, obtained with the NEA (dashed red) and with the vertical approximation (dotted red). | ||
In comparison to the cruder vertical treatment, the NEA redshifts the absorption bands, systematically approaching the measured ones, which is a well-documented effect.67–70 In addition, it changes the magnitude of the cross-sections, bringing them much closer to the experimental values. This is also in line with the few studies that have addressed the comparison between the cross-section magnitudes obtained with both models.52,70,71 Furthermore, we find that the NEA outperforms the vertical approximation for the high energy part of the spectrum, although the improvement is less striking than at the lower energies. This observation corroborates previous results that showed the consistently superior performance of the NEA across the wide spectral range of VUV.52,70 This is not only a merit of the NEA but, equally important, of the employed electronic structure method. However, the level of agreement at higher energies is less satisfactory, and we return to this point later.
In the following, we detail each of the absorption bands of 2ClPyr, while discussing the halogenation effect by comparing with the available results for 2BrPyr52 and pyrimidine.12 The text is organized according to energy ranges, each one loosely defining a band. The present labelling of the bands is such that the optically bright and analogous states of pyrimidine, 2ClPyr and 2BrPyr fall within the same band as much as possible. For this reason, notice that bands IV and V of 2BrPyr used in ref. 52 are grouped as band IV here. Similarly band VI in ref. 52 corresponds to band V in the present study. The theoretical assignment of the electronically excited states of 2ClPyr is given in Table 1, together with the computed vertical excitation energies and oscillator strengths, and the experimentally observed energy and cross-section at each maximum up to band IV. The orbitals relevant for the assignments are shown in Fig. 3.
| Experimental | Theoretical | |||||
|---|---|---|---|---|---|---|
| Band | Band max. | Peak cross-section | State | ΔE | f 0 | Character |
| I | 4.27 | 1.0 | 1B1 | 4.58 | 0.0043 | 0.99 n−(b2) π*(a2) |
| II | 4.91 | 7.8 | 1A2 | 5.05 | 0 | 0.99 n−(b2) π*(b1) |
| 1B2 | 5.50 | 0.0447 | 0.87 π(b1) π*(a2) | |||
| III | 6.18 | 33.5 | 1A2 | 6.29 | 0 | 0.97 n+(a1) π*(a2) |
| 1A1 | 6.29 | 0.2187 | 0.92 π(b1) π*(b1) | |||
| 1B1 | 6.71 | 0.0048 | 0.99 n+(a1) π*(b1) | |||
| IV | 7.15 | 77.6 | 1A2 | 6.98 | 0 | 1.00 nCl(b2) π*(b1) |
| 1B2 | 7.04 | 0.0004 | 0.98 n−(b2) 3s(a1) | |||
| 1B1 | 7.07 | 0.0010 | 0.99 nCl(b2) π*(a2) | |||
| 1B1 | 7.12 | 0.0270 | 0.98 π(b1) 3s(a1) | |||
| 1A1 | 7.41 | 0.3697 | 0.76 π(a2) π*(a2) | |||
| 1B2 | 7.44 | 0.0641 | 0.46 π(a2) π*(b1) + 0.32 πCl(b1) π*(a2) | |||
| 1B1 | 7.50 | 0.0068 | 0.99 π(b1)  |
|||
| 1B2 | 7.69 | 0.0051 | 0.73 n−/nCl(b2) 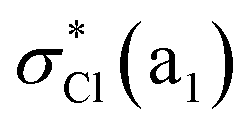 |
|||
| 1A1 | 7.69 | 0.0001 | 0.87 n−(b2) 3py(b2) | |||
| 7.93 | 60.4 | 1B1 | 7.83 | 0.0009 | 0.94 π(b1)  |
|
| 1B2 | 7.85 | 0.0113 | 0.91 n−/nCl(b2)  |
|||
| 1A2 | 7.88 | 0 | 0.95 n−(b2) 3px(b1) | |||
| 1A2 | 7.91 | 0 | 0.95 π(b1) 3py(b2) | |||
| 1A1 | 7.97 | 0.0094 | 0.90 π(b1) 3px(b1) | |||
| 1B2 | 8.15 | 0.2733 | 0.54 πCl(b1) π*(a2) + 0.29 π(a2) π*(b1) | |||
| 1B2 | 8.22 | 0.0246 | 0.56 n−(b2) 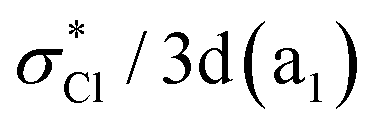 + 0.38 nCl(b2) + 0.38 nCl(b2)  |
|||
| 1A1 | 8.24 | 0.0674 | 0.75 πCl(b1) π*(b1) | |||
| 1B2 | 8.36 | 0.0294 | 0.71 n−(b2)  |
|||
| 1B1 | 8.38 | 0.0006 | 0.97 π(b1) 4s(a1) | |||
 | ||
| Fig. 3 The most relevant orbitals involved in the low-lying electronic transitions of 2-chloropyrimidine. | ||
3.1. The 3.7–4.7 eV photon energy range (Band I)
Fig. 4 focuses on the energetically lowest-lying band in the VUV spectrum of 2ClPyr. This band originates from a transition into the 1B1 first singlet excited state, of π*(a2) ← n−(b2) character, whereas the 1A2 second singlet excited state should have a much smaller contribution to the high-energy tail of the band based on the NEA results. This band has the lowest intensity in the energy range investigated here. | ||
| Fig. 4 Band I of the experimental photoabsorption cross-sections of 2-chloropyrimidine (green), compared, at the top, with those of pyrimidine12 (grey) and 2-bromopyrimidine52 (purple); in the middle, with TDDFT calculations performed within the FCHT approximation (solid red), the NEA (dashed red), and the vertical approximation (dotted red); and on the bottom, with the FCHT curve red-shifted (solid red) to match the experimental one, with selected νif transitions from the i-th to f-th level of the ν mode indicated. | ||
The spectrum of 2ClPyr is also compared with the available data for 2BrPyr52 and pyrimidine12 in Fig. 4. Whereas the overall shape and magnitude of the band are very similar across the three molecules, important differences can be seen in their fine structures. These are due to excitation of specific vibrational levels of the electronically excited state, which will be discussed in more detail later. Based on the FCHT results also discussed later, the sharp peak at 3.892 eV can be assigned to the 0–0 transition i.e. excitation from the vibrational ground state of the electronic ground state to the vibrational ground state of the electronic excited state. This value is slightly larger (by 0.02–0.04 eV) than the analogous 0–0 transitions identified in pyrimidine (3.854 eV)12 and 2BrPyr (3.869 eV).52 We also notice that pyrimidine presents the most sharp and intense peaks in this band, which are attenuated in 2ClPyr and even more so in 2BrPyr. In Fig. 4 we also compare the measured and calculated cross-sections of 2ClPyr, the latter obtained in the vertical approximation, the NEA, and the FCHT approximation. The vertical approximation yields a qualitatively correct band, although somewhat high in energy compared to the experiment (4.58 eV against ∼4.3 eV). In the NEA, the centre of the band is redshifted to 4.48 eV, thus reducing the gap to experiment by 0.1 eV, while in the FCHT approach, this discrepancy is further reduced by around 0.1 eV.
