
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effect of microhydration on the aromatic charge resonance interaction: the case of the pyrrole dimer cation†
Dashjargal
Arildii
a,
Yoshiteru
Matsumoto
 b and
Otto
Dopfer
*ac
b and
Otto
Dopfer
*ac
aInstitut für Optik und Atomare Physik, Technische Universität Berlin, Hardenbergstrasse 36, 10623 Berlin, Germany. E-mail: dopfer@physik.tu-berlin.de
bDepartment of Chemistry, Faculty of Science, Shizuoka University, 836 Ohya, Suruga, Shizuoka 422-8529, Japan
cInternational Research Frontiers Initiative, Institute of Science Tokyo, 4259 Nagatsuta-cho, Midori-ku, Yokohama 226-8503, Japan
First published on 25th April 2025
Abstract
Charge resonance (CR) interactions between aromatic molecules are amongst the strongest intermolecular forces and responsible for many phenomena in chemistry and biology. Microhydration of an aromatic radical dimer cation allows investigation of the strong effects of stepwise solvation on the charge distribution and strength of the CR. We characterise herein the microhydration process of the pyrrole dimer cation (Py2+), a prototypical aromatic homodimer with a strong CR. The NH and OH stretch vibrations (νNH/OH) of mass-selected bare and colder Ar-tagged hydrated clusters of Py2+, Py2+(H2O)nArm (n ≤ 3, m ≤ 1), recorded by infrared photodissociation (IRPD) spectroscopy provide detailed insight into the preferred cluster growth and strengths of the various intermolecular interactions by comparison to dispersion-corrected density functional theory calculations. The analysis of systematic frequency shifts, structural parameters, binding energies, and charge distributions allows for a quantitative evaluation of the drastic effects of stepwise hydration on the strength and symmetry of the aromatic CR, the strengths of the various hydrogen bonds (H-bonds), and the competition between slightly noncooperative interior ion hydration and strongly cooperative formation of a H-bonded solvent network. The most stable Py2+H2O structure exhibits a strong NH⋯O ionic H-bond of H2O to the antiparallel stacked Py2+(a) core, thereby breaking the symmetry of the CR. Py2+(H2O)2 prefers a highly symmetric C2h structure with two equivalent NH⋯O H-bonds of Py2+(a) and an optimised CR. Starting from n = 3, clusters with a parallel configuration, Py2+(p), are more stable than those with Py2+(a), further highlighting the strong impact of (micro-)solvation on the structural motif of the aromatic CR. The spectral and computational data demonstrate a linear correlation of νNH of the free Py unit with its partial charge, illustrating that IR spectroscopy is a powerful tool for probing the charge distribution in aromatic CR cluster cations. Comparison of Py2+(H2O)n with neutral Py2(H2O)n and Py+(H2O)n reveals the impact of the magnitude of positive charge and the number of acidic proton donors on the structure of the microhydration shell and strength of the various competing intermolecular bonds.
1. Introduction
Intermolecular interactions involving aromatic π electrons are highly relevant for a large number of chemical and biological phenomena.1–9 One of the most common interactions between aromatic molecules is π–π stacking, a weak interaction mostly based on London dispersion forces. Such π–π stacking is well-known to stabilise the DNA double-strand and to contribute to protein folding.10–12 In structural chemistry, π–π stacking often determines the geometry of supramolecules and the properties of solid or liquid crystals.13–15 In addition, π hydrogen bonds (π H-bonds) such as CH–π bonds are important weak interactions involving aromatic molecules.16–20 In contrast to these weak interactions, the charge resonance (CR) interaction in charged aromatic dimers is amongst the strongest intermolecular forces, with a strength of around 100 kJ mol−1.21–23 The CR interaction plays an important role for structure, energetics, and dynamics of intermolecular and intramolecular hole transport in stacked conjugated biological and organic semiconductor materials.24–31The CR in aromatic dimer cations (AB+) is formed by sharing the positive charge generated by removing a single π electron. This CR gives rise to two electronic states described by
| ψ+ = c1ϕ(A+)ϕ(B) + c2ϕ(A)ϕ(B+) |
| ψ− = c2ϕ(A+)ϕ(B) – c1ϕ(A)ϕ(B+) |
CR interactions were first detected in the condensed phase by electron spin resonance34–36 and optical absorption spectroscopy.37–40 However, these experiments suffer from environmental perturbations such as solvent and counter ions. In the gas phase, the binding energies of several isolated aromatic homo- and heterodimers were measured by mass spectrometric techniques.41–43 The spectra of the electronic CR transitions recorded for the aromatic homodimers of benzene,44–47 naphthalene,48–50 toluene,51 pyrene,52 and several heterodimers32,51,53 have proven that the splitting between the two electronic states ψ± depends on the size and ΔIE of the two monomer units. The strength of the CR interaction is controlled by the charge distribution on the respective monomers.32,51,53 However, the electronic CR bands observed by optical absorption in the spectral range around 1 eV are too broad even in the gas phase due to their transitions into the repulsive ψ− state, preventing a precise estimation of the partial charge distribution on the interacting monomers. To circumvent this problem, we previously demonstrated an efficient spectroscopic method to estimate the charge distribution in isolated aromatic A2+ and AB+ dimers by monitoring their vibrational transitions in the ground state (ψ+) utilising infrared photodissociation spectroscopy (IRPD).23 This study concentrated on the pyrrole dimer cation (Py2+), its clusters, and related heterodimers because of its strong CR interaction and the high sensitivity of the NH stretch frequency (νNH) of Py (C4H5N) to its partial charge.23 Besides being a fundamental building block of many important biomolecules such as porphyrins, amino acids, proteins, and DNA bases, Py has a single isolated, uncoupled, and strongly IR-active NH stretch oscillator, whose frequency is indeed rather sensitive to the charge state of Pyq (e.g., νNH = 3531 and 3447 cm−1 for qPy = 0 and 1e).54,55 The latter property differs from that of bare aromatic hydrocarbons, such as benzene and related polycyclic aromatic hydrocarbons, whose CH stretch frequencies (νCH) are less sensitive to charges and have weaker IR oscillator strengths in the cation ground state. Furthermore, often strong anharmonic or vibronic couplings further complicate the CH stretch range in these hydrocarbons (e.g., Fermi resonances in C6H6 and C6H4Cl2+ or Jahn–Teller effect in C6H6+).45,53,56–60
Because Py has an acidic NH group and aromatic π electrons, neutral Py2 dimer forms a weak NH⋯π H-bond,61–63 with an interaction energy of 0.07 eV.64 Its T-shaped structure has been confirmed by microwave,64 IR,63,65 and Raman spectroscopy,65 as well as quantum chemical calculations.61,66 Py2+ radical cations formed in a supersonic plasma expansion based on electron ionisation (EI) have a stacked sandwich structure stabilised by a strong CR interaction (1.01 eV), which has been characterised by IRPD and quantum chemical calculations.23 Further analysis of IRPD spectra of cold Py2+ dimers tagged with Ar or N2 reveals that the antiparallel Py2+(a) isomer with C2h symmetry is significantly more stable than the parallel Py2+(p) local minimum with Cs symmetry (by around 10 kJ mol−1) and thus dominates the population in the plasma expansion.67 A minor population of hot Py2+ ions (<15%) has been attributed to H-bonded clusters produced by intramolecular H-transfer and subsequent ring-opening reactions.67 The presence of these high-energy isomers has been inferred from additional transitions observed in the IRPD spectrum of hot Py2+ ions, which are absent in spectra recorded for cold Py2+Arn clusters.67 These additional transitions have also been observed in the IRPD spectra of Py2+ ions generated by VUV ionisation of neutral Pyn clusters but interpreted by isomers, in which the two heterocyclic units are connected by a chemical C–C or C–N bond.68,69 However, as the IR spectra computed for these chemically-bonded isomers are not compatible with the measured IRPD spectra of Py2+ for both EI67 and VUV68 generation, we currently prefer an assignment to H-bonded rather than chemically-bonded isomers.67
Py2+(a) is an ideal prototypical model to characterise in detail the aromatic CR interaction by IR spectroscopy of its bound ψ+(2Au) ground electronic state.23 First, its small five-membered ring causes the CR interaction to be very strong, in particular, stronger than any ionic NH⋯N H-bond or NH⋯π cation–π interaction.67 Moreover, the frequency of its rather isolated and strongly IR-active νNH mode provides a rather sensitive measure of the partial positive charge located on Py, which thus can precisely be measured by high-resolution IR spectroscopy.23 For example, the symmetric Py2+(a) dimer with a partial charge of qPy = 0.5e on each Py unit has its νNH (3480 cm−1) midway between those of Py (3531 cm−1) and Py+ (3447 cm−1).23 Indeed, we established a linear relation between νNH and qPy expressed as νNH(qPy)/cm−1 = −83.8 qPy/e + 3527.8.23 This linear correlation derived from data for Py, Py+, and Py2+ (qPy/e = 0, 1.0, and 0.5) was shown to hold for related clusters, in which the symmetry of Py2+ is reduced either by solvation (e.g., Py2+N2) or by substitution of functional groups (e.g., by replacing one Py by N-methyl-Py, NMPy).23 In both heterodimers (Py-PyN2, Py-NMPy), the IE of the aromatic binding partner is lowered compared to Py, leading to an asymmetric charge distribution in [Py-PyN2]+ and [Py-NMPy]+ with qPy < 0.5e and thus a blueshift in the free νNH of the remaining Py unit.23 In Py2+N2, N2 binds via an NH⋯N H-bond to one of the free acidic NH groups of Py2+(a).23,67 Therefore, stronger bases like H2O are expected to act as better H-bond acceptors with a larger reduction in symmetry of the geometric structure, the charge distribution, and thus the CR.
Herein, we characterise the microhydration process of Py2+ using IRPD of mass-selected Py2+(H2O)nArm clusters (n ≤ 3, m ≤ 1) in the CH, NH, and OH stretch ranges (νCH/NH/OH). Ar-tagging is applied to generate clusters with lower internal energy and to reduce the effective dissociation energy, leading to higher-resolution spectra and larger photodissociation efficiency.7 Complementary dispersion-corrected density functional theory (DFT) calculations at the B3LYP-D3/aug-cc-pVTZ level are employed to assign the measured IRPD spectra and to analyse the observed intermolecular interactions. The present study is motivated by the following main reasons. First, stepwise microhydration of Py2+ enables the investigation of breaking and restoring the symmetry of the CR interaction. Because H2O has a significantly higher proton affinity than N2 (PA = 691 vs. 494 kJ mol−1),70 it forms a much stronger NH⋯O ionic H-bond and thus causes a larger perturbation of the CR due to the larger difference in the IE of Py and PyH2O (computed herein as IE = 8.09 and 7.06 eV, respectively, vide infra). In addition, the magnitude of symmetry breaking can also be controlled by the degree of hydration (n). For example, the linear H-bonded (H2O)2 is an even stronger H-bond acceptor than a single H2O ligand, because of its larger PA (808 kJ mol−1),71 causing a larger perturbation of the CR in Py2+. Hence, we can address for the first time in detail the correlation between the CR in stacked π–π dimers and the strength of its H-bonds to the (dipolar) solvent molecules. In passing, we note that also only a few spectroscopic studies have been reported on the solvation effects of the related σ–σ72,73 and σ–π74 hemibonded dimer cation systems which reveal the dependence of the stability of the σ–σ and σ–π hemibonds upon solvation with various ligands. Second, microhydration of Py2+ is expected to show different hydration motifs than those of the neutral and cationic Py(+) monomers, because Py2+ offers two acidic NH groups as H-bond donors and an intermediate charge state (0.5e for each Py unit). Hence, we can follow the evolution of the Pyn(H2O)m cluster network (structure and interaction strength) as a function of (i) charge and (ii) number of available acidic proton donors in (aromatic solute)-(dipolar solvent) systems. Previous studies reveal that PyH2O20,55,75–79 and Py+H2O54 form conventional σ-type NH⋯O H-bonds, whereby the H-bond in the cation is much stronger than in the neutral (65.1 vs. 17.6 kJ mol−1), mainly due to the additional electrostatic and induction forces of the excess charge (charge-dipole and charge-induced dipole).54,80 Moreover, we have recently reported the hydration motif of Py+(H2O)2, in which the linear (H2O)2 chain is directly H-bonded to the NH group of Py+.80 Strong cooperativity of the nonadditive induction forces make the NH⋯O and OH⋯O H-bonds in the Py+(H2O)2 trimer much stronger than those in the respective Py+H2O and (H2O)2 dimers. In contrast to Py+, Py2+ has the potential to develop at least two principally different and competing H-bonding motifs in Py2+(H2O)2, such that either a (H2O)2 chain is H-bonded to one NH group (i.e., formation of a H-bonded solvent network) or two single H2O molecules are H-bonded to each NH group (i.e., interior ion solvation). Third, it is possible that the hydration process may change the preferred orientation of the two NH groups in π-stacked Py2+ from antiparallel (a) to parallel (p) and thus modify the structural motif, symmetry, and strength of the CR interaction. Our previous B3LYP-D3 calculations suggest that the Py2+(p) local minimum is less stable than the Py2+(a) global minimum by only 7.5 kJ mol−1.67 As this energy difference is much smaller than typical monohydration energies of aromatic ions with an NH⋯O ionic H-bond (∼50 kJ mol−1),80–83 stepwise hydration may indeed be able to induce a Py2+(a) → Py2+(p) transition of the CR core ion at a critical size (nc). Because Py2+(p) offers the possibility of forming cyclic hydration motifs in Py2+(p)(H2O)n clusters with one additional H-bond as compared to Py2+(a)(H2O)n, such a Py2+(a) → Py2+(p) switch in the preferred structural CR motif may indeed occur upon progressive hydration.
