Insights into triazole-based energetic material design from decomposition pathways of triazole derivatives†
Received
26th December 2024
, Accepted 31st January 2025
First published on 31st January 2025
Abstract
Nitrogen-rich heterocycles are of great interest for the design of high-energy materials (HEMs) because they offer high density, positive heat of formation, superior detonation properties, and high thermal stability. Among the different types of nitrogen-rich heterocyclic azoles, 1,2,4-triazole provides a remarkable framework for the development of green energetic materials. The presence of functional groups, such as nitro, amino, and nitramino groups, affects the stability, thermal decomposition behavior, and energetic properties of HEMs. In the present study, we chose amino- and nitramino substituted 1,2,4-triazole and triazole containing both amino and carboxymethyl groups to compare their decomposition mechanisms. The decomposition pathways of 3-amino-1,2,4-triazole (1), 2,4-dihydro-3H-1,2,4-triazol-3-ylidene-nitramide (2), and 5-amino-1,2,4-triazol-3-yl-acetic acid (3) were explored using thermal experiments and mass spectrometry. Kinetic parameters were evaluated using a nonlinear integral method, and decomposition pathways were elucidated based on mass fragmentation data obtained from mass spectrometry and tandem mass spectrometry. Furthermore, near-real-time identification of decomposition products that evolved in the form of gases was performed using the TG-FTIR technique. Based on kinetic analysis, mass fragmentation data, and TG-FTIR analysis, the possible degradation pathways of the HEMs following the introduction of different substituents were identified.
Introduction
The emergence of nitrogen-rich heterocycles has significantly advanced the field of high-energy materials (HEMs) due to their high nitrogen content, density, heat of formation, and compatibility with most explosophoric groups. Triazole (C2H3N3) belongs to the class of heterocyclic azoles, that exists in two isomeric forms, 1,2,3-triazole, and 1,2,4-triazole, and has a wide range of applications in pharmaceuticals, herbicides, and energetic materials.1 Triazoles have attracted significant attention in the design and synthesis of eco-friendly energetic materials because of their high nitrogen and energy content, high density, thermal stability, and smokeless combustion characteristics. The physicochemical, energetic, and safety parameters of HEMs can be modified by incorporating various explosophores such as nitro, nitramino, azido, etc., or groups that stabilize molecules such as amino, trifluoromethyl, carboxyl, etc.2 In addition, the presence of N–N single and double bonds contributes to the high heat of formation of the compounds.3 During an explosion, low-energy bonds (such as N–N, N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N, N–C, etc.) within the molecule rupture and reorganize, resulting in the formation of a significant amount of N
N, N–C, etc.) within the molecule rupture and reorganize, resulting in the formation of a significant amount of N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N, and the subsequent release of a substantial amount of energy.4 Among the different isomers of triazoles, 1,2,4-triazoles exhibit an ideal balance between thermal stability and high heat of formation, which are essential for their potential use as substrates for the design of HEMs. Hence, in recent years, 1,2,4-triazole has emerged as an excellent backbone for the development of green energetic materials.5 Examples of HEMs designed and synthesized based on the triazole ring include 5-amino-3-nitro-1,2,4-triazole (A),6 3-azido-N-nitro-1H-1,2,4-triazol-5-amine (B),7 5-nitro-3-trinitromethyl-1H-1,2,4-triazole (C),8 5-(dinitromethyl)-3-(trinitromethyl)-1,2,4-triazole (D),9 1,3-bis(trinitromethyl)-1H-1,2,4-triazole (E),10 3-trinitromethyl-5-nitramino-1H-1,2,4-triazole (F),11 and N-(3-nitro-1-(trinitromethyl)-1H-1,2,4-triazol-5-yl) nitramide (G)12 (Fig. 1). The incorporation of amino groups improves the thermal stability of energetic materials by enhancing hydrogen bonding.2 In the nitramino moiety, the presence of N–N bonds and nitro groups enhances the density, heat of formation, and energy of the system, making the nitramino group a highly promising explosophoric group. In addition, nitramino forms nitrimino via hydrogen transfer, leading to the formation of intramolecular hydrogen bonds. This, in turn, creates a larger conjugated system, thereby improving the thermal stability.13 Therefore, the thermal and kinetic stability of HEMs are largely influenced by the substituent group, which prompted us to investigate the change in decomposition kinetics and the mechanism of amino- and nitramino-substituted 1,2,4-triazole derivatives.