Once the FCHT spectrum is red-shifted by 0.110 eV (to correct for inaccuracies in the electronic structure calculations), the agreement with experiment at the low-energy tail of the band is remarkable (see Fig. 4). Besides the 0–0 peak at 3.892 eV, two prominent peaks can be seen at 3.945 eV and 3.998 eV, which are assigned to excitation of the ν7(a1) C–Cl stretching/ring deformation mode. Many of the weaker features in the spectrum, including the hot bands (occurring at lower energies than the 0–0 transition), are nicely reproduced by the FCHT calculations. The most intense transitions of the band involve the combined excitation of totally symmetric ring deformation and C–Cl stretching modes, mostly ν5(a1), ν6(a1), ν7(a1), and ν9(a1), in addition to the ν16(b1) Cl wagging mode. Although the shape of the band remains correct towards its maximum and high-energy tail, the agreement regarding the position and intensity of the peaks worsens, a known limitation of the FCHT model.
It is also worth mentioning that the 0–0 transition found at 3.892 eV in the present VUV spectrum perfectly matches (within our energy resolution) the value obtained with REMPI spectroscopy.44
3.2. The 4.7–5.6 eV photon energy range (Band II)
The second absorption band extends from 4.7 eV to 5.6 eV, peaking at 4.91 eV, as shown in Fig. 5. We attribute this band to the 1B2 third singlet excited state, a π*(a2) ← π(b1) dipole allowed transition, whereas the 1A2 [π*(b1) ← n−(b2)] dipole forbidden transition should contribute to a lesser extent. | ||
| Fig. 5 Band II of the experimental photoabsorption cross-sections of 2-chloropyrimidine (green), compared, at the top, with those of pyrimidine12 (grey) and 2-bromopyrimidine52 (purple), and, at the bottom, with TDDFT calculations performed within the NEA (dashed red) and the vertical approximation (dotted red). | ||
The band of 2ClPyr is similar in shape to the analogous ones of 2BrPyr52 and pyrimidine,12,53,54 especially with the former. Compared with the band of pyrimidine, there is a red-shift of 0.23 eV in 2ClPyr and of 0.29 eV in 2BrPyr.
The 0–0 transition of 2ClPyr is tentatively placed at 4.782 eV, which is followed by a clear vibrational progression with a reasonably constant spacing of around 0.12 eV. This progression is very similar for the three molecules, apart from the somewhat smaller spacing for pyrimidine (0.09–0.11 eV)12 and the displaced 0–0 position. We assign the progression of 2ClPyr to excitation of the ν7(a1) ring stretching mode, based on the following arguments. First, this mode has the closest energy (0.125 eV for the ground state and 0.119 eV for the first singlet excited state) to the observed spacing. Furthermore, the small difference between these two values suggests that the actual vibrational energy for the 1B2 [π*(a2) ← π(b1)] excited state is probably not too far. Moreover, the vibrational energies are virtually the same for 2BrPyr,52 where the same spacing has been observed. Finally, the ν7 (a1) mode is dominated by the C–N stretching, which would be expected to be activated based on the orbitals involved in this electronic excitation (see Fig. 3). A weaker secondary progression has also been identified for 2BrPyr, differing from the main progression by one quantum of the ν24(b2) Br rocking mode.52 For 2ClPyr, however, the analogous combination mode cannot be seen in the spectrum.
Compared to the experimental data, the vertical approximation significantly overestimates the centre of the band, by around 0.6 eV. Turning to the NEA results, the gap is reduced to 0.5 eV, showing that this discrepancy is not related to how the vibrational degrees of freedom are treated. A similar behaviour was also observed for the analogous states of 2BrPyr38 and benzene,64 suggesting this is a shortcoming of the CAM-B3LYP functional (used in the three cases) in accurately describing this excited state.
Excitation of 2ClPyr in solution at 254 nm (4.88 eV) has been shown to produce 2-hydroxypyrimidine and 2-chloro-4,2′-bipyrimidine.46 Although this was performed in solution and the present experiments were performed in gas-phase, it is likely that the 1B2 [π*(a2) ← π(b1)] excited state is involved in these photochemical reactions.
3.3. The 5.6–6.8 eV photon energy range (Band III)
The third absorption band of 2ClPyr can be seen in Fig. 6. It has a maximum cross-section at 6.18 eV, and originates from the 1A1 [π*(b1) ← π(b1)] excited state. | ||
| Fig. 6 Band III of the experimental photoabsorption cross-sections of 2-chloropyrimidine (green), compared, at the top, with those of pyrimidine12 (grey) and 2-bromopyrimidine52 (purple), and, at the bottom, with TDDFT calculations performed within the NEA (dashed red) and the vertical approximation (dotted red). | ||
The analogous state accounts for the very similar band of 2BrPyr,52 except that it is centred lower in energy (at 5.91 eV) and is slightly more intense. In contrast, between 5.6 and 6.2 eV, pyrimidine displays a flat profile having one order of magnitude smaller cross-section. At this energy, a 1B1 [π(b1) ← n+(a1)] transition takes place,12,54 which appears at around the same energy in 2ClPyr and 2BrPyr, but are masked by the stronger 1A1 [π*(b1) ← π(b1)] transition. The latter accounts for the feature between 6.3 and 7.0 eV in pyrimidine.12,54 Besides the substantial energy shift (0.5–0.8 eV), the analogous band in pyrimidine is less intense by a factor of two.