2. Experimental and computational details
IRPD spectra of mass-selected Py2+(H2O)nArm cluster cations (n = 1–3, m = 0–1) are measured in a tandem quadrupole mass spectrometer coupled to an EI source and an octupole ion guide.9,84 Cluster cations are produced in a pulsed supersonic plasma expansion of Py (Sigma-Aldrich, >98%, heated to 50 °C) seeded in Ar carrier gas. Hydrated cluster cations are generated by adding distilled H2O into the gas inlet system. In general, ionic clusters are formed by electron (and/or chemical) ionisation (EI) of Py close to the nozzle orifice and subsequent three-body aggregation reactions. Hydrated clusters may also be produced by EI of neutral Pyp≥2(H2O)q≥n clusters and subsequent fragmentation into Py2+(H2O)n. The cluster cations of interest are selected by the first quadrupole mass filter and irradiated in the adjacent octupole with a tunable IR laser pulse (νIR) generated by an optical parametric oscillator and amplifier (OPO/OPA) pumped by a Q-switched nanosecond Nd:YAG laser. The IR radiation is characterised by a pulse energy of 2–5 mJ, a bandwidth of <2 cm−1, and a repetition rate of 10 Hz. The IR laser frequency is calibrated to better than 1 cm−1 using a wavemeter. Resonant vibrational excitation induces the loss of the most weakly bonded ligand, i.e., either H2O or Ar. The generated fragment ions are selected by the second quadrupole mass filter and monitored by a Daly detector as a function of νIR to obtain the IRPD action spectrum of the parent cluster. The ion source is triggered at twice the laser frequency to subtract the metastable decay background from the total fragment ion signal to extract the laser-induced fragmentation signal. All IRPD spectra are normalised for frequency-dependent variations in the IR photon flux measured by a pyroelectric detector. The peak widths of the vibrational transitions observed in the IRPD spectra are mainly caused by unresolved rotational structure, sequence hot bands involving inter- and intramolecular modes, and possibly contributions of several isomers. In addition to laser-induced dissociation (IRPD and LID), low-energy collision-induced dissociation (CID) experiments are performed in the octupole ion guide to confirm the composition of the mass-selected parent clusters. For the latter mass spectra, the octupole is filled with N2 gas or air (10−5 mbar) resulting in collisions with 10 eV energy in the laboratory frame. The respective CID and LID spectra of the mass-selected parent clusters are shown in Fig. S1 in ESI.†Quantum chemical calculations are carried out at the dispersion-corrected B3LYP-D3/aug-cc-pVTZ level to determine the structural, energetic, vibrational, and electronic properties of the investigated cluster cations.85–89 This level of calculation has provided reliable results in our previous studies of related pyrrole cluster ions.23,54,67,80 Harmonic vibrational frequencies are scaled by factors of 0.9619 for NH and CH stretch vibrations and 0.9631 for OH stretch vibrations to optimise the agreement with the measured frequencies of bare Py and H2O (νNH = 3531 cm−1, ν1/3 = 3657/3756 cm−1), respectively.63,80,90 Intermolecular interaction energies (D0) and relative energies (E0) are corrected for zero-point vibrational energy. Relative free energies (G0) are calculated at 298 K. The counterpoise procedure for correcting energies for basis set superposition error is not included because such corrections are expected to be 1% or less for the large basis set employed, as has been shown for related microhydrated aromatic cluster cations.82,91 The natural charge distributions on each molecular component and the donor–acceptor interaction energies (E(2)) describing the strengths of H-bonds are obtained employing the natural bond orbital (NBO) analysis.92 Anharmonic vibrational calculations are carried out at the lower B3LYP-D3/aug-cc-pVDZ level to reduce computational costs and at the PBE0/aug-cc-pVDZ level to evaluate the dependence on the DFT functional.85,93 The reliability of the results obtained at the reduced basis set and the different DFT functional is evaluated by comparing the harmonic frequencies of each method to the frequencies obtained at the B3LYP-D3/aug-cc-pVTZ level. Harmonic vibrational frequencies calculated at the B3LYP-D3/aug-cc-pVDZ level are scaled by factors of 0.9628 for νCH/NH and 0.9621 for νOH and at the PBE0/aug-cc-pVDZ level by factors of 0.9522 for νCH/NH and 0.9474 for νOH. Vertical CR transition energies are obtained from time-dependent DFT (TD-DFT) calculations at the B3LYP-D385 level for the optimised ground state geometry and compared with results from the B3PW91-D3,94 CAM-B3LYP-D3,95 and M06-2X96 functionals using the aug-cc-pVTZ basis set. The evaluation of the strengths of the intermolecular H-bonds is facilitated by the noncovalent interaction (NCI) approach.97–99 This method is based on the analysis of the electron densities (ρ) and their reduced density gradients (RDG), s(ρ), and therefore highlights the interactions in the low-density region.99 The NCI analysis provides an index based on a plot of s(ρ) in the area in which s(ρ) is close to its minima of ρ. The final visualisation is then obtained by plotting RDG against ρ oriented by the sign of the second eigenvalue λ2 of the Hessian matrix, ρ* = sign (λ2)ρ.100,101 The plotted NCI surfaces cover the range −0.05 < ρ* < 0.0 a.u., with an isosurface value of 0.3 a.u. The NCI color code uses blue surfaces for attractive interactions (negative λ2), green surfaces for weak van der Waals contacts (λ2 near 0), and red surfaces for repulsive interactions (positive λ2). Cartesian coordinates of all considered isomers and their corresponding energies are provided in ESI.†
3. Results and discussion
3.1. Overview of IRPD spectra of Py2+(H2O)n
Fig. 1 compares the IRPD spectra of Py2+(H2O)n with n = 1–3 measured in the H2O loss channel. For comparison, the previously reported IRPD spectra of Py+Ar and Py2+ measured in the Ar and Py loss channels, respectively, are included as well.54,67 The positions and widths of the bands observed are listed in Table S1 (ESI†) along with the suggested vibrational and isomer assignments. The spectral range investigated (2600–3800 cm−1 for Py2+(H2O)n with n = 1–2 and 3000–3800 for n = 3) covers the free and bound OH and NH stretch bands of the H2O ligands and the Py2+ cation, as well as the aromatic CH stretch modes of Py2+. The IRPD spectra in Fig. 1 exhibit systematic shifts in the OH and NH stretch bands as a function of cluster size and composition, providing detailed information about the stepwise microhydration process around Py2+. The free OH stretch range (3600–3800 cm−1) contains the symmetric and antisymmetric OH stretch bands of H2O ligands acting as single/double acceptors (ν1 and ν3, B and A) and the free OH stretch bands of H2O ligands acting as single/double acceptor and single donor (νfOH, C). The corresponding H-bonded OH stretch modes of H2O involved as an H-bond donor occur near 3400 cm−1 (νbOH, D). The free NH stretch of Py2+ occurs near 3500 cm−1 (νfNH, E), while the H-bonded NH stretch shifts strongly to the red down to 3000–3300 cm−1 upon hydration (νbNH, F, F1). Peak G observed at ∼3130 cm−1 in all spectra is assigned to the aromatic CH stretch vibrations (νCH) of Py2+. Bands X, Y, and Y′ cannot arise from fundamentals of Py2+(H2O)n clusters with π-stacked Py2+(a/p) cores and are attributed at this initial stage to either overtone and/or combination bands or transitions of other minor isomers.102,103 The pronounced broad peak F* in the IRPD spectrum of Py2+ has been assigned in our previous study to νbNH of a minor population of H-bonded isomers, Py2(CC/OC)+.67 Its absence in the IRPD spectra of Py2+(H2O)n and their Ar-tagged clusters indicates that these isomers are below the detection limit for the hydrated clusters. In the following sections, the structures of the microhydrated Py2+ clusters are identified by comparing the measured IRPD spectra with linear IR absorption spectra computed for low-energy isomers. The IRPD spectra of their Ar-tagged clusters are also considered because their lower temperature yields narrower bands in the IRPD spectra, which provide more precise vibrational assignments.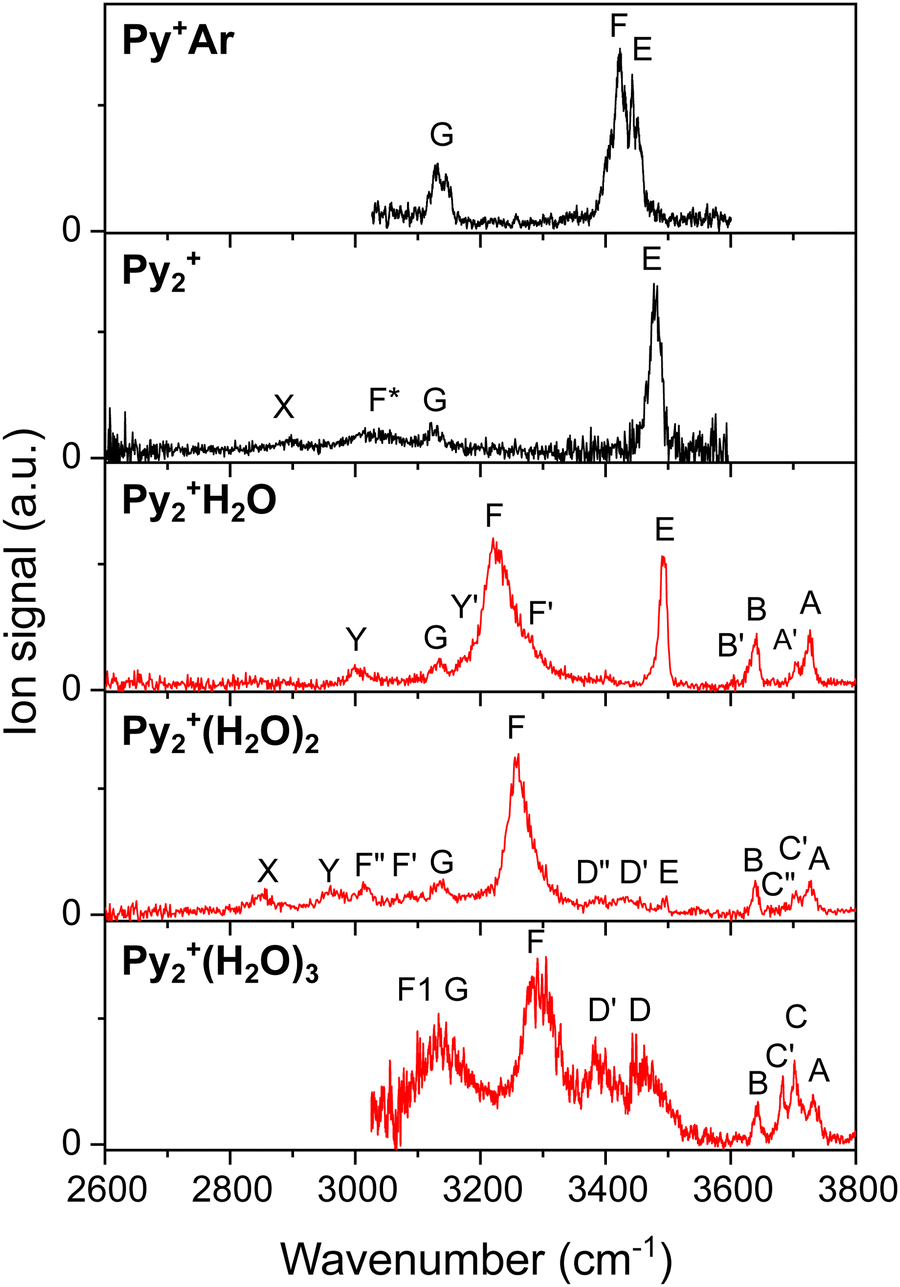 | ||
| Fig. 1 IRPD spectra of Py2+(H2O)n with n = 0–3 (and Py+Ar for comparison)54 recorded in the CH, NH, and OH stretch range. The positions, width, and assignments of the observed transitions are compiled in Table S1 (ESI†). | ||
3.2. Py1/2+ and (H2O)1/2
Before considering the Py2+(H2O)n clusters, we briefly review the available knowledge on the Py1/2+ and (H2O)1/2 subunits relevant to the current study. The structures of Py and Py+ in their 1A1 and 2A2 electronic ground states are planar (C2v), and the N–H bond length increases by 7 mÅ from rNH = 1.0031 to 1.0102 Å upon ionisation from the π(a2) HOMO orbital (Fig. 2). This geometry change results in a large calculated redshift from 3531 to 3459 cm−1 (ΔνfNH = −72 cm−1), which compares favourably with the measured values of 3531 and 3447 cm−1 (ΔνfNH = −84 cm−1), respectively.54,63 The IRPD spectrum of Py+Ar shown in Fig. 1 exhibits two bands E and F at 3450 and 3423 cm−1 assigned to νfNH of π-bonded Py+Ar(π) and νbNH of H-bonded of Py+Ar(H), respectively.54 Because the π-bonded local minimum with D0 = 6.6 kJ mol−1 shows a computed νfNH blueshift of only 3 cm−1 from that of bare Py+ (3459 vs. 3462 cm−1), an accurate experimental value can be estimated for bare Py+ as νfNH = 3447 cm−1.54 The formation of the NH⋯Ar H-bond in the planar Py+Ar(H) global minimum (C2v) with D0 = 9.4 kJ mol−1 and Re = 2.470 Å causes a larger redshift measured as ΔνbNH = −50 cm−1. | ||
| Fig. 2 Structures of (a) Py(+), (b) H2O and (H2O)2, (c) Py2+(a), and (d) Py2+(p) obtained at the B3LYP-D3/aug-cc-pVTZ level. Selected intra- and intermolecular bond lengths (in Å) are given in black and red, respectively. Values in dark red indicate NBO charges (in units of e). The dissociation energies (D0, in parentheses) are given in kJ mol−1. | ||