N, and the subsequent release of a substantial amount of energy.4 Among the different isomers of triazoles, 1,2,4-triazoles exhibit an ideal balance between thermal stability and high heat of formation, which are essential for their potential use as substrates for the design of HEMs. Hence, in recent years, 1,2,4-triazole has emerged as an excellent backbone for the development of green energetic materials.5 Examples of HEMs designed and synthesized based on the triazole ring include 5-amino-3-nitro-1,2,4-triazole (A),6 3-azido-N-nitro-1H-1,2,4-triazol-5-amine (B),7 5-nitro-3-trinitromethyl-1H-1,2,4-triazole (C),8 5-(dinitromethyl)-3-(trinitromethyl)-1,2,4-triazole (D),9 1,3-bis(trinitromethyl)-1H-1,2,4-triazole (E),10 3-trinitromethyl-5-nitramino-1H-1,2,4-triazole (F),11 and N-(3-nitro-1-(trinitromethyl)-1H-1,2,4-triazol-5-yl) nitramide (G)12 (Fig. 1). The incorporation of amino groups improves the thermal stability of energetic materials by enhancing hydrogen bonding.2 In the nitramino moiety, the presence of N–N bonds and nitro groups enhances the density, heat of formation, and energy of the system, making the nitramino group a highly promising explosophoric group. In addition, nitramino forms nitrimino via hydrogen transfer, leading to the formation of intramolecular hydrogen bonds. This, in turn, creates a larger conjugated system, thereby improving the thermal stability.13 Therefore, the thermal and kinetic stability of HEMs are largely influenced by the substituent group, which prompted us to investigate the change in decomposition kinetics and the mechanism of amino- and nitramino-substituted 1,2,4-triazole derivatives.
 |
| | Fig. 1 Molecular structures of some energetic materials featuring triazole ring. | |
In a comparative study, the influence of electron-donating substituents (amino and methyl) on the decomposition temperature of 1,2,4-triazole was determined using sealed cell-differential scanning calorimetry (SC-DSC) and molecular orbital calculations. The decomposition temperature was determined by analyzing the energy changes in the thermal decomposition pathway model of intramolecular proton transfer and bond cleavage.14 A study on the thermal behavior of 1,2,4-triazole-copper complexes with different substituents showed that incorporating substituents (amino, nitro, and chloro) and coordinating with copper improves the thermal stability of 1,2,4-triazole. The study also identified the various components of the decomposed gases at 1000 °C using Flash pyrolysis/FTIR (Fourier transform infrared spectroscopy).15 The thermal stability analysis of 3-amino-1,2,4-triazole was performed using simultaneous TG-DTA (thermogravimetry-differential thermal analysis) coupled with Fourier transform infrared spectrometer. 3-amino-1,2,4-triazole undergoes a two-stage decomposition, and the products after the first stage are identified using mass spectrometry. The results suggest that a mixture of compounds is produced, with the main component having a molecular weight of 126, together with 3-amino-1,2,4-triazole and the analysis of evolved gases suggests the liberation of N2, HCN, and NH3.16 A comparative molecular modelling study on the thermal decomposition of C and N-nitramino-1,2,4-triazole showed that the decomposition is favored at the NNO2 fragment rather than the triazole ring.17 A theoretical investigation into the effect of N-oxide groups on the density, formation enthalpy, detonation performance, and stability of azobistriazole compounds revealed that the presence of N-oxide groups enhances the density and the overall energetic performance of these compounds.18 Recently, our group explored the effect of incorporating an alkylidene bridge between the nitrogen heterocycles on the decomposition mechanism and pathway of different bridged tetrazoles, finding that the addition of various bridges between the two heterocyclic rings improved the thermal stability of compounds.19
In the present study, we investigated the effect of substituents on the decomposition mechanism of 1,2,4-triazole. Three different triazoles, namely 3-amino-1,2,4-triazole (compound 1), 2,4-dihydro-3H-1,2,4-triazol-3-ylidene-nitramide (compound 2), and 5-amino-1,2,4-triazol-3-yl-acetic acid (compound 3) (Fig. 2) have been synthesized and their decomposition kinetics have been analyzed using Vyazovkin's nonlinear integral isoconversional (model-free) method.20