Starting at around 6.42 eV, we also notice a series of subtle structures spaced by around 0.10 eV in the spectrum of 2ClPyr, which could be a vibrational progression of another excited state, possibly the 1B1 [π*(b1) ← n+(a1)]. No such structures are visible in the case of 2BrPyr. This can be related to the presence of excited states of the 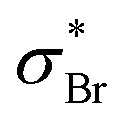 dissociative character in this region,52 whereas the analogous
dissociative character in this region,52 whereas the analogous  states only occur at higher energies. Thanks to non-adiabatic couplings to σ* dissociative states, otherwise sharp features associated with nearby states are broadened to a lesser or greater extent.
states only occur at higher energies. Thanks to non-adiabatic couplings to σ* dissociative states, otherwise sharp features associated with nearby states are broadened to a lesser or greater extent.
For this band we find a noticeable improvement from the vertical approximation to the NEA, as they yield a band maximum at 6.29 eV and 6.14 eV, respectively, the latter being very close to the experimental value of 6.18 eV. Furthermore, the cross-section at the band maximum calculated with the NEA lies within the ∼10% error bar of the experiment, whereas the vertical approximation largely overestimates it.
3.4. The 6.8–8.2 eV photon energy range (Band IV)
This region comprises the most intense absorption features of 2ClPyr, 2BrPyr, and pyrimidine, which are shown in Fig. 7. Two distinct features can be seen in the spectrum of 2ClPyr. The first extends from around 6.8 eV to 7.6 eV, with a maximum at 7.15 eV. The 1A1 [π*(a2) ← π(a2)] excited state has the largest oscillator strength in this region and should contribute the most to this feature, followed by a smaller contribution from the 1B2 [π*(b1) ← π(a2) + π*(a2) ← πCl(b1)] state (see Table 1). The second feature of 2ClPyr lies between 7.6 and 8.2 eV, peaking at 7.93 eV. It is dominated by the 1B2 [π*(a2) ← πCl(b1) + π*(b1) ← π(a2)] excited state, whereas other states, like the 1A1 [π*(b1) ← πCl(b1)], should contribute to a lesser extent. In this energy range, there are marked differences among the spectra of both halogenated molecules and pyrimidine. In the latter, a single broad band stands out, which originates from the superposition of two bright excited states. The 1A1 lower-lying bright state has a π*(a2) ← π(a2) dominant character with some π*(b1) ← π(b1) character, whereas the higher-lying 1B2 bright state has a π*(b1) ← π(a2) dominant character with some π*(a2) ← π(b1) contribution. Halogenation introduces excited configurations of π*(b1) ← πCl/Br(b1) and π*(a2) ← πCl/Br(b1) character, which strongly mix with the above configurations. The lower-lying 1A1 state of pyrimidine is redshifted upon halogenation, accounting for the first feature of 2ClPyr, centred at 7.15 eV, and of 2BrPyr, at 7.09 eV.52 It preserves its π*(a2) ← π(a2) dominant character while acquiring some π*(b1) ← πCl/Br(b1) character. In turn, the higher-lying 1B2 state of pyrimidine mixes more strongly with other configurations (see Table 1 and Table S2 of the ESI†), giving rise to the second feature observed in 2ClPyr (at 7.93 eV) and 2BrPyr (at 7.68 eV). For both halogenated molecules, the 1B2 [π*(a2) ← πCl/Br(b1) + π*(b1) ← π(a2)] excited state accounts for this feature, with the largest π*(a2) ← πCl(b1) contribution for 2ClPyr probably being related to the higher energy of this feature, in comparison to that of 2BrPyr. Furthermore, the higher vertical excitation energy calculated for 2ClPyr suggests that the second feature of 2BrPyr (lower-lying in energy) originates from the 1B2 bright state. The third and higher-lying feature, at 8.05 eV, would be explained by two 1A1 mixed Rydberg-valence excited states.52 | ||
| Fig. 7 Band IV of the experimental photoabsorption cross-sections of 2-chloropyrimidine (green), compared, at the top, with those of pyrimidine (grey)12 and 2-bromopyrimidine52 (purple), and, at the bottom, with TDDFT calculations performed within the NEA (dashed red) and the vertical approximation (dotted red). | ||
Besides the major differences in shape, another clear difference in the spectra lies in the absence of very sharp and pronounced peaks for the halogenated species, only seen for pyrimidine. These structures are signatures of Rydberg states, characterized by excitation into diffuse orbitals. For the halogenated compounds, however, much weaker features are observed embedded in their absorption bands.
Looking back at Table 1, the theoretical calculations for 2ClPyr predict a very weak 1B2 [3s(a1) ← n−(b2)] transition at 7.04 eV, and a stronger 1B1 [3s(a1) ← π(b1)] transition at 7.12 eV. Vibrational progressions of the latter Rydberg state might account for the several weak features seen between 7.0 and 7.4 eV. A similar conclusion had been reached for 2BrPyr,52 where the small peaks observed between 7.0 and 7.1 eV were suggested to arise from vibrational progressions of the 1B1 [3s(a1) ← π(b1)] Rydberg state (calculated at 7.00 eV). This is further supported by the difference in the corresponding vertical excitation energies, of 0.12 eV, consistent with the experimental difference between the peaks of each molecule. In pyrimidine, the analogous 1B2 [3s(a1) ← n−(b2)] state appears at 6.75 eV in our calculations and would account for some of the sharp peaks starting at 6.421 eV.12 In turn, the 0–0 transition of the 1B1 [3s(a1) ← π(b1)] state has been assigned to a peak at 7.442 eV,12 whereas its vertical energy is found at 7.32 eV in our calculations. The oscillator strengths for the two lower-lying Rydberg transitions of the three molecules are comparable and thus cannot account for the absence of sharp structures for the halogenated species.
Interestingly, the next two Rydberg transitions of pyrimidine display much larger oscillator strengths. The 1A1 [3py(b2) ← n−(b2)] and 1B2 [3pz(a1) ← n−(b2)] states were obtained at 7.35 eV and 7.39 eV in our calculations, with oscillator strengths of 0.11 and 0.05, whereas their 0–0 transitions would be at 6.977 eV and 7.056 eV according to ref. 12. Halogenation significantly reduces their oscillator strengths, most notably for the former state, which decreases by one or two orders of magnitude. This could help to explain the lack of sharp features in the spectra of 2ClPyr and 2BrPyr.