Because of the CR, the positive charge in Py2+(a) is equally shared between the two equivalent Py units and both N–H bond lengths and NH stretch frequencies of Py2+ are intermediate between those of Py and Py+ (Fig. 2).23 Indeed, for the Py2+(a) global minimum, rNH = 1.0061 Å is close to the average of Py and Py+ (rNH = 1.0031 and 1.0102 Å). As a result, the calculated frequencies of the antisymmetric IR-active and symmetric IR-forbidden NH stretch modes, νfNH = 3500 and 3501 cm−1, are midway between those of Py and Py+ computed as νfNH = 3531 and 3459 cm−1. These computational predictions agree well with band E observed at νfNH = 3479 cm−1 in the IRPD spectrum of Py2+ (Fig. 1), which is also midway between those of Py and Py+ observed at νfNH = 3531 and 3447 cm−1.23,67 The slightly less stable Py2+(p) local minimum structure with a parallel orientation of the two NH groups (Fig. 2) has two IR-active NH stretch modes calculated as νfNH = 3499 and 3510 cm−1 with a splitting of 11 cm−1 (Table S2, ESI†). As these frequencies are similar to those of the Py2+(a) global minimum, one cannot exclude the contribution of Py2+(p) to band E in the IRPD of bare Py2+ because its width (19 cm−1) is much larger than the νfNH shifts and splittings for the Py2+(a/p) rotamers.67 However, from the energetic point of view, the Py2+ population will be dominated by Py2+(a) because it is more stable than Py2+(p) by 7.5 kJ mol−1 (B3LYP-D3). Indeed, in our recent study on the internal energy dependence of the formation of Py2+ in our EI source, the higher-resolution IRPD spectra of cold Py2+Ln clusters tagged with L = Ar and N2 suggest that the population of Py2+(p) is at most 15% of the π-stacked Py2+ dimers.67 The high rotational barriers (Vb) for a → p and p → a isomerisation computed at different computational levels suggest that the Py2+(a/p) isomers are kinetically trapped in their potential well rather than isomerising between each other. Therefore, the estimated Py2+(p) abundance using a Boltzmann distribution based on ΔE0(B3LYP-D3) = 7.5 kJ mol−1 is 17% at 298 K, which is compatible with the experiment (≤15%). In the same study, we assigned the additional bands at 2888 (X) and 3028 (F*) cm−1 observed in the IRPD spectrum of bare Py2+ (but not in Py2+Ln) to a Fermi doublet (2βNH and νbNH) of a minor population (10%) of H-bonded Py2(OC)+ isomers containing one open-cycle (OC) Py unit. The latter dimers are characterized by an NH⋯N ionic H-bond between Py+ and a neutral noncyclic Py isomer formed by H-migration and subsequent ring opening in the EI source.67
The calculated O–H bond parameters of H2O in its 1A1 ground state (rOH = 0.9618 Å, ν1 = 3657 cm−1, ν3 = 3756 cm−1) agree well with the experimental data (0.9578 Å, 3657 and 3756 cm−1).90 Our computed (H2O)2 equilibrium geometry has the known trans-linear H-bonded structure (Fig. 2), with an O⋯O distance (rOO = 2.909 Å) and an angle of the acceptor H2O axis with respect to rOO (59.1°) consistent with corresponding experimental values (2.98 Å, 58°).104 The four calculated OH stretch frequencies of the H2O acceptor (ν1/3 = 3652/3747 cm−1) and donor (νb/fOH = 3540/3728 cm−1) are also close to their experimental data (ν1/3 = 3654/3746 cm−1, νb/fOH = 3601/3735 cm−1), respectively.105–107 The H-bond strength computed as D0 = 13.0 kJ mol−1 compares again favourably with the experimental determination of D0 = 13.2 ± 0.1 kJ mol−1 (1105 ± 10 cm−1),108 illustrating that the chosen B3LYP-D3 level reliably describes the H2O⋯H2O interaction. The IEs of (H2O)n calculated at the B3LYP-D3 level (12.60 and 10.73 eV for n = 1 and 2) are much higher than the IEs of both Py (8.09 eV) and Py2 (7.22 eV), indicating that the positive charge is mostly located on Py2 rather than on the H2O ligands, which justifies the notation of Py2+(H2O)n. These calculated IEs are close to available experimental values (IE = 12.615, 11.21, 8.207 for H2O, (H2O)2, Py, respectively).109–113
3.3. Py2+H2O
Based on the two stacked Py2+(a/p) minima, several low-energy Py2+H2O structures are obtained, and the four most stable ones, Py2+(a/p)H2O(I and II), are shown in Fig. 3 and Fig. S2 (ESI†). Their computed IR spectra are compared in Fig. 4 and Fig. S3 (ESI†) to the IRPD spectra recorded for Py2+H2O and Py2+H2OAr. In the most stable structure, Py2+(a)H2O(I), H2O is attached to Py2+(a) via a nearly linear NH⋯O H-bond, a binding motif similar to the NH⋯N H-bond reported previously for Py(2)+(a)N2.54,67 The H-bond in Py2+(a)H2O(I) is substantially weaker than that in Py+H2O(H)54 (RNH⋯O = 1.809 vs. 1.704 Å, D0 = 46.3 vs. 65.1 kJ mol−1, E(2) = 30.0 vs. 46.4 kJ mol−1), because of the strongly reduced charge on the proton-donor Py molecule upon attachment of the second Py unit (from 1.0 to 0.5e). As a result of the weaker H-bond, the charge transfer to H2O is smaller (qH2O = 0.033 vs. 0.051e). The N–H donor bond becomes less elongated (rNH = 1.0219 vs. 1.0363 Å, ΔrNH = 16 vs. 26 mÅ), leading to a smaller redshift in νbNH (ΔνbNH = −278 vs. −455 cm−1, νbNH = 3223 vs. 3004 cm−1). The free N–H bond of the second Py unit contracts slightly upon monohydration (1.0057 vs. 1.0061 Å), resulting in an observed blueshift of 6 cm−1 (νfNH = 3506 vs. 3500 cm−1). Monohydration completely decouples the two NH stretch oscillators of Py2+(a), and the free NH stretch is a pure local mode of the free Py unit. Its partial charge is smaller than that on the H-bonded Py molecule (qPy = 0.441 vs. 0.526e), which may be rationalised by the lower ionisation energy of PyH2O as compared to Py (calculated as IE = 7.60 vs. 8.09 eV). This asymmetry in the Py2+(a) charge distribution reduces the strength of the CR (vide infra). According to the qPy–νfNH relation, the lower charge is fully consistent with the blueshift of νfNH. As in Py2+(a)H2O(I) the H-bond to H2O is weaker than that in Py+H2O(H), the O–H bonds in H2O are less elongated (rOH = 0.9632/0.9627 vs. 0.9636 Å) and the computed OH stretch frequencies are correspondingly higher (ν1/3 = 3651/3739 vs. 3643/3729 cm−1). | ||
| Fig. 3 Structures of (a) and (b) Py2+(a)H2O(I and II), (c) Py2+(p)H2O(I), and (d) Py2+(a)H2O(I)Ar(I) obtained at the B3LYP-D3/aug-cc-pVTZ level. Selected intra- and intermolecular bond lengths (in Å) are given in black and red, respectively. Values in dark red indicate NBO charges (in units of e). Energies in parentheses are the relative energy, the dissociation energy of the most weakly bonded ligand, and the relative Gibbs free energy at 298 K (E0, D0, and G0 in kJ mol−1). | ||
 | ||
| Fig. 4 IRPD spectra of Py2+H2OArn (n = 0–1) recorded in the CH, NH, and OH stretch range are compared to linear IR absorption spectra calculated for Py2+(a)H2O(I and II) and Py2+(a)H2O(I)Ar(I and II) at the B3LYP-D3/aug-cc-pVTZ level (Tables S1 and S3, ESI†). | ||
Py2+(a)H2O(II) is the second most stable isomer with a Py2+(a) core. Its C2 symmetric structure has a bifurcated CH⋯O H-bond of H2O to two CH groups of the two Py units, with RCH⋯O = 2.467 Å and D0 = 28.5 kJ mol−1. It is substantially less stable than the global minimum by 17.8 kJ mol−1, because the two CH⋯O H-bonds are much weaker than the single NH⋯O H-bond, resulting in a smaller charge transfer to H2O (qH2O = 0.010 vs. 0.033e). The remaining charge on Py2+ is shared symmetrically (leading to an optimised and fully developed CR) but somewhat lower than in bare Py2+ (qPy = 0.495 vs. 0.500e) due to the minor charge transfer to H2O. In line with the qPy–νfNH relation, the N–H bonds are slightly shorter (1.0057 vs. 1.0061 Å), leading to a calculated blueshift of 5 cm−1 for the two νfNH frequencies (Table S3, ESI†).
The most stable isomer with a Py2+(p) core, Py2+(p)H2O(I), shown in Fig. 3 features an asymmetric bifurcated nonlinear H-bond of H2O (double acceptor) to both available NH donor groups (RNH⋯O = 1.988 and 2.292 Å) and thus the highest H2O dissociation energy (D0 = 50.1 kJ mol−1), although the individual NH⋯O H-bonds are much weaker than in Py2+(a)H2O(I). Hence, its energy gap to the Py2+(a)H2O(I) global minimum is reduced to only 3.7 kJ mol−1. The two Py units are not equivalent, and the relatively free Py unit carries less positive charge than the more strongly H-bonded bottom one (qPy = 0.456 vs. 0.517e). The weaker H-bond is strongly nonlinear and gives rise to an almost free νNH (E(2) = 4.4 kJ mol−1), while the stronger H-bond is more linear (E(2) = 17.6 kJ mol−1). As both NH groups are involved in H-bonding, we cannot readily apply the qPy–νfNH relation. The weaker H-bonded N–H bond is less elongated than the more strongly H-bonded one (rNH = 1.0082 and 1.0147 Å), which results in two redshifted and strongly IR-active νbNH modes at 3339 and 3466 cm−1, respectively (Table S4, ESI†). To enable bifurcated H-bonding, the H2O plane has to rotate by 90° providing direct access to both lone pairs of the O atom. In this way, H2O attachment strongly reduces the distance between the two Py rings (RN⋯N = 3.026 vs. 4.217 Å). Due to the charge transfer to H2O (qH2O = 0.027e), the O–H bonds become longer (rOH = 0.9646 Å) and the OH stretch frequencies are redshifted compared to H2O (ν1/3 = 3632/3713 vs. 3657/3756 cm−1). In contrast to Py2+(p)H2O(I), Py2+(p)H2O(II) has a single NH⋯O H-bond to the top Py unit. The geometric, energetic, and vibrational parameters of this NH⋯O H-bond are quite similar to those in Py2+(a)H2O(I), indicating that internal rotation of the free Py unit by 180° has only a small impact on the remote monohydration motif and interaction strength. As a result, the IR spectra predicted for Py2+(a)H2O(I) and Py2+(p)H2O(II) are very similar, too. The largest difference in both structures refers to the free N–H bond, which is shorter in Py2+(p)H2O(II) leading to a higher νfNH (rNH = 1.0048 vs. 1.0057 Å, νfNH = 3517 vs. 3506 cm−1), consistent with the modified CR, which is less asymmetric (0.485/0.483 vs. 0.526/0.441e).
Next, we briefly consider the predicted effects of Ar tagging on the properties of Py2+H2O by taking the example of the most stable Py2+(a)H2O(I) isomer (Fig. 3 and Fig. S4, ESI†). All considered Ar-tagged clusters have similar dissociation energies of D0 = 4.2–6.8 kJ mol−1, which are consistent with the experimentally determined Ar binding energy.67 Because the Ar binding energy is very low compared to ∼100 and ∼50 kJ mol−1 for the much stronger Py+⋯Py and Py2+⋯H2O interactions, respectively, the effects of Ar tagging are minor and thus we discuss only the two most stable Ar isomers, Py2+(a)H2O(I)Ar(I and II). They have slightly different N–H and O–H bond lengths because Ar affects their H-bonds in a different fashion. The NH⋯Ar H-bond of Ar to the free Py unit in Py2+(a)H2O(I)Ar(I) induces an N–H bond elongation by 0.8 mÅ to 1.0065 Å, resulting in a slight redshift of νfNH from 3506 to 3494 cm−1 (Table S1, ESI†). In Py2+(a)H2O(I)Ar(II), Ar forms an OH⋯Ar H-bond to the H2O ligand and interacts weakly with two adjacent CH groups. Its proton donor N–H bond is longer by 0.8 mÅ than in Py2+(a)H2O(I)Ar(I) (1.0225 vs. 1.0217 Å), as H2OAr has a larger PA than H2O, which leads to a slight redshift in νbNH from 3226 to 3212 cm−1 (Table S3, ESI†). For the structures, binding energies, and IR spectra of the Py2+(a)H2O(I)Ar(III–V) and Py2+(p)H2O(I)Ar(I) isomers, the reader is referred to Fig. S2–S4 and Tables S3 and S4 (ESI†).