 |
| | Fig. 2 Molecular structures of compounds 1, 2, and 3. | |
This method allows the calculation of the apparent activation energy and determination of the kinetics of the multistep decomposition processes. Furthermore, the individual fragmentation pathways of the compounds were identified using Tandem mass spectrometry (MS/MS) and gas chromatography-mass spectrometry (GC-MS). Hyphenated techniques such as thermogravimetry-Fourier transform infrared spectroscopy (TG-FTIR) and thermogravimetry-mass spectrometry (TG-MS) analysis have recently emerged as the major platforms for analyzing energetic materials as they provide better insights into the thermal degradation process.21 Hence, a TG-FTIR-evolved gas analysis system was employed to identify the gases that evolved during the thermal decomposition of the compounds. Our previous study on hexanitrohexaazaisowurtzitane (CL-20) revealed that the degradation pathway proceeds via a strained fragment elimination route rather than conventional NO2 or HONO elimination. TG-FTIR analysis of CL-20 confirmed the presence of NO2, N2O, NO/CO, H2O, and HCN as the gaseous decomposition products.22
Experimental
Materials and methods
3-Amino-1,2,4-triazole (1) was purchased from Sigma-Aldrich. 2,4-Dihydro-3H-1,2,4-triazol-3-ylidene-nitramide (2) was synthesized from 3-amino-1,2,4-triazole using fuming nitric acid and concentrated sulphuric acid.23 5-Amino-1,2,4-triazole-3-yl-acetic acid (3) was synthesized from malonic acid and aminoguanidine bicarbonate using water as the solvent.24 All the compounds were characterized using multinuclear NMR (nuclear magnetic resonance) techniques (1H and 13C) and high-resolution mass spectrometry (HRMS) (Fig. S1–S9, ESI†).
Thermal analysis
Thermogravimetric (TG) analysis was performed using a PerkinElmer Simultaneous Thermal Analyzer (STA 6000). In all experiments, approximately 0.6–0.7 mg of the sample was loaded in an open 100 μL alumina sample pan and heated under nitrogen flow maintained at 20 mL min−1. Non-isothermal TG runs were performed at heating rates (β) of 2, 4, 6, and 8 °C min−1. All samples were stable below 100 °C, and thermal data were collected from 100 to 250 °C at different heating rates. DSC analysis was carried out on a TA Instruments Q20 Differential Scanning Calorimeter, and nitrogen flow was maintained at 50 mL min−1 by loading about 0.6–0.7 mg of sample in a sealed aluminium pan and heating at 5 °C min−1.
Evolved gas analysis using simultaneous TG-FTIR
TG-FTIR experiments were performed using a Netzsch 209 F1 Libra TGA instrument coupled to a TG-FTIR cell placed in a Bruker Vertex 80 FTIR Spectrometer.25 The TGA experiments were conducted at a heating rate of 10 °C min−1. The transfer line and IR cell temperatures were maintained at 210 and 190 °C, respectively. IR data were collected at a spectral resolution of 4 cm−1 over a wavenumber range of 650–3600 cm−1.
HRMS analysis
All samples were analyzed using a Waters Synapt XS high-resolution mass spectrometer. The samples were dissolved in LC–MS grade acetonitrile/water to obtain a concentration of the order of 100 ppb. Data acquisition was performed using positive and negative electrospray ionization (ESI) modes. Leucine enkephalin (200 pg μL−1) was used as the lock mass for mass correction. Further details of HRMS data acquisition are provided in the ESI.†
Gas chromatography-mass spectrometry (GC-MS) analysis
GC-MS was performed using a Pegasus BT 4D GCxGC time-of-flight mass spectrometer (Leco Corporation, USA). The ionization temperature was set to 250 °C. The mass spectrometer was operated in the electron impact ionization mode at a voltage of 70 eV. The mass scan range was 30–300 Da and the flow rate of the helium carrier gas was 1 mL min−1. DB-5 column (30 m × 0.25 mm ID, 0.25 μm film thickness) was used and the column was held at 50 °C for 0.2 min and then increased from 50 °C to 270 °C at a rate of 10 °C min−1. The analysis was performed in the splitless mode with an injector temperature of 250 °C.