Moreover, while halogenation preserves the character of the 1A1 [3py(b2) ← n−(b2)] state, the 1B2 state acquires some  valence contribution to its 3pz(a1) ← n−(b2) Rydberg character. This dissociative element is expected to suppress vibrational progressions, thus broadening the signature of the transition in the spectrum. In addition to this Rydberg-valence mixed state, several other states having
valence contribution to its 3pz(a1) ← n−(b2) Rydberg character. This dissociative element is expected to suppress vibrational progressions, thus broadening the signature of the transition in the spectrum. In addition to this Rydberg-valence mixed state, several other states having  or nCl/nBr dominant character can be seen in the calculations (see Table 1 and Table 1 of ref. 52), as low as 6.98 eV in 2ClPyr and 6.34 eV in 2BrPyr.52 These states clearly have no parallel in pyrimidine. Vibronic couplings to these halogen-specific states would be expected to broaden the typically pronounced peaks associated with Rydberg transitions, which could also play a role to explain the absence of sharp peaks in the spectra.
or nCl/nBr dominant character can be seen in the calculations (see Table 1 and Table 1 of ref. 52), as low as 6.98 eV in 2ClPyr and 6.34 eV in 2BrPyr.52 These states clearly have no parallel in pyrimidine. Vibronic couplings to these halogen-specific states would be expected to broaden the typically pronounced peaks associated with Rydberg transitions, which could also play a role to explain the absence of sharp peaks in the spectra.
A comparison between the experimental and calculated spectra is also shown in Fig. 7. Compared to the vertical approximation, the superior accuracy of the NEA is striking. Regarding the energy of the maximum, the first feature is displaced from 7.41 eV (vertical) to 7.28 eV (NEA), reducing the gap to the experiment (7.15 eV) by a factor of two. A similar improvement is observed for the second feature, which shifts from 8.19 eV (vertical) to 8.04 eV (NEA), with experiment at 7.93 eV. Furthermore, the cross-sections decrease in magnitude with the NEA, in comparison with the vertical result, thus bringing the calculated spectrum substantially closer to the experimental one.
3.5. The 8.2–10.8 eV photon energy range (Band V)
The results for the last energy range are compared in Fig. 8. The measured spectrum of 2ClPyr is extremely rich. A few features that stand out include the peak at 8.95 eV, the broad enhancement between 9.2 and 9.8 eV, and the two very narrow and closely-lying peaks at 9.609 and 9.618 eV. The many excited states in this energy range and the overall worse comparison with the calculated spectrum make any assignment of the structures extremely challenging. While the narrow features are likely due to Rydberg states, their relatively high energies do not allow us to confidently confirm the nature of the transition.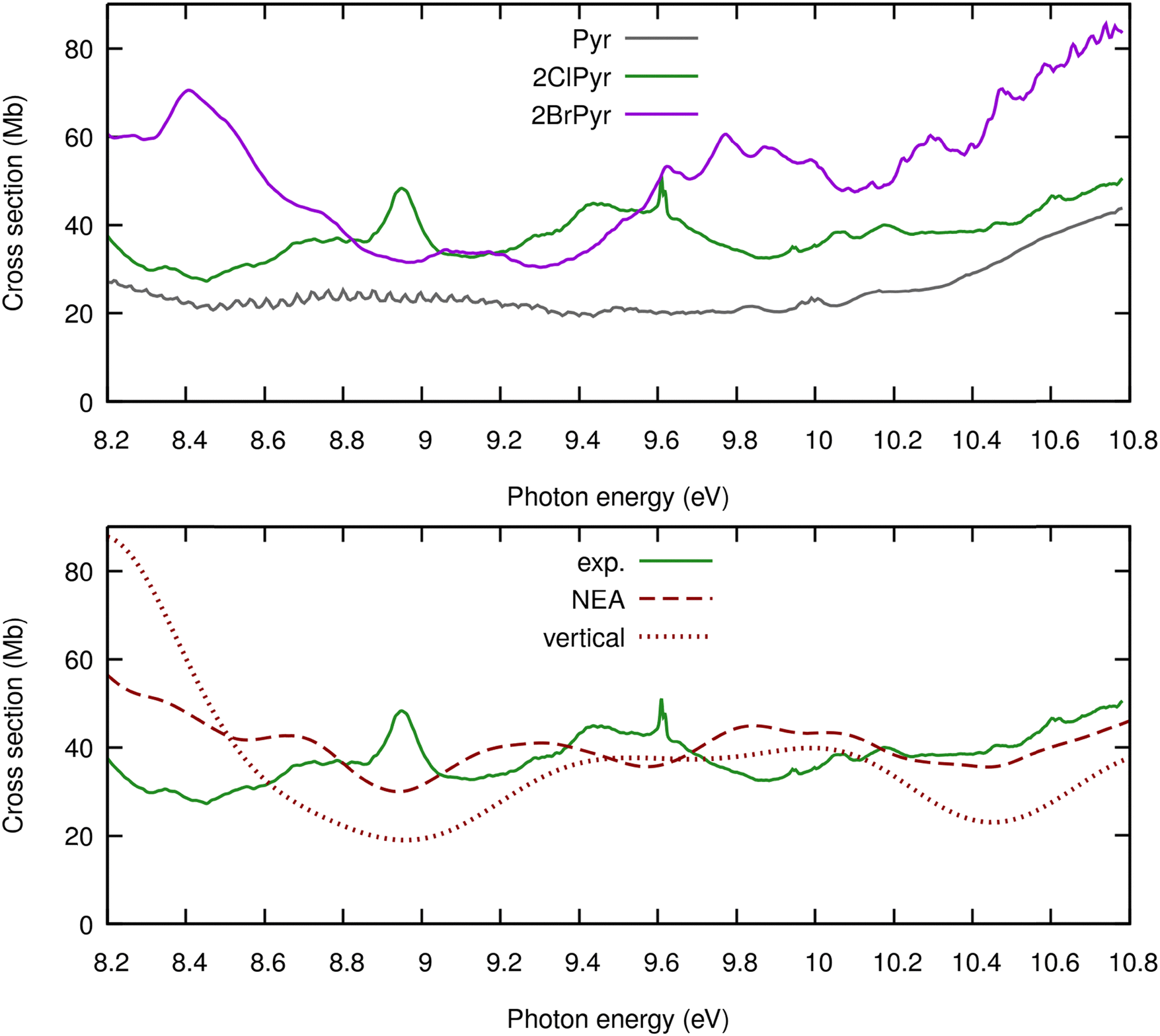 | ||
| Fig. 8 Band V of the experimental photoabsorption cross-sections of 2-chloropyrimidine (green), compared, at the top, with those of pyrimidine12 (grey) and 2-bromopyrimidine52 (purple), and, at the bottom, with TDDFT calculations performed within the NEA (dashed red) and the vertical approximation (dotted red). | ||
In this region one finds major differences among the spectra of the three molecules. Pyrimidine has the lowest cross-sections, which increase in magnitude with the halogen atomic number. In addition, the very fine structures seen for pyrimidine, assigned to overlapping series of Rydberg excited states12 are suppressed upon halogenation. This probably reflects a stronger mixing between Rydberg and valence excitations in the halogenated species, which is supported by the present calculations for 2ClPyr and previous results for 2BrPyr,52 in addition to vibronic couplings to σ* dissociative states. In turn, the spectra of the halogenated species are overall richer, displaying many features of varying characteristics, whereas the spectrum of pyrimidine is significantly flatter. Transitions involving σ* orbitals centred at the carbon–halogen bond are found in the calculations, which have no parallel in pyrimidine. Even though they certainly play a role, we do not attempt to interpret the origin of the many peaks.