The IRPD spectra of Py2+H2O and Py2+H2OAr are compared in Fig. 4 to the IR spectra computed for the Py2+(a)H2O(I and II) and Py2+(a)H2O(I)Ar(I and II) isomers. The positions and widths of the transitions observed are listed in Table S1 (ESI†) along with vibrational and isomer assignments. The IRPD spectrum of Py2+H2O exhibits ten main transitions A, A′, B, B′, E, F, F′, G, Y, and Y′ at 3725, 3705, 3639, 3605, 3492, 3228, 3282, 3134, 3006, and 3175 cm−1 with widths of 13–58 cm−1. The IRPD spectrum of Py2+H2OAr shows corresponding, but better resolved, transitions at 3727, 3706, 3642, 3607, 3488, 3210, 3274, 3133, 3010, and 3175 cm−1, respectively, with similar intensity ratios but much smaller widths (4–28 cm−1), because the lower binding energy leads to colder clusters. The small shifts of ≤10 cm−1 imply that Ar tagging has indeed little impact on the Py2+H2O structure and resulting IR spectrum apart from the substantially improved spectral resolution. An even colder IRPD spectrum of Py2+H2OAr measured in the range of 3100–3800 cm−1 exhibits only six main transitions (A, B, E, F, G, and Y′ at 3727, 3642, 3489, 3213, 3141, and 3178 cm−1) with even narrower widths and the absence of the minor peaks A′, B′, and F′ present in the IRPD spectra of Py2+H2O and Py2+H2OAr recorded under warmer conditions. This difference suggests a minor contribution of less stable isomers that are responsible for A′, B′, and F′. The colder IRPD spectrum of Py2+H2OAr also shows that the most abundant and thus most stable isomer can be probed by tagging under the coldest conditions. The major transitions in the IRPD spectra of Py2+H2O(Ar) can be explained by the most stable Py2+(a)H2O(I) isomer. Indeed, good agreement is observed between measured and computed IR spectra concerning both the positions and relative intensities of the vibrational transitions. Bands A and B of Py2+H2O at 3725 and 3639 cm−1 correspond well with the predicted transitions of ν3 = 3739 and ν1 = 3651 cm−1. Band E at 3492 cm−1 can be assigned to νfNH predicted at 3506 cm−1. Its modest blueshift of 13 cm−1 upon monohydration from 3479 to 3492 cm−1 is well reproduced by the calculations (from 3500 to 3506 cm−1). The intense band F at 3228 cm−1 is readily assigned to νbNH predicted at 3223 cm−1. Its blue-shaded band contour confirms the assignment to a proton-donor stretch vibration, consistent with the strong NH⋯O ionic H-bond.54,80,81,114 This band shows again drastic narrowing and a small redshift (−18 cm−1) as a result of cooling by Ar tagging.54,80–82 Band G observed at 3134 cm−1 is attributed to the eight unresolved and nearly degenerate νCH modes with a predicted spread of only 24 cm−1. The colder IRPD spectrum of Py2+H2OAr exhibits only the major peaks A, B, E, F, G, and Y′ assigned to Py2+H2O(I)Ar(I). Therefore, the absent minor peaks A′, B′, and F′ observed in the IRPD spectrum of Py2+H2O and warmer Py2+H2OAr must come from a higher-energy isomer. Although the substantially less stable Py2+(a)H2O(II) can at first glance be excluded as a major carrier of the spectrum because of its high relative energy (E0 = 17.8 kJ mol−1) and the lack of the intense νbNH transition (band F), it can be still considered as a candidate for the minor contribution from its predicted (scaled) harmonic frequencies (vide infra). Next, Py2+(p)H2O(I) can be excluded as the predominant carrier (Fig. S3a, ESI†) because of the lack of its intense νbNH band predicted at 3339 cm−1 in the observed IRPD spectrum although its relative energy is higher by only 3.7 kJ mol−1 compared to Py2+(a)H2O(I). However, the predicted harmonic ν1/3 of Py2+(p)H2O(I) are lower than those of Py2+(a)H2O(I) (3651/3713 vs. 3651/3739) and νbNH is higher (3339 vs. 3223 cm−1), indicating that Py2+(p)H2O(I) may contribute to the minor peaks A′, B′, and F′. The slightly less stable Py2+(p)H2O(II) isomer will not be considered any further because of the close similarity of its predicted spectrum to that of the assigned Py2+(a)H2O(I) global minimum. Although the scaled harmonic spectra of Py2+(a/p)H2O(I) explain most of the peaks (A–G), none of the structures can explain peaks Y and Y′. Therefore, we consider anharmonic calculations for Py2+(a)(H2O)(I) conducted at the reduced B3LYP-D3/aug-cc-pVDZ level (Fig. S3b, ESI†). These calculations suggest an assignment of band Y to the NH bend overtone (2βNH) of the N–H⋯O H-bonded Py unit, which gains intensity via Fermi resonance with the nearby and strongly IR-active νbNH fundamental.82,102,103,115 Shoulder Y′ may be assigned to the OH bend overtone (2βOH) of H2O.116–119 Although the B3LYP-D3/aug-cc-pVDZ level explains the anharmonicity of Py2+(a) well, it overestimates the anharmonicity of ν1/3 of NH⋯O H-bonded H2O. Therefore, we also consider the PBE0/aug-cc-pVDZ level to support our anharmonic predictions. Fig. S3b (ESI†) shows the reliability of the PBE0/aug-cc-pVDZ level by comparison to the harmonic and anharmonic spectra calculated at the B3LYP-D3/aug-cc-pV(D/T)Z levels. The anharmonic frequencies of Py2+(a/p)H2O(I) calculated at the PBE0/aug-cc-pVDZ level are again in good agreement with our assignment.
The higher-resolved IRPD spectra of the colder Ar-tagged ions reveal small additional bands under certain experimental conditions, indicating the minor presence of higher-energy isomers. These may arise from different Py2+H2O isomers or different Ar tagging sites of the major assigned Py2+(a)H2O(I) core. Band E observed at 3489 cm−1 and assigned to νfNH of Py2+H2OAr in the cold spectrum is redshifted by 3 cm−1 from that of Py2+H2O (3492 cm−1), suggesting that Ar is attached to the free NH group of Py2+(a)H2O(I) in the major Py2+H2OAr isomer, with a computed redshift of 13 cm−1. The other four Py2+(a)H2O(I)Ar(II–V) isomers, in which Ar does not bind to the NH group, do not reproduce this νfNH redshift (Fig. S4 and Table S3, ESI†). The somewhat warmer Py2+H2OAr spectrum in Fig. 4 shows additional small bands A′, B′, and F′ at 3706, 3607, and 3274 cm−1, which are similar to those of the bare Py2+H2O spectrum at 3705, 3605, and 3282 cm−1, indicating a small contribution of Py2+(p)H2O(I)Ar. The observed redshift in F′ from 3282 to 3274 cm−1 suggests that the Ar may bind to the NH group of Py2+(p)H2O(I) as shown in Fig. S2 and S3 (ESI†). The abundance of Py2+(p)H2O(I)Ar(I) is estimated to be below 25% when considering its computed νbNH intensity (Fig. S3e, ESI†). The IRPD spectrum of colder Py2+H2OAr exhibits sharp bands with no splitting in the range of ν1/3, revealing the sole presence of Py2+(a)H2O(I), while the other high-energy isomers are below the detection limit of 5% by considering the signal-to-noise ratio of peak F (νbNH).
3.4. Py2+(H2O)2
Our B3LYP-D3 calculations yield seven dihydrated isomers with a Py2+(a) core, Py2+(a)(H2O)2(I–VII) (Fig. 5 and Fig. S5, ESI†). These can be divided into two groups depending on whether two individual H2O ligands or an H-bonded (H2O)2 dimer are attached to Py2+(a). In the highly-symmetric global minimum (C2h), Py2+(a)(H2O)2(I), which belongs to the first group, two individual H2O ligands form equivalent NH⋯O H-bonds to the two available acidic NH groups of Py2+(a). The next two isomers II and III belong to the second group, in which a (H2O)2 dimer chain is attached to one of the NH groups either with or without a second CH⋯O contact, respectively, while the other NH group remains free. The latter structure is similar to that of Py+(H2O)2 reported previously.80 The relative energies calculated for isomers I–III are E0 = 0.0, 5.0, and 5.5 kJ mol−1, and their relative free energies amount to G0 = 0.0, 11.1, and 8.2 kJ mol−1, respectively. The energy gap between I and both II and III increases significantly with temperature due to the reduced entropy contribution for the less flexible isomers II and III resulting from the structural constraints of the (H2O)2 unit. From the energetics, I–III are expected to dominate the Py2+(H2O)2 population in the molecular beam. The other isomers IV–VII, which also feature NH⋯O and CH⋯O H-bonds, are substantially higher in energy (E0 = 8.5–37.2 and G0 = 15.4–40.6 kJ mol−1) and thus not considered in detail further. | ||
| Fig. 5 Structures of Py2+(a)(H2O)n with n = 2 (a)–(c) and n = 3 (d)–(g) obtained at the B3LYP-D3/aug-cc-pVTZ level. Selected intra- and intermolecular bond lengths (in Å) are given in black and red, respectively. Values in dark red indicate NBO charges (in units of e). Energies in parentheses indicate the relative energy, the dissociation energy of the most weakly bonded ligand, and the relative Gibbs free energy at 298 K (E0, D0, and G0 in kJ mol−1). | ||
The NH⋯O H-bonds in the dihydrated Py2+(a)(H2O)2(I) global mininum are slightly weaker than that of the related monohydrated Py2+(a)H2O(I) isomer (RNH⋯O = 1.826 vs. 1.809 Å, D0 = 44.2 vs. 46.3 kJ mol−1, E(2) = 37.7 vs. 30.0 kJ mol−1), with a smaller perturbation of the N–H proton donor bond (rNH = 1.0202 vs. 1.0219 Å, νbNH = 3247 vs. 3223 cm−1). This is the result of the slightly noncooperative three-body induction forces for interior ion solvation with individual ligands due to extended delocalisation of the positive charge. The asymmetric CR in Py2+(a)H2O(I) becomes symmetric again in Py2+(a)(H2O)2(I) (qPy = 0.526/0.441 vs. 0.461/0.461e). Although the individual H-bonds are weaker, the charge transfer to the H2O ligands is increased in Py2+(a)(H2O)2(I) (qH2O = 0.039 vs. 0.033e) because of the restored symmetry and optimised CR interaction.
Both Py2+(a)(H2O)2(II and III) local minima have a similar solvent configuration with one free NH group and one NH group solvated by (H2O)2, resulting in a similar charge distribution. Their major difference is that the terminal H2O in II has an additional weak CH⋯O contact with Py2+(a), forming a four-membered cyclic ring. This additional H-bond causes, however, strain on the other H-bonds which become weaker in II compared to III, so that the net energy gain by increasing the size of the H-bonded network is only 0.5 kJ mol−1. The intermolecular Py⋯H2O and H2O⋯H2O distances in isomer II are longer than in III, and the N–H proton donor bond elongates less (rNH = 1.0283 vs. 1.0308 Å, νbNH = 3107 vs. 3068 cm−1) due to the weakened NH⋯O H-bond. As (H2O)2 is attached to Py2+ in II and III, the resulting NH⋯O H-bond is much stronger than in I (R = 1.742 and 1.720 vs. 1.826 Å), because of the higher PA of (H2O)2 compared to H2O and the enhanced charge transfer to the solvent (qH2O = 0.050 and 0.059 vs. 0.039e). As a result, the elongation of the N–H proton donor bond is larger (rNH = 1.0283 and 1.308 vs. 1.0202 Å), causing larger NH stretch redshifts (νbNH = 3107 and 3068 vs. 3247 cm−1). The same PA argument explains the stronger NH⋯O H-bond in dihydrated Py2+(a)(H2O)2(III) compared to that in monohydrated Py2+(a)H2O(I). The high cooperativity of three-body induction forces typical for H-bonded solvent networks also strongly strengthens the H-bond of (H2O)2 (Fig. 2) by the presence of the positive charge of the nearby Py2+ ion (R = 1.773 vs. 1.946 Å, D0 = 38.8 vs. 13.0 kJ mol−1). Nonetheless, these H-bonds are in Py2+(a)(H2O)2(III) with a Py2+(a) core substantially weaker than those in Py+(H2O)2(I) with a Py+ core (RNH⋯O = 1.720 vs. 1.609 Å, ROH⋯O = 1.773 vs. 1.732 Å) due to the increased charge delocalisation arising from the CR (qPy = 0.528 vs. 0.926e). The free NH group of Py2+(a) becomes progressively less acidic with mono- and dihydration (rNH = 1.0061, 1.0057, 1.0054 Å for n = 0–2), resulting in small incremental blueshifts of νfNH as n increases from 0 to 2 (3500, 3506, and 3510 cm−1).
Before comparing the computed IR spectra to the IRPD spectra, we briefly discuss the effects of Ar tagging on the structure and energy of Py2+(a)(H2O)2 (Fig. S6, ESI†), with particular focus on the Py2+(a)(H2O)2(I) global minimum with C2h symmetry. The three most stable Py2+(a)(H2O)2(I)Ar(I–III) isomers have Ar binding energies of D0 = 6.7, 4.3, and 3.2 kJ mol−1. In the most stable Ar(I) isomer, Ar binds to the OH group of H2O and one CH group of Py. In the Ar(II) isomer, Ar interacts with the π electron ring of Py and thus has the least perturbation on the structure and IR spectrum of Py2+(a)(H2O)2. In Ar(III), Ar forms a linear OH⋯Ar H-bond to one of the H2O ligands, which causes redshifts of 7 and 5 cm−1 in ν3 and ν1 of Py2+(a)(H2O)2(I), respectively. In Ar complexes of the higher-energy isomer Py2+(a)(H2O)2(II), Ar preferentially binds to the free NH group, resulting in its slight elongation of 0.2 mÅ. Nevertheless, νfNH redshifts by only 2 cm−1 (3508 vs. 3510 cm−1). In Py2+(a)(H2O)2(III)Ar(I), Ar is part of a four-membered ring with OH⋯Ar⋯HC bonds, whereas in isoenergetic Py2+(a)(H2O)2(III)Ar(II), Ar binds to the free NH group of Py and elongates its N–H bond by 0.7 mÅ, resulting in a slight redshift of νfNH by 8 cm−1 (from 3508 to 3500 cm−1). At higher temperature, Py2+(a)(H2O)2(III)Ar(II) is more favourable than Py2+(a)(H2O)2(III)Ar(I) due to entropy (G0 = 2.4 vs. 10.3 kJ mol−1). For the structures, binding energies, and IR spectra of the less stable Py2+(a)(H2O)2(IV-VI)Ar isomers, the reader is referred to Fig. S6 and Table S5 (ESI†). As expected from the weak Ar interaction energies (D0 ≤ 7 kJ mol−1), the overall impact of Ar tagging on the computed IR spectra (<10 cm−1) and energetic order is very minor for all considered Py2+(a)(H2O)2 isomers and thus not considered in detail further.