Computational studies
All calculations were performed using the Gaussian 09 program suite.26 Geometry optimization of the structures was achieved using the B3LYP functional with the 6-311G++(d,p) basis set.
Kinetic computations
Conventionally, model-fitting methods or model-free (isoconversional) methods are used for the computation of kinetic parameters.27 However, isoconversional methods minimize the possibility of errors for both homogeneous and heterogeneous kinetic data as it does not follow any assumptions for the reaction mechanism.28 In this study, Vyazovkin's nonlinear integral isoconversional method was employed for the computations of apparent activation energy values by performing numerical integration.20 The mass loss data obtained from the non-isothermal TG runs were converted to the extent of conversion (α) using the standard eqn (1),| |  | (1) |
where m0 is the initial mass, mf the final mass, and mt the mass at a given temperature. An increment of 0.025 in α was used to compute the Eα values using Vyazovkin's nonlinear integral isoconversional method. Further details of the kinetic analysis are provided in the ESI.†
Results and discussion
Thermal analysis
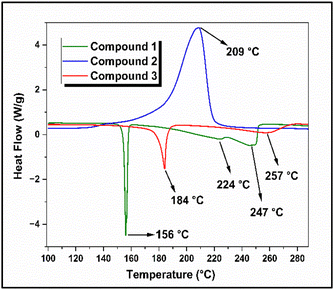
Among the TG analysis curves of compounds 1, 2, and 3, the data obtained at a heating rate of 6 °C min−1 were considered representative curves and are shown in Fig. 3(a). The corresponding differential thermogravimetric (DTG) curves are shown in Fig. 3(b) and the thermal analysis data is consolidated in Table S1 (ESI†). Compound 1 remained stable up to 168 °C, after which it slowly began to decompose. A decomposition peak was observed at 223 °C, and the mass loss continued until 234 °C, with an overall weight loss of 90%. For compound 2, the onset of decomposition was observed at 164 °C, and the decomposition temperature was observed at 209 °C (Fig. 3(b)). The mass loss continued until 232 °C, and an overall weight loss of 60% was observed. The DTG curve of compound 3 showed two stages of decomposition. The onset of decomposition was observed at 166 °C and the first-stage decomposition peak temperature was observed at 188 °C. The second stage decomposition temperature was 212 °C, and the mass loss continued until 232 °C. The compound exhibited an overall mass loss of 67%. The DSC curves of compounds 1, 2, and 3, obtained at a heating rate of 5 °C min−1, are shown in Fig. 4.
 |
| | Fig. 3 TGA (a) and DTG (b) curve of compounds 1, 2, and 3 at a heating rate of 6 °C min−1. | |
 |
| | Fig. 4 DSC curve of compounds 1, 2, and 3 at a heating rate of 5 °C min−1. | |
Compound 1 exhibited three endothermic peaks in its DSC thermogram. The first endothermic peak at 156 °C corresponds to the melting point of the compound. The first-stage decomposition was observed at 224 °C, and the second-stage decomposition was observed at 247 °C. For compound 2, an exothermic decomposition peak at 209 °C was observed, which was also reflected in the DTG curve of the compound. The DSC thermogram of compound 3 showed an endothermic peak at 184 °C, which corresponded to the melting of the compound. The second endothermic peak in the DSC thermogram was observed at 257 °C, corresponding to the second-stage decomposition of the compound, which was observed at 212 °C in the DTG curve (Fig. 3(b)). The variation in the decomposition temperature of compound 3 observed during DSC analysis is due to the different experimental conditions under which the TG (open crucibles) and DSC (closed sample pans) experiments were conducted. During the TG experiment (open crucibles), the possibility of secondary reactions was limited because the flowing nitrogen gas immediately carried away the decomposition products. In contrast, during the DSC experiment (closed sample pans), the decomposition products were retained and remained in contact with the decomposed solid compound. From the DSC curves, it was observed that compounds 1 and 3 exhibited endothermic decomposition, whereas compound 2 decomposed exothermically. This indicates that the introduction of the nitro (–NO2) group to the 1,2,4-triazole substrate significantly altered the decomposition behaviour of 3-amino-1,2,4-triazole.