Looking again at Fig. 8, we can see that the NEA produces cross-sections in overall closer agreement with experiments than the vertical approximation, as far as the magnitude is concerned. However, the improvement is not as noticeable as for the lower-lying bands, since the shape of the band is not very well reproduced. On the one hand, this could be due to limitations of the present TDDFT calculations in describing the excited states found at this energy range, including higher-lying Rydberg states and doubly-excited states. On the other hand, the less satisfactory agreement at higher energies could reflect a worse performance of the NEA. In this energy range, the high density of states should favour significant vibronic coupling effects, only partially accounted for in the NEA. It is not obvious whether the observed discrepancies arise mostly from limitations of the electronic structure method or of the nuclear model.
4. Conclusions
In summary, here we have reported a combined experimental–theoretical investigation on the VUV spectroscopy of 2ClPyr. From the experimental side, absolute photoabsorption cross-sections are reported for the first time, in the 330 nm (3.757 eV) to 115 nm (10.781 eV) spectral range. From the theoretical side, the excited states were described with TDDFT calculations, which were employed within the vertical approximation and the NEA to provide calculated photoabsorption cross-sections.Going beyond the vertical approximation through the NEA considerably improved the agreement with the experimental result. This was observed for the shape and magnitude of the cross-section profile. Importantly, we found that the NEA outperforms the vertical approximation throughout the wide energy range covered here. Yet, the region above ∼8 eV remains challenging and would require a more accurate description of the underlying excited states. Furthermore, the NEA may become less reliable at higher energies due to the increasingly congested vibronic couplings, only approximately described in this approach. More systematic comparisons across different electronic structure levels of theory and different treatments for the nuclear degrees of freedom are left as perspectives for future work.
A key finding is that the halogen atom plays an increasingly larger role in the photoabsorption spectra as the photon energy increases. Starting with band I (3.7–4.6 eV), we found very similar profiles for 2ClPyr, 2BrPyr, and pyrimidine, with differences limited to the fine vibrational structures, relatively well reproduced with FCHT calculations. Band II (4.6–5.7 eV) of the three molecules is also similar in shape, but is red-shifted upon halogenation. Even larger red-shifts are seen for band III (5.7–6.7 eV), which are also more intense in the halogenated species. Photoabsorption is most likely at band IV (6.7–8.2 eV), where the spectra of the three species become qualitatively different. Because of specific mixing of π* ← π configurations, the single broad feature of pyrimidine splits into two in 2ClPyr and 2BrPyr. Moreover, the very rich sharp structures observed for pyrimidine disappear in the halogenated species, which could arise from a broadening induced by vibronic couplings with halogen-specific excited states and from the reduced oscillator strength of some Rydberg transitions. At the highest energies surveyed here, labelled band V (8.2–10.8 eV), the cross-section profiles no longer share any obvious resemblance. Comparing the spectra of the two halogenated pyrimidines, they also become progressively more dissimilar as the energy increases. At lower energies, the difference is less striking than that found between pyrimidine and either 2ClPyr or 2BrPyr. In contrast, the three spectra are similarly uncorrelated at higher energies. Therefore, not only halogenation plays a larger role at higher energies, but also photoabsorption becomes remarkably halogen-specific.
Data availability
Data for this article, including experimental cross-sections and theoretical cross-sections (obtained with the vertical approximation, nuclear ensemble approach, and Franck–Condon Herzberg–Teller approximation) are available at Zenodo at https://doi.org/10.5281/zenodo.15068291.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The research leading to these results was supported by the project CALIPSOplus under the Grant Agreement 730872 from the EU Framework Programme for Research and Innovation HORIZON 2020. MM also acknowledges the Portuguese National Funding Agency FCT-MCTES from the research project (2023.14518.PEX). The authors acknowledge the Portuguese National Funding Agency FCT-MCTES through the research grant number UID/FIS/00068/2020 (10.54499/UIDP/00068/2020) (CEFITEC).References
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, The Halogen Bond, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- P. Metrangolo, F. Meyer, T. Pilati, G. Resnati and G. Terraneo, Halogen Bonding in Supramolecular Chemistry, Angew. Chem., Int. Ed., 2008, 47, 6114–6127 CrossRef CAS PubMed.
- T. L. Ellington, P. L. Reves, B. L. Simms, J. L. Wilson, D. L. Watkins, G. S. Tschumper and N. I. Hammer, Quantifying the Effects of Halogen Bonding by Haloaromatic Donors on the Acceptor Pyrimidine, Chem. Phys. Chem., 2017, 18, 1267–1273 CrossRef CAS PubMed.
- R. Wilcken, M. O. Zimmermann, A. Lange, A. C. Joerger and F. M. Boeckler, Principles and Applications of Halogen Bonding in Medicinal Chemistry and Chemical Biology, J. Med. Chem., 2013, 56, 1363–1388 CrossRef CAS PubMed.
- L. C. Blasiak and C. L. Drennan, Structural Perspective on Enzymatic Halogenation, Acc. Chem. Res., 2009, 42, 147–155 CrossRef CAS PubMed.
- C. M. Harris, R. Kannan, H. Kopecka and T. M. Harris, The role of the chlorine substituents in the antibiotic vancomycin: preparation and characterization of mono- and didechlorovancomycin, J. Am. Chem. Soc., 1985, 107, 6652–6658 CrossRef CAS.