The IRPD spectra of Py2+(H2O)2 and Py2+(H2O)2Ar are compared in Fig. 6 to the IR spectra computed for the three most stable isomers Py2+(a)(H2O)2(I–III) with a Py2+(a) core. The positions and widths of the transitions observed are listed in Table S1 (ESI†), along with vibrational and isomer assignments. The IRPD spectrum of Py2+(H2O)2 exhibits 13 major transitions at 3726 (A), 3639 (B), 3703 (C′), 3684 (C′′), 3430 (D′), 3388 (D′′), 3493 (E), 3260 (F), 3093 (F′), 3014 (F′′), 3136 (G), 2852 (X), and 2967 (Y) cm−1, with widths of 12–64 cm−1. The IRPD spectrum of Py2+(H2O)2Ar recorded in a smaller spectral range has corresponding transitions at 3730 (A), 3643 (B), 3706 (C′), 3700 (C′′), 3424 (D′), 3376 (D′′), 3493 (E), 3242 (F), and 3135 (G) cm−1, respectively, with similar intensity ratios but much smaller widths (3–19 cm−1). Ar tagging shifts of those peaks are less than 18 cm−1 and on average only 7 cm−1. Thus, Ar tagging has essentially no impact on the intermolecular structure and vibrational signatures of the observed Py2+(H2O)2 isomers. As Py2+(a)(H2O)2 has only six NH/OH stretch oscillators (2νNH, 4νOH), a single isomer cannot explain all observed features detected in this spectral range. Therefore, several isomers need to be considered. The major three bands A, B, and F observed for Py2+(H2O)2 and Py2+(H2O)2Ar are fully reproduced by the most stable isomer of Py2+(a)(H2O)2(I) concerning both band positions and relative intensities. The most intense and strongly blueshaded band F is readily assigned to νbNH, while the weak bands A and B are assigned to ν3 and ν1, respectively. Because of C2h symmetry of Py2+(a)(H2O)2(I), only three out of the six OH/NH stretch fundamentals are IR-active, namely the out-of-phase linear combinations of νbNH, ν1, and ν3. Band G at 3136 cm−1 assigned to νCH is not sensitive to isomeric structure. Band E at 3493 cm−1 is close to νfNH of Py2+ and well reproduced by Py2+(a)(H2O)2(II and III), both of which exhibit a free NH group. In addition, the (H2O)2 unit of isomers II and III give rise to νbOH and νfOH bands of the H2O donor, which are predicted to be close to the D′/D′′ and C′/C′′ pairs. Their corresponding νbNH bands may be attributed to bands F′/F′′. The interpretation of the weak bands Y and X is less certain, as no fundamentals are predicted in this range for any of the considered isomers. Similar to the n = 1 cluster, they may arise from Fermi resonance of 2βNH/νbNH.54,67 In summary, Py2+(a)(H2O)2(I) is concluded as the major carrier of the IRPD spectrum of Py2+(H2O)2, while Py2+(a)(H2O)2(II and III) account for all remaining weaker transitions. When comparing the computed and measured intensities of the assigned νbNH bands (Fig. 6a), the relative abundances of isomers I–III can roughly be estimated as 65, 10, and 25%, consistent with their relative free energies of G0 = 0.0, 11.1, and 8.2 kJ mol−1, respectively.
 | ||
| Fig. 6 IRPD spectra of (a) Py2+(H2O)2Arn (n = 0–1) and (b) Py2+(H2O)3 recorded in the CH, NH, and OH stretch range are compared to the linear IR absorption spectra calculated for Py2+(a)(H2O)n(I–III) (n = 2–3) at the B3LYP-D3/aug-cc-pVTZ level (Tables S1, S5, and S7, ESI†). | ||
Interestingly, microhydration reduces the energy gap ΔE0(a–p) between Py2+(a) and Py2+(p) due to the more favourable formation of H-bonded solvent networks with two NH⋯O ionic H-bonds. For example, ΔE0(a–p) = 7.5 and 3.7 kJ mol−1 for n = 0 and 1, respectively. Indeed, for n = 2 the most stable Py2+(p)(H2O)2(I) isomer with a Py2+(p) core is computed to be even more stable than Py2+(a)(H2O)2(I) by ΔE0(a–p) = −1.0 kJ mol−1 (Fig. S7, ESI†). However, from an entropic point of view, ΔG0(a–p) = +6.0 kJ mol−1 for n = 2 due to the strain imposed by the H-bonded network. The two most stable Py2+(p)(H2O)2(I and II) isomers form tetra- and tricyclic rings involving Py2+(p) and (H2O)2, with ΔE0 = −1.0 and +3.4 kJ mol−1, respectively. In Py2+(p)(H2O)2(I), (H2O)2 acts as a dual H-bond acceptor with two individual NH⋯O H-bonds, while in Py2+(p)(H2O)2(II) both NH groups attack the donor of (H2O)2via a bifurcated O⋯(HN)2 configuration, similar to monohydrated Py2+(p)H2O(I). The larger PA of (H2O)2 results in a larger cooperative effect, thereby reducing ΔE0(a–p). Although Py2+(p)(H2O)2(I) is predicted as the most stable Py2+(H2O)2 structure at T = 0 K and the OH stretches (ν3, νfOH, ν1, νbOH) seem to be consistent with the observed bands A, B, and E, the predicted νbNH bands disagree with the IRPD spectrum (Fig. S8 and Table S6, ESI†). The same conclusion applies also to the next stable Py2+(p)H2O(II) isomer with a Py2+(p) core. Therefore, the population of Py2+(p)(H2O)2 isomers is concluded to be below the detection limit of 15%, considering the achieved signal-to-noise ratio of band F and the computed IR oscillator strengths of νbNH bands of Py2+(a/p)(H2O)2(I).
3.5. Py2+(H2O)3
The optimised geometries of Py2+(a)(H2O)3(I–III) based on the most abundant structure of Py2+(a)(H2O)2(I) are shown in Fig. 5d–f. All isomers are constructed through two NH⋯O H-bonds, in which one NH group is solvated by (H2O)2 and the other one by a single H2O. They differ only by the orientation of the linear (H2O)2, which is close to that in isomers II–IV of Py2+(a)(H2O)2, and their energies increase as E0 = 0.0, 0.4, and 3.2 kJ mol−1, respectively. Similar to Py2+(a)(H2O)2(II and III), Py2+(a)(H2O)3(II) with the free dangling (H2O)2 is more stable than Py2+(a)(H2O)3(I) at higher temperature (G0 = 0.0 vs. 3.5 kJ mol−1). In general, the intermolecular distances in Py2+(a)(H2O)3(I–III) are longer compared to those of Py2+(H2O)2(II–IV) due to the weaker H-bonds, resulting in slight blueshifts of νbNH/OH. The weaker H-bonds in n = 3 are due to increased delocalisation of the excess positive charges on the Py units compared to n = 2, resulting again from an increased perturbation of the CR by the third H2O ligand.The IRPD spectrum of Py2+(H2O)3 is compared in Fig. 6 to the linear IR spectra computed for Py2+(a)(H2O)3(I–III). The positions and widths of the transitions observed are listed in Tables S1 and Fig. S7 (ESI†) along with vibrational and isomer assignments. The isomers based on the Py2+(p) core are shown in Fig. S9 (ESI†) and their IR spectra are compared in Fig. S10 (ESI†). Compared to the n = 1–2 clusters, the IRPD spectrum of n = 3 is significantly more congested with broader bands (9–85 cm−1), thus complicating the assignment. It exhibits nine transitions at 3732 (A), 3642 (B), 3702 (C′), 3683 (C), 3293 (F), 3139 (F′), 3459 (D), 3388 (D′), and 3139 (G) cm−1. The trend in the IRPD spectra of mono- and dihydrated Py2+(a) in Fig. 1 provides hints about potential Py2+(H2O)3 structures. First, the absence of a sharp peak E (νfNH) indicates that both NH groups of Py2+ are hydrated. Second, bands C/C′ and D/D′, which are typical for νfOH and νbOH modes of single-donor H2O ligands are enhanced compared to those in the IRPD spectrum of Py2+(H2O)2. This observation suggests an increased abundance of structures with an NH⋯O H-bond of the NH groups to a (H2O)2 chain. Band F (νbNH) at 3293 cm−1 appears similarly to νbNH of n = 1–2, indicating the presence of a NH group which is H-bonded to a single H2O ligand. The intense and broad feature attributed to F1/G at 3139 cm−1 covers νCH and has a comparable intensity to band F assigned to the more strongly H-bonded νbNH of an NH group binding to (H2O)2. Band F of the n = 3 cluster is blueshifted compared to those of n = 1–2, indicating that the H-bond strength weakens with increasing number of H2O ligands around Py2+. This observation is typical for noncooperative effects of progressive interior ion solvation and indicates that the partial positive charge on the Py units of Py2+ decreases with increasing n, thereby weakening the charge-dipole interaction of the NH⋯O H-bonds. Overall, the IRPD spectrum of Py2+(H2O)3 is consistent with the computed Py2+(a)(H2O)3(I–III) isomers, although only I and II are required to explain all observed transitions, in particular in the νbOH range.
Because I and II are nearly isoenergetic, both isomers should be considered. Py2+(a)(H2O)3(I) has three intense bands (νbNH, νbOH) predicted below 3500 cm−1 and five weak bands (ν1, νfOH, ν3) above 3600 cm−1. The lowest νbNH band arising from the H-bond to (H2O)2 is strongly coupled with the aromatic νCH modes (local mode mixing). This coupled νbNH and the second νbNH at 3268 cm−1, which comes from the H-bond to H2O, are consistent with the observed bands at 3139 and 3293 cm−1 (F1 and F), respectively. The νbOH mode of the OH⋯O H-bond of (H2O)2 reproduces the observed band at 3459 cm−1 (D). The free OH stretches ν3, νfOH, and ν1 correspond to the observed bands A and B. However, the bands C and C′ at 3683 and 3702 cm−1, which are more intense and sharper than A and B and correspond to νfOH of a single-donor H2O, are not predicted well by isomer I. On the other hand, isomer II offers the same assignment as Py2+(a)(H2O)3(I), except that νbOH corresponds to the observed band D′. However, again bands C and C′ cannot be fully explained by II in terms of both their intensities and positions.
In an effort to assign bands C and C′, isomers based on Py2+(p) are considered because the most stable structure found for n = 3, Py2+(p)(H2O)3(I), is indeed more stable than Py2+(a)(H2O)3(I), D0 = 43.3 vs. 36.6 kJ mol−1 (Fig. S9, ESI†). In this structure, the third H2O expands the H-bonded solvent network of the four-membered Py2+(p)(H2O)2(I) ring at its surface, resulting in two ionic NH⋯O and two neutral OH⋯O H-bonds. Compared to Py2+(a)(H2O)2(I), all H-bonds become stronger because of cooperativity. Due to the same cooperative effect, Py2+(p)(H2O)3(I) is more stable than Py2+(a)(H2O)3(I) by ΔE0 = 7.7 kJ mol−1. However, due to the strain of its cyclic H-bond network, ΔG0 is still in favour of Py2+(a)(H2O)3(I) by 1.4 kJ mol−1. Its predicted IR spectrum differs slightly in the νOH range, exhibiting two νfOH and two νbOH at 3720/3706 and 3458/3363 cm−1, and ν3/1 = 3727/3641 cm−1, respectively. Comparing this predicted IR spectrum with the measured IRPD spectrum, peaks A and B may also be assigned to ν3 and ν1, C/C′ to νfOH, and D/D′ to νbOH, while F/F′ match with νbNH (Fig. S10 and Table S8, ESI†). A further local minimum, Py2+(p)(H2O)3(II), in which the third H2O is attached at another binding site in the (H2O)3 network is also predicted to be more stable than Py2+(a)(H2O)3(I) by ΔE0 = 5.0 kJ mol−1 (Fig. S9, ESI†). Its predicted IR spectrum is, however, not compatible with the IRPD spectrum in the νbNH range. Hence, the two most stable antiparallel Py2+(a)(H2O)3(I and II) isomers and the most stable parallel Py2+(p)(H2O)3(I) isomer can fully account for the measured IRPD spectrum. Due to the broad widths of the bands observed in the IRPD spectrum, a more definitive determination of their individual contributions is not feasible at the current spectral resolution. Unfortunately, the achieved Py2+(H2O)3Ar ion yield has been insufficient to record a cold spectrum with better resolution for the n = 3 cluster.