TG-FTIR studies
TG-FTIR analysis was employed to analyze the gaseous decomposition products of compounds 1, 2, and 3. The FTIR spectra of the evolved gases for the compounds at various temperatures are shown in Fig. 5–7 and the elapsed temperature FTIR plots are shown in Fig. S10–S12 (ESI†). The temperatures were primarily chosen to display the evolved gases at the onset temperatures and around the decomposition peaks in the TG analysis at a heating rate of 10 °C min−1 for all the compounds. Fig. 5 shows the FTIR spectra acquired at 186 °C (onset temperature), 243 °C (decomposition peak), and 260 °C (end temperature) during the thermal decomposition of compound 1. At all temperatures, the predominant species identified was H2O, confirmed by its characteristic bands at 3946–3501 cm−1 (OH stretching) and 1869–1313 cm−1 (OH bending) vibrations. Additionally, the possible evolution of IR-inactive homonuclear diatomic gases such as N2 is anticipated. FTIR spectra acquired at 193 °C (onset temperature), 200 °C (decomposition peak), and 205 °C (end temperature) during the thermolysis of compound 2 are shown in Fig. 6. The analysis confirmed the presence of N2O (3341, 3273, 2239, 2202, 1304, 1269 cm−1), CO2 (2380–2260, 669 cm−1), HN3 (2111 cm−1), NO (1966–1800 cm−1), and HCN (713 cm−1) at 193 °C, 200 °C, and 205 °C. From the spectra, it was observed that the concentration of evolved gases increased significantly near the decomposition peak temperature (200 °C) (Fig. S11, ESI†). Among the released gases, the predominant species identified were CO2, N2O, and HCN.
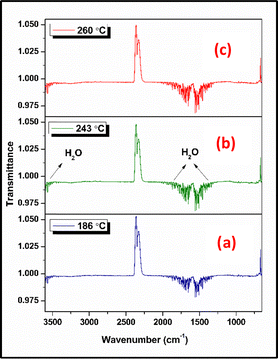 |
| | Fig. 5 Comparison of vibrational spectra of compound 1 decomposition products at (a) 186 °C, (b) 243 °C, and (c) 260 °C. | |
 |
| | Fig. 6 Comparison of vibrational spectra of compound 2 decomposition products at (a) 193 °C, (b) 200 °C, and (c) 205 °C. | |
 |
| | Fig. 7 Comparison of vibrational spectra of compound 3 decomposition products at (a) 124 °C, (b) 193 °C, (c) 205 °C, (d) 249 °C, and (e) 286 °C. | |
Compound 3 exhibited two stages of decomposition; therefore, two decomposition peaks were considered in TG-FTIR analysis. Fig. 7 shows the FTIR spectra acquired at 124 °C (onset temperature), 193 °C (first decomposition peak), 205 °C (onset temperature), 249 °C (second decomposition peak), and 286 °C (end temperature) during the thermal decomposition of compound 3. From the spectra, the presence of evolved gases was confirmed at 193 and 205 °C, the first stage of decomposition. The major species identified was CO2, which was confirmed by its characteristic bands at 2400–2210 cm−1 and 669 cm−1. Furthermore, no species were identified at 249 °C, which is the second decomposition peak, and no species were observed following the second stage of decomposition. In addition to these species, the possible evolution of IR-inactive homonuclear diatomic gases, such as N2 and O2 (for compounds 2 and 3), is anticipated.
GC/MS and MS/MS analysis
The ESI-MS data for the compounds are listed in Table 1. The positive mode ESI-MS spectrum of compound 1 is shown in Fig. S3 (ESI†). The base peak obtained at m/z 85.0518 corresponds to the (M + H)+ ion. Fig. S6 (ESI†) represents the negative mode ESI-MS spectrum of compound 2, and the base peak was obtained at m/z 128.0211, corresponding to the (M − H)− ion. Similarly, the negative mode ESI-MS spectrum of compound 3 is shown in Fig. S9 (ESI†). A base peak was obtained at m/z 141.0418, corresponding to the (M − H)− ion.