- D. Cantillo and C. O. Kappe, Halogenation of organic compounds using continuous flow and microreactor technology, React. Chem. Eng., 2017, 2, 7–19 RSC.
- J. Martí-Rujas, L. Colombo, J. Lü, A. Dey, G. Terraneo, P. Metrangolo, T. Pilati and G. Resnati, Hydrogen and halogen bonding drive the orthogonal self-assembly of an organic framework possessing 2D channels, Chem. Commun., 2012, 48, 8207 RSC.
- D. Lin-Vien, N. B. Colthup, W. G. Fateley and J. G. Grasselli, The Handbook of Infrared and Raman Characteristic Frequencies of Organic Molecules, Elsevier, 1st edn, 1991 Search PubMed.
- N. Kerru, L. Gummidi, S. Maddila, K. K. Gangu and S. B. Jonnalagadda, A Review on Recent Advances in Nitrogen-Containing Molecules and Their Biological Applications, Molecules, 2020, 25, 1909 CrossRef CAS PubMed.
- I. Baccarelli, I. Bald, F. A. Gianturco, E. Illenberger and J. Kopyra, Electron-induced damage of DNA and its components: Experiments and theoretical models, Phys. Rep., 2011, 508, 1–44 CrossRef CAS.
- F. F. da Silva, D. Almeida, G. Martins, A. R. Milosavljević, B. P. Marinković, S. V. Hoffmann, N. J. Mason, Y. Nunes, G. Garcia and P. Limão-Vieira, The electronic states of pyrimidine studied by VUV photoabsorption and electron energy-loss spectroscopy, Phys. Chem. Chem. Phys., 2010, 12, 6717 RSC.
- F. F. da Silva, C. Matias, D. Almeida, G. García, O. Ingólfsson, H. D. Flosadóttir, B. Ómarsson, S. Ptasinska, B. Puschnigg, P. Scheier, P. Limão-Vieira and S. Denifl, NCO –, a Key Fragment Upon Dissociative Electron Attachment and Electron Transfer to Pyrimidine Bases: Site Selectivity for a Slow Decay Process, J. Am. Soc. Mass Spectrom., 2013, 24, 1787–1797 CrossRef PubMed.
- D. Almeida, F. Ferreira da Silva, G. García and P. Limão-Vieira, Selective Bond Cleavage in Potassium Collisions with Pyrimidine Bases of DNA, Phys. Rev. Lett., 2013, 110, 023201 CrossRef PubMed.
- M. Mendes, B. Pamplona, S. Kumar, F. F. da Silva, A. Aguilar, G. García, M.-C. Bacchus-Montabonel and P. Limao-Vieira, Ion-Pair Formation in Neutral Potassium-Neutral Pyrimidine Collisions: Electron Transfer Experiments, Front. Chem., 2019, 7, 1–10 CrossRef PubMed.
- E. Itälä, S. Granroth, D. T. Ha, K. Kooser, H. Levola, E. Rachlew, K. Tanzer and E. Kukk, Fragmentation of imidazole, pyrimidine and purine induced by core ionization: Significance of small-scale chemical environment, J. Photochem. Photobiol., A, 2018, 356, 283–289 CrossRef.
- R. Pandey, M. Ryszka, T. da Fonseca Cunha, M. Lalande, M. Dampc, P. Limão-Vieira, N. J. Mason, J. C. Poully and S. Eden, Threshold behavior in metastable dissociation of multi-photon ionized thymine and uracil, Chem. Phys. Lett., 2017, 684, 233–238 CrossRef CAS.
- A. E. W. Knight, C. M. Lawburgh and C. S. Parmenter, n,π* fluorescence from selected vibronic levels of pyrimidine vapor: Franck–Condon factors and excited state anharmonic coupling, J. Chem. Phys., 1975, 63, 4336–4351 CrossRef CAS.
- Y. Luo, H. Ågren, S. Knuts and P. Jørgensen, The two-photon spectrum of pyrimidine. Role of vibronic coupling, Chem. Phys. Lett., 1993, 213, 357–362 CrossRef.
- O. Plekan, M. Coreno, V. Feyer, A. Moise, R. Richter, M. de Simone, R. Sankari and K. C. Prince, Electronic state resolved PEPICO spectroscopy of pyrimidine, Phys. Scr., 2008, 78, 058105 CrossRef.
- P. Wardman, Chemical Radiosensitizers for Use in Radiotherapy, Clin. Oncol., 2007, 19, 397–417 CrossRef CAS PubMed.
- H. Wang, X. Mu, H. He and X.-D. Zhang, Cancer Radiosensitizers, Trends Pharmacol. Sci., 2018, 39, 24–48 CrossRef CAS PubMed.
- M. M. Poggi, C. N. Coleman and J. B. Mitchell, Current Problems in Cancer, Elsevier Science Ltd, 2001 Search PubMed.
- T. J. Kinsella, P. P. Dobson, J. B. Mitchell and A. J. Fornace, Enhancement of X ray induced DNA damage by pre-treatment with halogenated pyrimidine analogs, Int. J. Radiat. Oncol., Biol., Phys., 1987, 13, 733–739 CrossRef CAS PubMed.
- S. E. Berry and T. J. Kinsella, Targeting DNA mismatch repair for radiosensitization, Semin. Radiat. Oncol., 2001, 11, 300–315 CrossRef CAS PubMed.
- C. J. McGinn and T. S. Lawrence, Recent advances in the use of radiosensitizing nucleosides, Semin. Radiat. Oncol., 2001, 11, 270–280 CrossRef CAS PubMed.
- B. Pauwels, A. E. C. Korst, F. Lardon and J. B. Vermorken, Combined Modality Therapy of Gemcitabine and Radiation, Oncologist, 2005, 10, 34–51 CrossRef CAS PubMed.
- L. Chomicz, M. Zdrowowicz, F. Kasprzykowski, J. Rak, A. Buonaugurio, Y. Wang and K. H. Bowen, How to Find Out Whether a 5-Substituted Uracil Could Be a Potential DNA Radiosensitizer, J. Phys. Chem. Lett., 2013, 4, 2853–2857 CrossRef CAS.
