
Evaluating and interpreting the exchange correlation contributions to the total static and interelectron interaction force densities, with a force-field perspective on the exchange charge density†
 a
and
Robert R.
Fayzullin
*a
a
and
Robert R.
Fayzullin
*a
Abstract
A crucial aspect of the conservative exchange force Fx(r) is the possible existence of nonnuclear basins in the corresponding vector field, which encompass internuclear binding regions for various covalent bonds. The intricate behavior of Fx(r) can be evaluated through its component contributions to the total static force density 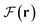 and the interelectron interaction force density Fee(r). The sought-after contributions are expressed as the scalar projection of the partial force (SPPF) onto the corresponding cumulative force. The resulting scalar component distribution permits the differentiation of the regions of physical space, exhibiting directional divergence or convergence between the two considered force fields. The scalar projections of Fx(r) onto
and the interelectron interaction force density Fee(r). The sought-after contributions are expressed as the scalar projection of the partial force (SPPF) onto the corresponding cumulative force. The resulting scalar component distribution permits the differentiation of the regions of physical space, exhibiting directional divergence or convergence between the two considered force fields. The scalar projections of Fx(r) onto 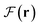 and Fee(r) enable the identification of shared electrons and lone electron pairs. Concurrently, the SPPF discloses the counteracting mechanism for the charge-transfer-induced quantum chemical response, which is implicated in a negative contribution of Fx(r) to
and Fee(r) enable the identification of shared electrons and lone electron pairs. Concurrently, the SPPF discloses the counteracting mechanism for the charge-transfer-induced quantum chemical response, which is implicated in a negative contribution of Fx(r) to 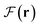 for the interatomic transferred density. Furthermore, Fx(r) typically positively contributes to Fee(r) within a part of each binding region, thereby facilitating the retention of electron “glue” between nuclei. Its negative contribution outside the binding regions highlights the role of Fx(r) in mitigating interelectron repulsion. These findings demonstrate the mechanical role of the exchange correlation in chemical bonding and the interatomic electron transfer in many-electron multinuclear systems. Additionally, the exchange charge density ∇·Fx(r)/(4π), which signifies the effective electron density distribution that arises from the influence of Fx(r), has been shown to accurately reproduce the details of subatomic structure, with negative values indicating electron cloud thickening. The underlying procedures are readily implementable and offer reliable chemical structure descriptors.
for the interatomic transferred density. Furthermore, Fx(r) typically positively contributes to Fee(r) within a part of each binding region, thereby facilitating the retention of electron “glue” between nuclei. Its negative contribution outside the binding regions highlights the role of Fx(r) in mitigating interelectron repulsion. These findings demonstrate the mechanical role of the exchange correlation in chemical bonding and the interatomic electron transfer in many-electron multinuclear systems. Additionally, the exchange charge density ∇·Fx(r)/(4π), which signifies the effective electron density distribution that arises from the influence of Fx(r), has been shown to accurately reproduce the details of subatomic structure, with negative values indicating electron cloud thickening. The underlying procedures are readily implementable and offer reliable chemical structure descriptors.

 Please wait while we load your content...
Please wait while we load your content...