3.6. Impact of stepwise hydration on the CR
Inspection of Fig. 1 reveals that the free NH stretch band νfNH (band E) of the free Py units shows small but systematic shifts as a function of solvation. Our previous study demonstrates a linear correlation between νfNH of the free Py unit in Pyn+ with n = 0–2 and its partial charge qPy, νfNH(qPy)/cm−1 = −83.8 qPy/e + 3527.8 as indicated by the dashed line in Fig. 7.23 Using this correlation, νfNH measured for Py2+N2 has been successfully used to determine small changes in the charge distribution of Py2+(a) upon N2 complexation due to symmetry breaking and charge delocalisation.23 H2O solvation induces a stronger perturbation of Py2+(a) because of its large dipole moment,54,102 leading to a stronger H-bond and larger charge delocalisation. As a result, νfNH measured for Py2+H2O is slightly more blueshifted than νfNH of Py2+N2 (3492 vs. 3490 cm−1), corresponding to a lower partial charge on free Py (qPy = 0.427 vs. 0.451e). This experimentally determined charge, qPy = 0.427e, agrees reasonably well with the NBO charge computed for Py2+(a)H2O(I), qPy = 0.441e. For Py2+(H2O)2, νfNH observed at 3493 cm−1 is assigned to the minor Py2+(a)(H2O)2(III) isomer. The partial charge of its free Py unit estimated as qPy = 0.415e from the correlation is well reproduced by its NBO charge of 0.413e. Its NH⋯O H-bond is stronger than in Py2+(a)H2O(I) because the PA of (H2O)2 is higher than the PA of H2O (808 vs. 691 kJ mol−1).70,71,80 Because the IE of the Py(H2O)2 unit in Py2+(a)(H2O)2(III) is lower than the IE of PyH2O in Py2+(a)H2O(I), the partial charge on free Py is lowered in Py2+(a)(H2O)2(III) compared to Py2+(a)H2O(I). Interestingly, a comparison between the higher-energy isomers III and IV of Py2+(a)(H2O)2, which differ by the way the (H2O)2 dimer binds to Py2+(a) (linear or cyclic) reveals that subtle differences in the solvation network also have an impact on the charge of the free Py unit (qPy = 0.413 vs. 0.425e, Fig. S5, ESI†). The smaller charge transfer to (H2O)2 in IV compared to III (qH2O = 0.046 vs. 0.059e) may be an indication of the weaker H-bonds in the solvent network. Therefore, the charge distribution and CR in Py2+(a) can be affected by the H-bond strength even for similar hydration motifs. As can be seen from Fig. 7, the two available experimental νfNH data points for isomer I of n = 1 and isomer III of n = 2 are compatible with the initially derived linear qPy–νfNH dependence. | ||
| Fig. 7 (a) Observed νfNH values for Py, Py+, and Py2+ (open circles) and Py2+N2 and Py2+(H2O)1,2 (filled circles) as a function of the calculated NBO charge on the free Py unit (qPy). The dashed line is a linear fit obtained by using νfNH of Py, Py+, and Py2+. (b) Expanded view in the vicinity of Py2+ and its solvated clusters. | ||
To systematically analyse the impact of stepwise hydration on the CR, we consider the related Py2+(a)(H2O)n isomers for n = 1–3, in which a linear (H2O)n chain is attached to one Py unit via a linear NH⋯O ionic H-bond. These are isomers I (n = 1), III (n = 2), and linear (n = 3) shown in Fig. 3 and 5, respectively. As the PA of (H2O)n clusters increases with n (691 < 808 < 862 kJ mol−1 for n = 1–3)70,71,120,121 the NH⋯O H-bond becomes stronger and shorter (1.809 > 1.720 > 1.685 Å). The gradually increasing negative ρ* values with increasing n (−0.034, −0.043, −0.047 a.u.) from the NCI analysis (Fig. S11, ESI†) and E(2) donor–acceptor interaction energies (30.0 < 54.6 ≈ 50.5 kJ mol−1) support the increased H-bond strengths, which are well reproduced by the strongly redshifted νbNH available from the experiment (3228 > 3014 cm−1 for n = 1–2) and the calculation (3223 > 3068 > 2937 cm−1 for n = 1–3, Table S9, ESI†). The increased H-bond strengths of the NH⋯O and OH⋯O H-bonds as a function of n are also visible in the increased cooperativity calculated from the difference between the total interaction energy and the sum of the individual interactions (13% for n = 2 and 20% for n = 3, Table S10, ESI†). This result is in line with the increasing charge transfer to the solvent (0.033, 0.059, 0.056e). As a result of the stronger H-bond, the N–H bond of the free Py units contracts slightly (1.0057 > 1.0054 > 1.0053 Å), leading to increasing computed νfNH (3506 < 3510 < 3512 cm−1 for n = 1–3), which are in good agreement with available experimental values (3492 < 3493 for n = 1–2). As νfNH increases with n, the computed NBO charges on the free Py unit decrease accordingly (0.441 > 0.413 > 0.402e). These values agree well with those derived from the qPy–νfNH relation using the experimental νfNH values (0.427 > 0.415 > 0.403e). Interestingly, the total charge transfer to (H2O)3 of 0.056e is close to that of (H2O)2, 0.059e, due to the similar IEs of Py(H2O)2 and Py(H2O)3 computed as 7.46 and 7.47 eV, which relates to the similar IEs of (H2O)2 and (H2O)3 reported as 10.73 and 10.06 eV.122 Therefore, the critical hydration number for the partial charge transfer from Py2+(a) to the H2O solvent cluster may be determined as n = 2.
Next, we discuss the impact of the CR interaction on the H-bond motifs. Due to its C2h symmetry, the two N–H bonds in Py2+(a) have the same acidity and strength (rNH = 1.0061 Å). In Py2+(a)H2O(I), H2O forms an NH⋯O H-bond (RNH⋯O = 1.809 Å, D0 = 46.3 kJ mol−1) to one of the two NH groups and elongates it to rNH = 1.0219 Å. Symmetry reduction upon monohydration causes an unequal charge distribution on the two Py units, qPy = 0.526 and 0.441e. Due to the decreased positive partial charge on the free Py unit, its N–H bond contracts to rNH = 1.0057 Å. In Py2+(a)(H2O)2(I), the second H2O ligand prefers to solvate the remaining free NH group and restores C2h symmetry and consequently the maximal CR in Py2+(a) (qPy = 0.461e), rather than forming a (H2O)2 solvent network. This preference for interior ion solvation over the formation of the H-bonded network is observed because the additional strong charge-dipole interaction, although slightly noncooperative, is still more favourable than the significantly cooperative dipole–dipole interaction in the (H2O)2 chain. As a result of restored C2h symmetry, the two H-bonds in Py2+(a)(H2O)2(I) have equal strength with RNH⋯O = 1.826 Å, rNH = 1.0202 Å, and D0 = 44.2 kJ mol−1, although slightly weaker than the single one in monohydrated Py2+(a)H2O(I). These relative H-bond strengths are also obtained from the NCI analysis (ρ* = −0.034 > −0.033 a.u. for n = 1 and 2, Fig. S11, ESI†). The weaker H-bond in dihydrated Py2+(a)(H2O)2(I) is rationalised by the reduced charge on the H-bond donor Py units (0.461 vs. 0.526e, Fig. 3 and 5), resulting in weaker local charge-dipole interactions between Py and H2O.
As a next step, we analyse the impact of hydration on the CR of Py2+(a), Py2+(a)H2O(I), and Py2+(a)(H2O)2(I) by comparing the vertical excitation energies for the CR transition (ΔE) using the TD-DFT approach. First, the reliability of the considered dispersion-corrected DFT functionals for predicting the CR transition, namely B3LYP, CAM-B3LYP, B3PW91, and M06-2X with the aug-cc-pVTZ basis set is evaluated by comparing the TD-DFT transition energies of the cations of benzene dimer (Bz2+), benzene-naphthalene (BzNp+), and benzene-toluene (BzTol+) in their optimised ground state geometries to their experimentally reported values (Table S11, ESI†).32,44,45,51,53 The CAM-B3LYP-D3 and M06-2X functionals yield results closest to the experimental values. Therefore, the ΔE values, obtained for the optimised ground state geometries of Py2+(a), Py2+(a)H2O(I), and Py2+(a)(H2O)2(I) at the B3LYP-D3 level, are supported by those obtained with the CAM-B3LYP-D3 and M06-2X functionals (Fig. S12 and Table S12, ESI†). To this end, ΔE values increase slightly with n (1.7917 < 1.7982 < 1.8015 eV, Fig. S12, ESI†). The CR transition energy of the dimer cation is roughly given by the difference in the ionisation energies of the two units and its binding energy, ΔE ≈ ΔIE + 2D0.51 Within this approximation, D0 corresponds to the orbital coupling term (2D0 = 2V). For the Py2+(a) homodimer (i.e., ΔIE = 0), the coupling is maximised and given by D0 (107.4 kJ mol−1 or 1.11 eV), in line with roughly half of the computed excitation energy (ΔE = 1.79 eV). For monohydrated Py2+(a), the nonvanishing ΔIE value of 0.49 eV for PyH2O and Py (7.60 and 8.09 eV) reduces the orbital overlap and lowers D0 of Py2+H2O → Py+H2O + Py compared to that of Py2+(a) from 107.4 to 88.2 kJ mol−1 (1.11 < 0.91 eV). However, the contribution of ΔIE to ΔE roughly compensates for the reduced D0 value caused by poorer coupling arising from symmetry reduction, leading to an ΔE value comparable to that of Py2+(a). Because ΔIE = 0 for the symmetrically dihydrated Py2+(a)(H2O)2(I) cluster, the CR interaction becomes stronger again due to optimised overlap of the two HOMOs. The ΔE of Py2+(a)(H2O)2(I) and D0 for dissociating the core according to Py2+(a)(H2O)2 → Py+H2O + PyH2O are predicted to be slightly higher than that of Py2+(a) (1.8015 vs. 1.7917 eV and 114.4 vs. 107.4 kJ mol−1, 1.19 vs. 1.11 eV, respectively), suggesting that charge delocalisation into the H2O molecules (0.922 vs. 1.0e) might strengthen the CR interaction in the core.
Interestingly, the energy gap ΔE0(a–p) of Py2+(H2O)n with n = 0–3 decreases with increasing degree of hydration as 7.5 > 3.7 > −1.0 > −7.7 kJ mol−1 for the most stable isomers (Fig. 8). Hence, stepwise hydration is predicted to reverse the energetic order of the Py2+(a) and Py2+(p) cores between n = 2 and 3, suggesting a hydration-induced switch in the structural CR motif. A similar result has been reported for the formation of the most stable Py3+ trimer cation, in which Py solvates a Py2+(p) core whereas Py2+(a) is most stable for Py2+.68 For Py2+(H2O)n with n = 2, the Py2+(p) core isomer becomes more stable because of cooperative NH⋯O and OH⋯O H-bonds and increased H-bond strengths as compared to n = 1 while at the same time the Py2+(a) core isomer suffers from slight noncooperativity arising from internal ion solvation and slightly reduced H-bond strengths (Fig. S13, ESI†). Both effects are further enhanced for n = 3 (Table S10, ESI†), thereby changing the ΔE0(a–p) gap even more in favour of the Py2+(p) core. While the measured IRPD spectra are consistent with this computational prediction, better-resolved IRPD spectra are required to provide definitive experimental proof for this drastic impact of solvation on the structure motif of the aromatic CR.
 | ||
| Fig. 8 Relative energy differences (ΔE0 in kJ mol−1) of the most stable structures of Py2+(a)(H2O)n and Py2+(p)(H2O)n (n = 0–3) depending on the hydration number (n) calculated at the B3LYP-D3/aug-cc-pVTZ level. | ||
3.7. Impact of partial charge of Py on strength of H-bonds
The current study offers the unique opportunity to evaluate the strengths of the NH⋯O H-bonds between H2O and Py as a function of the Py charge in PyH2O, Py2+(a)H2O(I), and Py+H2O because in all three charge states (qPy = 0.0, 0.5, 1.0e) a similar linear H-bond motif is observed.54,55,80 Indeed, with increasing charge, the experimental νbNH values shift progressively to the red (3448 > 3228 > 2945 cm−1)54,55 due to the increasing strength of the NH⋯O H-bond. This trend is well supported by the computations, according to which the H-bonds become shorter and stronger with increasing charge (RNH⋯O = 1.991 > 1.809 > 1.704 Å, D0 = 17.6 < 46.3 < 65.1 kJ mol−1, ρ* = −0.023 > −0.034 > −0.045 a.u., Fig. S14, ESI†), mostly due to the additional charge-dipole forces. The H-bond in Py2+(a)H2O is quite a bit stronger than the average of PyH2O and Py+H2O (by 5.0 kJ mol−1) mainly due to the reorganisation of the charge resonance in Py2+(a) upon monohydration, causing the charge of the proton donor Py (0.526e) to be slightly higher than 0.5e.Next, we consider the preferred dihydration motifs of Py, Py+, and Py2+ to discuss the evolution of the H-bonded solvent network as a function of charge and the number of available NH proton donor groups. Neutral Py(H2O)2 has a cyclic (σ–π bridge) structure, in which a linear OH⋯O H-bonded (H2O)2 dimer binds to Py by NH⋯O and OH⋯π H-bonds.55 In contrast, Py+(H2O)2 has a linear chain structure with adjacent NH⋯O and OH⋯O H-bonds because of the strongly anisotropic charge-dipole interaction and the repulsive interaction between Py+ and the π-bonded OH hydrogen.80 On the other hand, Py2+(a) with its two NH groups offers two strongly acidic H-bond attractors for individual H2O ligands so that in the Py2+(a)(H2O)2(I) global minimum both positive partial charges are solvated, thereby restoring the CR. Nonetheless, the Py2+(a)(H2O)2(II and III) local minima with an NH⋯O H-bond of one NH group to a linear (H2O)2 are energetically competitive with Py2+(a)(H2O)2(I). Isomer III is more stable than II at higher temperatures due to entropy and exhibits a similar binding motif as Py+(H2O)2. Because of the CR interaction and the strongly reduced positive charge, the NH⋯O and OH⋯O H-bonds in Py2+(a)(H2O)2(III) are weaker than those in Py+(H2O)2 (RNH⋯O = 1.720 vs. 1.609 Å, ROH⋯O = 1.773 vs. 1.732 Å, D0 = 38.8 vs. 46.1 kJ mol−1 for loss of H2O, ρ*NH⋯O = −0.043 vs. −0.050 a.u., ρ*OH⋯O = −0.037 vs. −0.041 a.u., Fig. S14, ESI†) which is well supported by the measured νbOH values (3306 vs. 3388 cm−1 for qPy = 1.0 and 0.5e).80 The H-bond cooperativity is drastically reduced from 30% to 13% (Table S10, ESI†), which is nearly the same as that of the neutral cluster (12%),80 although the H-bonds are much stronger in Py2+(H2O)2(III) (Fig. S14, ESI†). Therefore, although the excess charge of 0.5e substantially increases the H-bond strength due to the additional charge-dipole interaction, it does not increase the degree of cooperativity in the H-bonded network.