- J. Ameixa, E. Arthur-Baidoo, R. Meißner, S. Makurat, W. Kozak, K. Butowska, F. Ferreira da Silva, J. Rak and S. Denifl, Low-energy electron-induced decomposition of 5-trifluoromethanesulfonyl-uracil: A potential radiosensitizer, J. Chem. Phys., 2018, 149, 164307 CrossRef CAS PubMed.
- S. Wang, P. Zhao, C. Zhang and Y. Bu, Mechanisms Responsible for High Energy Radiation Induced Damage to Single-Stranded DNA Modified by Radiosensitizing 5-Halogenated Deoxyuridines, J. Phys. Chem. B, 2016, 120, 2649–2657 CrossRef CAS PubMed.
- V. I. Berezin and V. V. Berezin, Characteristic vibrations of 2-monosubstituted pyrimidines, Chem. Heterocycl. Compd., 1984, 20, 102–106 CrossRef.
- Y. Ikari, H. Sakamoto, S. Nakama, Y. Nibu, H. Shimada and R. Shimada, Normal Vibrations of 5-Chloro-, 5-Bromo-, 5-Methyl-, and 2-Methylpyrimidines, Bull. Chem. Soc. Jpn., 1990, 63, 2891–2898 CrossRef CAS.
- A. Joshi, J. Tonannavar, N. H. Ayachit, M. A. Shashidhar and K. S. Rao, Infra-red and electronic absorption spectra of 2-bromopyrimidine, Spectrochim. Acta, Part A, 1987, 43, 1017–1021 CrossRef.
- S. Nakama, H. Shimada and R. Shimada, Polarized Raman and Infrared Spectra of 2-Chloro- and 2-Bromopyrimidines, Bull. Chem. Soc. Jpn., 1984, 57, 2584–2590 CrossRef CAS.
- P. Bolognesi, J. A. Kettunen, A. Cartoni, R. Richter, S. Tosic, S. Maclot, P. Rousseau, R. Delaunay and L. Avaldi, Site- and state-selected photofragmentation of 2Br-pyrimidine, Phys. Chem. Chem. Phys., 2015, 17, 24063–24069 RSC.
- M. H. Palmer, T. Ridley, S. V. Hoffmann, N. C. Jones, M. Coreno, M. De Simone, C. Grazioli, M. Biczysko, A. Baiardi and P. Limão-Vieira, Interpretation of the vacuum ultraviolet photoabsorption spectrum of iodobenzene by ab initio computations, J. Chem. Phys., 2015, 142, 134302 CrossRef PubMed.
- M. H. Palmer, T. Ridley, S. V. Hoffmann, N. C. Jones, M. Coreno, M. de Simone, C. Grazioli, T. Zhang, M. Biczysko, A. Baiardi and K. Peterson, Interpretation of the photoelectron, ultraviolet, and vacuum ultraviolet photoabsorption spectra of bromobenzene by ab initio configuration interaction and DFT computations, J. Chem. Phys., 2015, 143, 164303 CrossRef PubMed.
- P. Bolognesi, G. Mattioli, P. O’Keeffe, V. Feyer, O. Plekan, Y. Ovcharenko, K. C. Prince, M. Coreno, A. Amore Bonapasta and L. Avaldi, Investigation of Halogenated Pyrimidines by X-ray Photoemission Spectroscopy and Theoretical DFT Methods, J. Phys. Chem. A, 2009, 113, 13593–13600 CrossRef CAS PubMed.
- P. Bolognesi, P. O’Keeffe, Y. Ovcharenko, M. Coreno, L. Avaldi, V. Feyer, O. Plekan, K. C. Prince, W. Zhang and V. Carravetta, Pyrimidine and halogenated pyrimidines near edge x-ray absorption fine structure spectra at C and N K-edges: experiment and theory, J. Chem. Phys., 2010, 133, 034302 CrossRef CAS PubMed.
- L. Storchi, F. Tarantelli, S. Veronesi, P. Bolognesi, E. Fainelli and L. Avaldi, The Auger spectroscopy of pyrimidine and halogen-substituted pyrimidines, J. Chem. Phys., 2008, 129, 154309 CrossRef CAS PubMed.
- P. O’Keeffe, P. Bolognesi, A. R. Casavola, D. Catone, N. Zema, S. Turchini and L. Avaldi, An experimental and computational study of the valence photoelectron spectra of halogenated pyrimidines, Mol. Phys., 2009, 107, 2025–2037 CrossRef.
- M. C. Castrovilli, P. Bolognesi, A. Cartoni, D. Catone, P. O’Keeffe, A. R. Casavola, S. Turchini, N. Zema and L. Avaldi, Photofragmentation of Halogenated Pyrimidine Molecules in the VUV Range, J. Am. Soc. Mass Spectrom., 2014, 25, 351–367 CrossRef CAS PubMed.
- M. C. Castrovilli, P. Bolognesi, A. Casavola, A. Cartoni, D. Catone, P. O’Keeffe and L. Avaldi, Insights into 2-Chloropyrimidine fragmentation through a thermochemical analysis of the ionic fragments, Eur. Phys. J. D, 2014, 68, 253 CrossRef.
- Y. He, C. Wu and W. Kong, Two-color two-photon REMPI and ZEKE photoelectron spectroscopy of jet-cooled 2-chloropyrimidine, Chem. Phys. Lett., 2004, 391, 38–43 CrossRef CAS.
- M. P. V. Boarland and J. F. W. McOmie, 712. Pyrimidines. Part II. The ultra-violet absorption spectra of some monosubstituted pyrimidines, J. Chem. Soc., 1952, 3716 RSC.
- L. Lindqvist, B. Czochralska, M.-P. Fontaine-Aupart, W. Kawczynski, F. Tfibel and T. Douki, Photochemistry of 2-chloropyrimidine, Photochem. Photobiol. Sci., 2002, 1, 600–606 CrossRef CAS PubMed.
- L. N. Short and H. W. Thompson, 38. Infra-red spectra of derivatives of pyrimidine, J. Chem. Soc., 1952, 168 RSC.
- A. Modelli, P. Bolognesi and L. Avaldi, Temporary Anion States of Pyrimidine and Halopyrimidines, J. Phys. Chem. A, 2011, 115, 10775–10782 CrossRef CAS PubMed.
- A. S. Barbosa and M. H. F. Bettega, Shape resonances in low-energy-electron collisions with halopyrimidines, J. Chem. Phys., 2013, 139, 214301 CrossRef PubMed.