Table 1 Major ion species found in the ESI-MS analysis of compounds 1, 2, and 3
| Compound |
Molecular weight |
Possible species |
Elemental composition |
Observed mass (m/z) |
Calculated mass (Da) |
Error (ppm) |
| Compound 1 |
84.0820 |
(M + H)+ |
C2H5N4+ |
85.0518 |
85.0509 |
10 |
| Compound 2 |
129.0790 |
(M − H)− |
C2H2N5O2− |
128.0211 |
128.0214 |
2.3 |
| Compound 3 |
142.1180 |
(M − H)− |
C4H5N4O2− |
141.0418 |
141.0418 |
0 |
Since compound 1 has a low molecular weight, MS/MS analysis failed to produce any prominent peaks in the spectrum. Hence, GC-MS was employed to investigate the fragmentation pattern of the compound. The GC-MS spectrum of compound 1 showed three major peaks at m/z 84.04, 57.03, and 43.04 (Table 2 and Fig. S13, ESI†). Using this data, a plausible fragmentation pattern for compound 1 was identified, as shown in Scheme 1. The corresponding exact and observed masses are abbreviated as EM and OM, respectively. Compound 1 eliminated HCN to produce (CH3N3)+ (1A) at m/z 57.03, which then underwent fragmentation to form (CH3N2)+ (methyl diazonium) ion (1B) at m/z 43.04. MS/MS analysis of the (M − H)− ions observed at m/z 128.03 and 141.06 Da respectively, of compounds 2 and 3 were performed to understand the fragmentation pathways of these compounds. The MS/MS spectra of these compounds are shown in Fig. S14 and S15 (ESI†), respectively. MS/MS data are presented in Table 3. The spectrum of compound 2 showed two major daughter ions at m/z 111.0171 and 83.0119 (Fig. S14 and Table 3, ESI†). Based on this data, a possible fragmentation pattern was investigated, as shown in Scheme 2. Initially, compound 2 eliminated hydrogen to form (M − H)− ion (2A) at m/z 128.0211 (C2H2N5O2)−. Subsequently, the (M − H)− ion underwent hydroxyl radical elimination to form 2B (C2HN5O)˙− at m/z 111.0171. This intermediate, 2B underwent a nitrogen extrusion reaction to form 2C (C2HN3O)˙− at m/z 83.0119. A 65% weight loss was observed during the transition between 2A and 2C. In addition, the TG-DTG curves (Fig. 3(a) and (b)) of compound 2 showed an overall weight loss of 60%. This finding supports the formation of 2C from 2A. Additionally, the TG-FTIR data showed the possible elimination of N2O and NO from the compound. This is further supported by the mass fragmentation pattern, which suggests the possible evolution of N2O from 2B and NO from 2A and 2B. Similarly, the spectrum of compound 3 showed two major daughter ions at m/z 97.0522 and 66.0120 (Fig. S15 and Table 3, ESI†), and the possible fragmentation pattern is given in Scheme 3. Compound 3 eliminated hydrogen to afford the product ion (M − H)− ion (3A) at m/z 141.0418 (C4H5N4O2)−. The (M − H)− ion (3A) underwent carbon dioxide (CO2) elimination to form 3B (C3H5N4)− at m/z 97.0522, 3B eliminates CH5N (methylamine) to afford 3C (C2N3)− at m/z 66.0120. The conversion of 3A to 3B was associated with a weight loss of 69%. In addition, the TG-DTG curves (Fig. 3(a) and (b)) of compound 3 showed an overall weight loss of 67%. This finding supports the formation of 3B from 3A. The fragmentation pattern showed the possible elimination of CO2 during the transition from 3A to 3B, which is corroborated by the findings of the TG-FTIR analysis, which also showed CO2 evolution during the decomposition of the compound.