In the following, we compare the mono- and dihydration motifs of neutral and cationic Py2(+) by considering Py2(H2O)n and Py2+(H2O)n with n = 1–2. Neutral Py2 has a T-shaped structure with an NH⋯π H-bond.61,63,66 In Py2H2O(I), H2O forms a cyclic structure with T-shaped Py2via NH⋯OH⋯π bonds (Fig. S15, ESI†).55,123 Upon ionisation, the cyclic Py2H2O structure opens toward linear Py2+(a)H2O(I) due to the repulsive interaction between Py2+ and the π-bonded H2O ligand arising from the excess positive charge.80 Interestingly, due to the stable cyclic structure with two neutral H-bonds, D0 of Py2H2O is almost comparable to that of Py2+H2O featuring a single ionic H-bond (38.3 vs. 46.3 kJ mol−1). All minima calculated for neutral Py2(H2O)2 have cyclic structures and differ depending on whether one or two H2O molecules are present in the cycle (Fig. S15, ESI†). The global minimum, Py2(H2O)2(I), has a similar structure as Py2H2O(I) with a (H2O)2 dimer replacing H2O. In Py2(H2O)2(II), higher in energy by only 3.3 kJ mol−1, both H2O molecules form individual NH⋯O and OH⋯π H-bonds to the two Py units. Upon π electron emission, Py2(H2O)2(I) likely converts to Py2+(a)(H2O)2(II and III), while Py2(H2O)2(II) probably generates Py2+(a)(H2O)2(I) due to the repulsive interactions between the positive excess charge and the π-bonded OH hydrogens, thus breaking the OH⋯π H-bonds. In the cationic clusters, Py2+(a)(H2O)2(I) becomes more stable because it forms a stronger CR than Py2+(a)(H2O)2(II and III). Hence, ionisation changes the order of the isomeric structures of dihydrated Py2.
Py2+(a) offers two spatially separated acidic functional NH groups. Their hydration motifs, interior ion solvation or formation of a single H-bonded solvent network, become competitive starting from two H2O molecules, as demonstrated for Py2+(a)(H2O)2(I) and Py2+(a)(H2O)2(II and III). Similar competing hydration motifs are observed for the previously studied microsolvation of the 5-hydroxyindole cation (5HI+), a bicyclic aromatic ion offering an NH group in the pyrrole ring and an OH group at the phenyl ring.83 5HI+(H2O)2(OH/NH), in which both acidic functional groups are solvated by a single H2O ligand, corresponding to interior ion solvation, is found as the dominant isomer, whereas 5HI+(H2O)2(OH/H2O or NH/H2O) isomers with the (H2O)2 dimer chain attached to either the NH or OH group provide only a minor contribution to the population. The high abundance of 5HI+(H2O)2(OH/NH) with slightly noncooperative H-bonds originates from two strongly acidic functional groups.83 Similarly, the presence of two acidic NH groups is mainly responsible for the high abundance of Py2+(a)(H2O)2(I) with interior ion solvation, in addition to its favourable CR interaction.
4. Concluding remarks
Herein, we investigate the effects of stepwise microhydration on the CR-stabilised Py2+ homodimer cation by IRPD spectroscopy of mass-selected bare and Ar-tagged Py2+(H2O)n clusters and complementary quantum chemical calculations at the B3LYP-D3/aug-cc-pVTZ level including NCI, NBO, and TD-DFT analysis. Systematic shifts observed for the νNH and νOH stretch bands provide detailed information about the structure and bonding of the observed cluster isomers and the preferred cluster growth, including charge distributions, cooperativity, and the strengths of the CR and H-bonds as a function of the degree and site of hydration. Significantly, the results provide for the first time information about the solvation effects on the fundamental aromatic CR interaction in isolated clusters. The salient results may be summarised as follows.(1) The preferred cluster growth of Py2+(a) begins with hydrating the two available acidic NH protons (n = 1–2) followed by the formation of a H-bonded solvent network (n ≥ 3). In general, interior ion hydration is slightly noncooperative, leading to slightly weaker NH⋯O H-bonds for n = 2 compared to n = 1. On the other hand, formation of the H-bonded solvent network via OH⋯O H-bonds is strongly cooperative due to nonadditive induction forces. To this end, for n = 2 the local minina with a (H2O)2 attached to a single NH group become energetically competitive with the most stable symmetrically hydrated global minimum, with an observed population ratio of around 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 in our plasma expansion. When considering the n = 1–3 isomers in which one of the Py units of Py2+(a) is solvated by a linear (H2O)n chain, the NH⋯O ionic H-bond to the solvent cluster becomes progressively stronger due to increasing proton affinity of (H2O)n, resulting in larger charge transfer to solvent and stronger νbNH redshifts, an effect that seems to converge around n = 2–3.
:2 in our plasma expansion. When considering the n = 1–3 isomers in which one of the Py units of Py2+(a) is solvated by a linear (H2O)n chain, the NH⋯O ionic H-bond to the solvent cluster becomes progressively stronger due to increasing proton affinity of (H2O)n, resulting in larger charge transfer to solvent and stronger νbNH redshifts, an effect that seems to converge around n = 2–3.
(2) The new experimental and computational data for the various Py2+(H2O)n clusters allow us to further confirm the roughly linear qPy–νfNH correlation between the free NH stretch frequency of the Py unit and its partial positive charge developed earlier from data of Py(+) and Py2+.23 The experimental qPy values obtained for Py2+(H2O)n with n = 1–3 from this qPy–νfNH correlation are in good agreement with the computed NBO charges, once again validating our approach of characterising the CR interaction in aromatic dimers utilising the IRPD technique in the ground electronic state (ψ+).
(3) Our characterisation of the Py2+(H2O)n clusters offers the unique opportunity to determine the effects of sequential hydration on the CR interaction as a function of isomeric structure and cluster size n. According to a rough approximation, two competing factors have to be considered, namely the difference in the ionisation energies (ΔIE) and the coupling constant (V) approximated by the binding energy D0. Monohydration of Py2+(a) reduces the symmetry of the cluster and thus the CR interaction for n = 1. Symmetric dihydration for n = 2 restores the symmetry and increases the CR again. However, when taking ΔIE into account, the splitting between ψ+ and ψ− (ΔE) giving rise to the CR transition is only weakly affected upon mono- and dihydration, as confirmed by TD-DFT calculations.
(4) Moreover, our experimental IRPD spectra, along with the computational results, provide the first evidence that stepwise hydration induces a change in the preferred CR core ion from the symmetric Py2+(a) structure (n ≤ 2) to the parallel Py2+(p) core (n ≥ 3) due to the strong cooperativity favouring the formation of single-sided H-bonded hydration networks. However, IR spectra at higher resolution are required for a definitive proof of this scenario. To this end, IR spectra for clusters with n > 3 may shed further light on this preliminary conclusion, while at the same time further expanding the correlation between the CR interaction and the H-bond motifs.
(5) Comparison of microhydration of Py, Py+, and Py2+ offers the valuable opportunity to quantitatively characterise the H-bond strength of the aromatic solute to the (dipolar) solvent network as a function of its positive partial charge. Due to the increasing charge-dipole interaction, the NH⋯O H-bond becomes progressively stronger the larger the charge on the Py unit.
As an outlook, we are currently expanding these studies on the CR interaction in several directions using the same combined experimental and computational approach, including (i) Py2+ dimers solvated by molecules with different proton affinities (e.g., CH3OH), (ii) larger Pyn+ clusters, and (iii) clusters of [PyA]+ heterodimers with aromatic molecules A with different IEs.
Data availability
The data supporting this article has been included as part of the ESI.† ESI† includes LID and CID mass spectra, additional isomeric structures and energies, measured and computed harmonic and anharmonic IR spectra of Py2(+)(H2O)n and Py2+(H2O)nAr, NCI analysis of Pyq(H2O)n, and frontier MOs and computed CR transition energies of Py2+(H2O)n.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by Deutsche Forschungsgemeinschaft (DFG, project DO 729/10) and the Japan Society for the Promotion of Science (JSPS) Core-to-Core Program (Grant JPJSCCA20210004, JPJSCCA20240002). YM is grateful for travel support from the Open Partnership Joint Research Project of JSPS (JPJSBP120229917). YM also appreciates the financial support from the Cooperative Research Program of “Network Joint Research Centre for Material and Devices” (No. 20221199). OD acknowledges support from the World Research Hub Initiative (WRHI) of the Institute of Science Tokyo. DA was also partly supported by the International Max Planck Research School for Elementary Processes in Physical Chemistry.Notes and references
- C. A. Hunter, K. R. Lawson, J. Perkins and C. J. Urch, J. Chem. Soc., Perkin Trans. 2, 2001, 651 RSC.
- S. E. Wheeler, Acc. Chem. Res., 2013, 46, 1029 CrossRef CAS PubMed.
- K. E. Riley and P. Hobza, Acc. Chem. Res., 2013, 46, 927 CrossRef CAS PubMed.
- P. Hobza, H. L. Selzle and E. W. Schlag, Chem. Rev., 1994, 94, 1767 CrossRef CAS.
- K. Müller-Dethlefs and P. Hobza, Chem. Rev., 2000, 100, 143 CrossRef PubMed.
- A. S. Mahadevi and G. N. Sastry, Chem. Rev., 2016, 116, 2775 CrossRef CAS PubMed.
- O. Dopfer and M. Fujii, Chem. Rev., 2016, 116, 5432 CrossRef CAS PubMed.
- M. Fujii and O. Dopfer, Int. Rev. Phys. Chem., 2012, 31, 131 Search PubMed.
- O. Dopfer, Z. Phys. Chem., 2005, 219, 125 CrossRef CAS.
- L. M. Salonen, M. Ellermann and F. Diederich, Angew. Chem., Int. Ed., 2011, 50, 4808 CrossRef CAS PubMed.
- E. A. Meyer, R. K. Castellano and F. Diederich, Angew. Chem., Int. Ed., 2003, 42, 1210 CrossRef CAS PubMed.
- E. T. Kool, Annu. Rev. Biophys. Biomol. Struct., 2001, 30, 1 CrossRef CAS PubMed.
- R. Bu, Y. Xiong and C. Zhang, Cryst. Growth Des., 2020, 20, 2824 CrossRef CAS.
- N. Gospodinova and E. Tomšík, Prog. Polym. Sci., 2015, 43, 33 CrossRef CAS.
- K. Binnemans, Chem. Rev., 2005, 105, 4148 CrossRef CAS PubMed.
- S. Suzuki, P. G. Green, R. E. Bumgarner, S. Dasgupta, W. A. Goddard and G. A. Blake, Science, 1992, 257, 942 CrossRef CAS PubMed.
- D. A. Dougherty, Science, 1996, 271, 163 CrossRef CAS PubMed.
- J. C. Ma and D. A. Dougherty, Chem. Rev., 1997, 97, 1303 CrossRef CAS PubMed.
- K. S. Kim, P. Tarakeshwar and J. Y. Lee, Chem. Rev., 2000, 100, 4145 CrossRef CAS PubMed.
- R. E. Dawson, A. Hennig, D. P. Weimann, D. Emery, V. Ravikumar, J. Montenegro, T. Takeuchi, S. Gabutti, M. Mayor, J. Mareda, C. A. Schalley and S. Matile, Nat. Chem., 2010, 2, 533 CrossRef CAS PubMed.
- B. Badger and B. Brocklehurst, Nature, 1968, 219, 263 CrossRef CAS.
- A. K. Chandra, K. Bhanuprakash, V. J. Bhasu and D. Srikanthan, Mol. Phys., 1984, 52, 733 CrossRef CAS.
- K. Chatterjee, Y. Matsumoto and O. Dopfer, Angew. Chem., Int. Ed., 2019, 58, 3351 CrossRef CAS PubMed.
- V. Coropceanu, J. Cornil, D. A. da Silva Filho, Y. Olivier, R. Silbey and J.-L. Brédas, Chem. Rev., 2007, 107, 926 CrossRef CAS PubMed.
- A. J. Heeger, J. Phys. Chem. B, 2001, 105, 8475 CrossRef CAS.
- J. L. Brédas, J. P. Calbert, D. A. da Silva Filho and J. Cornil, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5804 CrossRef PubMed.