- H.-Y. Cheng, Y.-C. Chen, C.-J. Lin, W.-C. Liu and S.-H. Hsieh, Temporary anion states of radiosensitive halopyrimidines: Shape and core-excited resonances, Comput. Theor. Chem., 2016, 1075, 18–29 CrossRef CAS.
- B. Żywicka and P. Możejko, Cross-Section Calculations for Electron-Impact Ionization of Pyrimidine Molecule and Its Halogenated Derivatives: 2-Chloropyrimidine, 5-Chloropyrimidine, 2-Bromopyrimidine and 5-Bromopyrimidine, Molecules, 2024, 30, 6 CrossRef PubMed.
- M. Mendes, F. Kossoski, A. I. Lozano, J. Pereira-da-Silva, R. Rodrigues, J. Ameixa, N. C. Jones, S. V. Hoffmann and F. Ferreira da Silva, Excited States of Bromopyrimidines Probed by VUV Photoabsorption Spectroscopy and Theoretical Calculations, Int. J. Mol. Sci., 2021, 22, 6460 CrossRef CAS PubMed.
- A. Bolovinos, P. Tsekeris, J. Philis, E. Pantos and G. Andritsopoulos, Absolute vacuum ultraviolet absorption spectra of some gaseous azabenzenes, J. Mol. Spectrosc., 1984, 103, 240–256 CrossRef CAS.
- M. H. Palmer, I. C. Walker, M. F. Guest and A. Hopkirk, The electronic states of the azines. III. Pyrimidine, studied by VUV absorption, near-threshold electron energy-loss spectroscopy and ab initio multi-reference configuration calculations, Chem. Phys., 1990, 147, 19–33 CrossRef CAS.
- D. Duflot, S. V. Hoffmann, N. C. Jones and P. Limão-Vieira, Synchrotron radiation UV-VUV photoabsorption of gas phase molecules, in Radiation in bioanalysis: spectroscopic techniques and theoretical methods, ed. A. S. Pereira, P. Tavares and P. Limão-Vieira, Springer, 2019, pp. 43–82 DOI:10.1007/978-3-030-28247-9.
- S. Eden, P. Limão-Vieira, S. V. Hoffmann and N. J. Mason, VUV photoabsorption in CF3X (X = Cl, Br, I) fluoro-alkanes, Chem. Phys., 2006, 323, 313–333 CrossRef CAS.
- T. Yanai, D. P. Tew and N. C. Handy, A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP), Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- D. Jacquemin, V. Wathelet, E. A. Perpète and C. Adamo, Extensive TD-DFT Benchmark: Singlet-Excited States of Organic Molecules, J. Chem. Theory Comput., 2009, 5, 2420–2435 CrossRef CAS PubMed.
- R. Sarkar, M. Boggio-Pasqua, P.-F. Loos and D. Jacquemin, Benchmarking TD-DFT and Wave Function Methods for Oscillator Strengths and Excited-State Dipole Moments, J. Chem. Theory Comput., 2021, 17, 1117–1132 CrossRef CAS PubMed.
- J. Bloino, M. Biczysko, F. Santoro and V. Barone, General Approach to Compute Vibrationally Resolved One-Photon Electronic Spectra, J. Chem. Theory Comput., 2010, 6, 1256–1274 CrossRef CAS.
- M. Piccardo, J. Bloino and V. Barone, Generalized vibrational perturbation theory for rotovibrational energies of linear, symmetric and asymmetric tops: Theory, approximations, and automated approaches to deal with medium-to-large molecular systems, Int. J. Quantum Chem., 2015, 115, 948–982 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Wallingford CT, 2016 Search PubMed.
- J. P. Bergsma, P. H. Berens, K. R. Wilson, D. R. Fredkin and E. J. Heller, Electronic spectra from molecular dynamics: a simple approach, J. Phys. Chem., 1984, 88, 612–619 CrossRef CAS.
- R. Crespo-Otero and M. Barbatti, Spectrum simulation and decomposition with nuclear ensemble: formal derivation and application to benzene, furan and 2-phenylfuran, Theor. Chem. Acc., 2012, 131, 1237 Search PubMed.
- M. Barbatti, M. Bondanza, R. Crespo-Otero, B. Demoulin, P. O. Dral, G. Granucci, F. Kossoski, H. Lischka, B. Mennucci, S. Mukherjee, M. Pederzoli, M. Persico, M. Pinheiro Jr, J. Pittner, F. Plasser, E. Sangiogo Gil and L. Stojanovic, Newton-X Platform: New Software Developments for Surface Hopping and Nuclear Ensembles, J. Chem. Theory Comput., 2022, 18, 6851–6865 CrossRef CAS PubMed.
- F. Santoro, A. Lami, R. Improta, J. Bloino and V. Barone, Effective method for the computation of optical spectra of large molecules at finite temperature including the Duschinsky and Herzberg–Teller effect: The Qx band of porphyrin as a case study, J. Chem. Phys., 2008, 128(22), 224311 CrossRef PubMed.
- F. J. Avila Ferrer, J. Cerezo, E. Stendardo, R. Improta and F. Santoro, Insights for an Accurate Comparison of Computational Data to Experimental Absorption and Emission Spectra: Beyond the Vertical Transition Approximation, J. Chem. Theory Comput., 2013, 9, 2072–2082 CrossRef CAS PubMed.
- F. Kossoski and M. Barbatti, Nuclear Ensemble Approach with Importance Sampling, J. Chem. Theory Comput., 2018, 14, 3173–3183 CrossRef CAS PubMed.
- S. Bai, R. Mansour, L. Stojanović, J. M. Toldo and M. Barbatti, On the origin of the shift between vertical excitation and band maximum in molecular photoabsorption, J. Mol. Model., 2020, 26, 107 CrossRef CAS PubMed.
- M. Barbatti and K. Sen, Effects of different initial condition samplings on photodynamics and spectrum of pyrrole, Int. J. Quantum Chem., 2016, 116, 762–771 CrossRef CAS.
- J. Pereira-da-Silva, M. Mendes, F. Kossoski, A. I. Lozano, R. Rodrigues, N. C. Jones, S. V. Hoffmann and F. Ferreira da Silva, Perfluoro effect on the electronic excited states of para -benzoquinone revealed by experiment and theory, Phys. Chem. Chem. Phys., 2021, 23, 2141–2153 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cp00198f |
| This journal is © the Owner Societies 2025 |