Table 2 Major fragment ion species found in the GC-MS analysis of compound 1
| Compound |
Observed mass (m/z) |
Molecular formula |
Exact mass (Da) |
| Compound 1 |
84.04 |
C2H4N4 (1) |
84.0436 |
| 57.03 |
CH3N3 (1A) |
57.0327 |
| 43.04 |
CH3N2+ (1B) |
43.0291 |
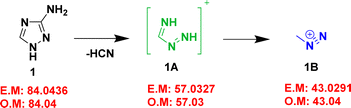 |
| | Scheme 1 Possible fragmentation pathway of compound 1. | |
Table 3 Major fragment ion species found in the MS-MS analysis of compounds 2 and 3
| Compound |
Peak selected for MS/MS analysis |
Observed mass (m/z) (daughter ions) |
Molecular formula |
Exact mass (Da) |
| Compound 2 |
128.03 (C2H2N5O2)− (2A) |
111.0171 |
(C2HN5O)˙− (2B) |
111.0187 |
| 83.0119 |
(C2HN3O)˙− (2C) |
83.0125 |
|
|
| Compound 3 |
141.06 (C4H5N4O2)− (3A) |
97.0522 |
(C3H5N4)− (3B) |
97.0520 |
| 66.0120 |
(C2N3)− (3C) |
66.0098 |
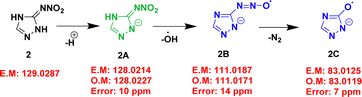 |
| | Scheme 2 Possible fragmentation pathway of compound 2. | |
 |
| | Scheme 3 Possible fragmentation pathway of compound 3. | |
Computational studies
Fig. 8(a)–(c) show the optimized molecular structures of compounds 1, 2, and 3, respectively, and the bond lengths for the different bonds are listed in Table S2 (ESI†) (Fig. S16, ESI†). As illustrated in Fig. 8, compounds 1 and 2 exhibited planar structures, whereas compound 3 exhibited a non-planar structure. For compound 2, there was a possibility of intramolecular hydrogen bonding between the oxygen atom in the nitro group and the hydrogen atom at the C5 position, as these atoms were close to each other. The weakest bonds identified were C–NH2 (1.38 Å) in 1, N–NO2 (1.38 Å) in 2, and CH2–COOH (1.52 Å) in 3 (Table S2, ESI†). The mass fragmentation pathway of compound 1 also supported the cleavage of the C–NH2 bond. From the mass fragmentation pattern observed for compound 2, the N–O bond initially breaks instead of the N–NO2 bond (weakest bond). This may be because the bond energy calculation was performed for a single molecule, whereas in a 3D structure, the bond energies of different bonds can be altered by the packing arrangement. Hence, rather than breaking the theoretically weakest bond, the weakest bond in the solid-state arrangement broke. The fragmentation pattern of compound 3 suggested that the weakest bond, CH2–COOH, was cleaved.
 |
| | Fig. 8 Optimized molecular structures of (a) compounds 1, (b) compounds 2, and (c) compound 3 (blue, grey, white, and red spheres represent nitrogen, carbon, hydrogen, and oxygen atoms, respectively). | |
Kinetic analysis
The kinetic parameters for compounds 1, 2, and 3 were determined by employing the TG data obtained at heating rates of 2, 4, 6, and 8 °C min−1. After performing TG experiments at four different heating rates, the α values were determined from the mass loss data. The α–T curves of 1 are shown in Fig. S17 (ESI†). The Eα values were obtained using the Vyazovkin method and plotted against α, as shown in Fig. 9 (Eα values obtained using integral isoconversional method is shown in Fig. S20, ESI†). The average activation energy for the decomposition of compound 1 is 86 kJ mol−1 within the 0.05–0.95 region of α, and the linear relationship between Eα and α shows that the decomposition process for the compound is defined by a single activation energy or the unification of multiple activation energies. The lower activation energy is attributed to the cleavage of weak bonds, such as the C–NH2 bond (1.38 Å, Fig. S16 and Table S2, ESI†), which was also observed in the fragmentation pattern obtained from the GC-MS analysis (Scheme 1).
 |
| | Fig. 9
E
α
–α curve of compound 1. | |
The α–T curves for compound 2 were computed from the TG data at four different heating rates, 2, 4, 6, and 8 °C min−1, and are shown in Fig. S18 (ESI†). Subsequently, the Eα values were computed, and the activation energy for the decomposition of compound 2 varied with the extent of conversion (α) (Fig. 10). The activation energy rises consistently from 69 kJ mol−1 at low conversion to 200 kJ mol−1 at 95% conversion. The initial lower activation energy values were due to the cleavage of some weak bonds, such as the N–NO2 bond (1.38 Å, Fig, S16 and Table S2, ESI†). During the initial stage of decomposition, some strong bonds remained intact, and thus, a greater amount of activation energy is required for the decomposition of the molecules. A slight decrease in activation energy is observed in the 0.3–0.4 region. This corresponds to a change in the mass loss rate, as evident in the 180–200 °C temperature range on the DTG curve of compound 2. The slight increase in the mass loss rate is reflected as a small decrease in activation energy. As the decomposition progressed, the value of the activation energy increased up to a conversion of 95%, which was associated with the breaking of some strong linkages, such as the CN bond (1.3–1.37 Å) in the triazole ring.