- M. Kitamura and Y. Arakawa, J. Phys.: Condens. Matter, 2008, 20, 184011 CrossRef.
- B. Giese, Acc. Chem. Res., 2000, 33, 631 CrossRef CAS PubMed.
- B. Giese, Annu. Rev. Biochem., 2002, 71, 51 CrossRef CAS PubMed.
- B. Giese, J. Amaudrut, A. K. Köhler, M. Spormann and S. Wessely, Nature, 2001, 412, 318 CrossRef CAS PubMed.
- S. Giannini, A. Carof, M. Ellis, H. Yang, O. G. Ziogos, S. Ghosh and J. Blumberger, Nat. Commun., 2019, 10, 3843 CrossRef PubMed.
- M. Matsumoto, Y. Inokuchi, K. Ohashi and N. Nishi, J. Phys. Chem. A, 1997, 101, 4574 CrossRef CAS.
- Y. Tsujii, A. Tsuchida, S. Ito and M. Yamamoto, Macromolecules, 1991, 24, 4061 CrossRef CAS.
- I. C. Lewis and L. S. Singer, J. Chem. Phys., 1965, 43, 2712 CrossRef CAS.
- H. van Willigen, E. de Boer, J. T. Cooper and W. F. Forbes, J. Chem. Phys., 1968, 49, 1190 CrossRef CAS.
- O. W. Howarth and G. K. Fraenkel, J. Am. Chem. Soc., 1966, 88, 4514 CrossRef CAS.
- B. Badger and B. Brocklehurst, Trans. Faraday Soc., 1969, 65, 2582 RSC.
- B. Badger, B. Brocklehurst and R. D. Russell, Chem. Phys. Lett., 1967, 1, 122 CrossRef CAS.
- J. K. Kochi, R. Rathore and P. L. Magueres, J. Org. Chem., 2000, 65, 6826 CrossRef CAS PubMed.
- B. Badger and B. Brocklehurst, Trans. Faraday Soc., 1970, 66, 2939 RSC.
- M. S. El-Shall and M. Meot-Ner, J. Phys. Chem., 1987, 91, 1088 CrossRef CAS.
- M. Meot-Ner, P. Hamlet, E. P. Hunter and F. H. Field, J. Am. Chem. Soc., 1978, 100, 5466 CrossRef CAS.
- M. Rusyniak, Y. Ibrahim, E. Alsharaeh, M. Meot-Ner (Mautner) and M. S. El-Shall, J. Phys. Chem. A, 2003, 107, 7656 CrossRef CAS.
- K. Ohashi, Y. Nakai, T. Shibata and N. Nishi, Laser Chem., 1994, 14, 3 CrossRef CAS.
- K. Ohashi and N. Nishi, J. Phys. Chem., 1992, 96, 2931 CrossRef CAS.
- K. Ohashi, Y. Inokuchi and N. Nishi, Chem. Phys. Lett., 1996, 263, 167 CrossRef CAS.
- Y. Inokuchi and N. Nishi, J. Chem. Phys., 2001, 114, 7059 CrossRef CAS.
- Y. Inokuchi, K. Ohashi, M. Matsumoto and N. Nishi, J. Phys. Chem., 1995, 99, 3416 CrossRef CAS.
- J. Bernard, A. Al-Mogeeth, A.-R. Allouche, L. Chen, G. Montagne and S. Martin, J. Chem. Phys., 2019, 150, 54303 CrossRef CAS PubMed.
- J. Bernard, S. Martin, A. Al-Mogeeth, C. Joblin, M. Ji, H. Zettergren, H. Cederquist, M. H. Stockett, S. Indrajith, L. Dontot, F. Spiegelman, D. Toublanc and M. Rapacioli, Phys. Chem. Chem. Phys., 2024, 26, 18571 RSC.
- K. Ohashi, Y. Nakane, Y. Inokuchi, Y. Nakai and N. Nishi, Chem. Phys., 1998, 239, 429 CrossRef CAS.
- J. Bernard, A. Al-Mogeeth, S. Martin, G. Montagne, C. Joblin, L. Dontot, F. Spiegelman and M. Rapacioli, Phys. Chem. Chem. Phys., 2021, 23, 6017 RSC.
- Y. Inokuchi, K. Ohashi, H. Sekiya and N. Nishi, J. Chem. Phys., 2002, 117, 10648 CrossRef CAS.
- M. Schütz, Y. Matsumoto, A. Bouchet, M. Öztürk and O. Dopfer, Phys. Chem. Chem. Phys., 2017, 19, 3970 RSC.
- Y. Matsumoto and K. Honma, J. Chem. Phys., 2009, 130, 54311 CrossRef PubMed.
- S. Chakraborty, A. Patzer, A. Lagutschenkov, J. Langer and O. Dopfer, Int. J. Mass Spectrom., 2010, 297, 85 CrossRef CAS.
- A. Fujii, E. Fujimaki, T. Ebata and N. Mikami, J. Chem. Phys., 2000, 112, 6275 CrossRef CAS.
- K. Chatterjee and O. Dopfer, Phys. Chem. Chem. Phys., 2017, 19, 32262 RSC.
- P. A. Pieniazek, S. E. Bradforth and A. I. Krylov, J. Chem. Phys., 2008, 129, 74104 CrossRef PubMed.
- R. Lindner, K. Müller-Dethlefs, E. Wedum, K. Haber and E. R. Grant, Science, 1996, 271, 1698 CrossRef CAS.
- H. Park and S. Lee, Chem. Phys. Lett., 1999, 301, 487 CrossRef CAS.
- V. Stefov, L. Pejov and B. Šoptrajanov, J. Mol. Struct., 2003, 649, 231 CrossRef CAS.
- Y. Matsumoto and K. Honma, J. Chem. Phys., 2007, 127, 184310 CrossRef PubMed.
- G. Columberg and A. Bauder, J. Chem. Phys., 1997, 106, 504 CrossRef CAS.
- I. Dauster, C. A. Rice, P. Zielke and M. A. Suhm, Phys. Chem. Chem. Phys., 2008, 10, 2827 RSC.
- V. Profant, V. Poterya, M. Fárník, P. Slavícek and U. Buck, J. Phys. Chem. A, 2007, 111, 12477 CrossRef CAS PubMed.
- D. Arildii, Y. Matsumoto and O. Dopfer, J. Phys. Chem. A, 2024, 128, 3993 CrossRef CAS PubMed.
- C. Wei, X. Chen, Y. Wang, Y. Li, J. Gao, M. Xie and Y. Hu, Phys. Chem. Chem. Phys., 2023, 25, 11368 RSC.
- X. Chen, C. Wei, M. Xie and Y. Hu, J. Phys. Chem. A, 2023, 127, 8272 CrossRef CAS PubMed.
- E. P. L. Hunter and S. G. Lias, J. Phys. Chem. Ref. Data, 1998, 27, 413 CrossRef CAS.
- D. J. Goebbert and P. G. Wentold, Eur. J. Mass Spectrom., 2004, 10, 837 CrossRef CAS PubMed.
- K. Hattori, D. Wang and A. Fujii, Phys. Chem. Chem. Phys., 2019, 21, 16064 RSC.
- M. Xie, H.-R. Tsai, A. Fujii and Y.-P. Lee, Phys. Chem. Chem. Phys., 2019, 21, 16055 RSC.
- T. Kato and A. Fujii, J. Phys. Chem. A, 2023, 127, 742 CrossRef CAS PubMed.
- M. J. Tubergen, A. M. Andrews and R. L. Kuczkowski, J. Phys. Chem. Lett., 1993, 97, 7451 CAS.
- J. E. Del Bene and I. Cohen, J. Am. Chem. Soc., 1978, 100, 5285 CrossRef CAS.
- P. I. Nagy, G. Durant and D. A. Smith, J. Am. Chem. Soc., 1993, 115, 2912 CrossRef CAS.
- A. Kumar, M. Kołaski and K. S. Kim, J. Chem. Phys., 2008, 128, 34304 CrossRef PubMed.
- A. L. Sobolewski and W. Domcke, Chem. Phys. Lett., 2000, 321, 479 CrossRef CAS.
- D. Arildii, Y. Matsumoto and O. Dopfer, J. Phys. Chem. A, 2023, 127, 2523 CrossRef CAS PubMed.
- J. Klyne, M. Schmies, M. Fujii and O. Dopfer, J. Phys. Chem. B, 2015, 119, 1388 CrossRef CAS PubMed.
- J. Klyne, M. Schmies, M. Miyazaki, M. Fujii and O. Dopfer, Phys. Chem. Chem. Phys., 2018, 20, 3148 RSC.
- J. Klyne, M. Miyazaki, M. Fujii and O. Dopfer, Phys. Chem. Chem. Phys., 2018, 20, 3092 RSC.
- O. Dopfer, Int. Rev. Phys. Chem., 2003, 22, 437 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, GAUSSIAN 16 (Revision C.01), Gaussian Inc., Wallingford, CT, 2016.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372 CrossRef CAS.
- R. A. Kendall, T. H. Dunning and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796 CrossRef CAS.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- A. G. Császár, G. Czakó, T. Furtenbacher, J. Tennyson, V. Szalay, S. V. Shirin, N. F. Zobov and O. L. Polyansky, J. Chem. Phys., 2005, 122, 214305 CrossRef PubMed.
- K. Chatterjee and O. Dopfer, Chem. Sci., 2018, 9, 2301 RSC.
- E. D. Glendening, C. R. Landis and F. Weinhold, J. Comput. Chem., 2013, 34, 1429 CrossRef CAS PubMed.
- J. P. Perdew, M. Ernzerhof and K. Burke, J. Chem. Phys., 1996, 105, 9982 CrossRef CAS.
- J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244 CrossRef PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 Search PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498 CrossRef CAS PubMed.
- J. Contreras-García, W. Yang and E. R. Johnson, J. Phys. Chem. A, 2011, 115, 12983 CrossRef PubMed.
- J. Contreras-García, E. R. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625 CrossRef PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- J. Klyne, M. Schmies and O. Dopfer, J. Phys. Chem. B, 2014, 118, 3005 CrossRef CAS PubMed.
- C.-K. Lin, R. Shishido, Q.-R. Huang, A. Fujii and J.-L. Kuo, Phys. Chem. Chem. Phys., 2020, 22, 22035 RSC.
- T. R. Dyke, K. M. Mack and J. S. Muenter, J. Chem. Phys., 1977, 66, 498 CrossRef CAS.
- F. Huisken, M. Kaloudis and A. Kulcke, J. Chem. Phys., 1996, 104, 17 CrossRef CAS.
- Z. S. Huang and R. E. Miller, J. Chem. Phys., 1989, 91, 6613 CrossRef CAS.
- K. Kuyanov-Prozument, M. Y. Choi and A. F. Vilesov, J. Chem. Phys., 2010, 132, 14304 CrossRef PubMed.
- B. E. Rocher-Casterline, L. C. Ch'ng, A. K. Mollner and H. Reisler, J. Chem. Phys., 2011, 134, 211101 CrossRef PubMed.
- P. J. Derrick, L. Åsbrink, O. Edqvist and E. Lindholm, Spectrochim. Acta, Part A, 1971, 27, 2525 CrossRef CAS.
- A. J. van den Brom, M. Kapelios, T. N. Kitsopoulos, N. H. Nahler, B. Cronin and M. N. R. Ashfold, Phys. Chem. Chem. Phys., 2005, 7, 892 RSC.
- S. Tomoda, Y. Achiba and K. Kimura, Chem. Phys. Lett., 1982, 87, 197 CrossRef CAS.
- C. Y. Ng, D. J. Trevor, P. W. Tiedemann, S. T. Ceyer, P. L. Kronebusch, B. H. Mahan and Y. T. Lee, J. Chem. Phys., 1977, 67, 4235 CrossRef CAS.
- L. Karlsson, L. Mattsson, R. Jadrny, R. G. Albridge, S. Pinchas, T. Bergmark and K. Siegbahn, J. Chem. Phys., 1975, 62, 4745 CrossRef CAS.
- K. Sakota, Y. Kouno, S. Harada, M. Miyazaki, M. Fujii and H. Sekiya, J. Chem. Phys., 2012, 137, 224311 CrossRef PubMed.
- Q.-R. Huang, R. Shishido, C.-K. Lin, C.-W. Tsai, J. A. Tan, A. Fujii and J.-L. Kuo, Angew. Chem., Int. Ed., 2021, 60, 1936 CrossRef CAS PubMed.
- T. Seki, K.-Y. Chiang, C.-C. Yu, X. Yu, M. Okuno, J. Hunger, Y. Nagata and M. Bonn, J. Phys. Chem. Lett., 2020, 11, 8459 CrossRef CAS PubMed.
- A. A. Kananenka and J. L. Skinner, J. Chem. Phys., 2018, 148, 244107 CrossRef PubMed.
- K. Chatterjee and O. Dopfer, J. Phys. Chem. A, 2019, 123, 7637 CrossRef CAS PubMed.
- B. M. Auer and J. L. Skinner, J. Chem. Phys., 2008, 128, 224511 CrossRef CAS PubMed.
- R. Knochenmuss and S. Leutwyler, J. Chem. Phys., 1989, 91, 1268 CrossRef CAS.
- A. Courty, M. Mons, J. Le Calvé, F. Piuzzi and I. Dimicoli, J. Phys. Chem. A, 1997, 101, 1445 CrossRef CAS.
- R. N. Barnett and U. Landman, J. Phys. Chem. A, 1997, 101, 164 CrossRef CAS.
- S. Sarkar, N. Ramanathan and K. Sundararajan, J. Phys. Chem. A, 2018, 122, 2445 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cp00067j |
| This journal is © the Owner Societies 2025 |