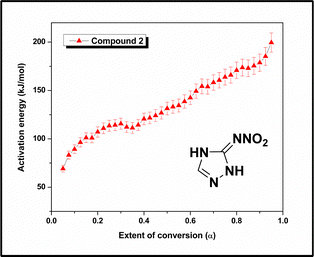 |
| | Fig. 10
E
α
–α curve of compound 2. | |
Similarly, the α versus T curves for compound 3 were plotted (Fig. S19, ESI†), and the Eα values were computed for the decomposition process. Fig. 11 shows the change in the activation energy with the decomposition progress (α) for compound 3. The curve shows that the activation energy of the decomposition process varies with α. The activation energy increases in the 0.15–0.39 region of α, which is characterized by an average activation energy of 169 kJ mol−1. The melting point of the compound was 184 °C (Fig. 4), and the corresponding α value was 0.22 (Fig. S19, ESI†). In the DTG curve (Fig. 3(b)), the first-stage decomposition peak was observed at 188 °C, indicating that the initial decomposition began immediately after the compound melted. Subsequently, a decrease in Eα (193–99 kJ mol−1) was observed in the 0.39–0.72 region of α. To better understand the decomposition mechanism, the residue obtained from the TG analysis after the first stage of decomposition (210 °C) was characterized using proton NMR and FTIR spectroscopy techniques (Fig. S21 and S22, ESI†). The analysis revealed that the product formed was 3-methyl-1H-1,2,4-triazole-5-amine, and the initial increase in the activation energy was attributed to the formation of this stable intermediate. Subsequently, the activation energy decreases as the intermediate compound decomposes, forming less stable products. This region is characterized by an average activation energy of 146 kJ mol−1. After the first stage of decomposition, the CO2 evolution stopped because of the absence of a carboxylic acid group in the intermediate compound. This finding is supported by the TG-FTIR data (Fig. S12, ESI†), indicating that the CO2 concentration was halted after 207 °C. The fragmentation pathway deduced using MS/MS analysis (Scheme 3) also showed the formation of 3-methyl-1H-1,2,4-triazol-5-amine (3B) from compound 3 by cleavage of the CH2–COOH bond, the weakest bond identified in computational studies. It was found that the Eα remains practically constant at ∼98 kJ mol−1 within the conversion range of 0.72–0.95.
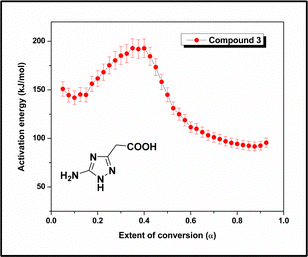 |
| | Fig. 11
E
α
–α curve of compound 3. | |
Among the three molecules, the average activation energy for compound 1 was 86 kJ mol−1, which was associated with the rupture of the weak C–NH2 bond. The mass fragmentation pattern obtained using GC-MS data also showed cleavage of the C–NH2 bond during the decomposition process. The activation energy of compound 2 rises consistently in the 0.05–0.95 region of α and the initial lower value are associated with the rupture of the weak N–NO2 bond. The mass loss associated with the conversion between 2A and 2C was corroborated by the mass loss obtained from TG analysis. The decomposition of compound 3 followed a multi-step process, with the formation of 3-methyl-1H-1,2,4-triazol-5-amine, as indicated by the residue analysis and mass fragmentation pattern. The mass loss observed during the transition from 3A to 3B aligns with the weight loss detected by the TG analysis. TG-FTIR showed the presence of CO2 as the evolved gas during decomposition, and the mass fragmentation pattern further suggested the possible elimination of CO2 from compound 3. The introduction of a nitro group to amino triazole (compound 1) enhanced the thermal stability of the compound (compound 2), as indicated by the high activation energy values. This is explained by the fact that, in compound 2, triazole possessing a protonated imino nitrogen atom is resonance-stabilized by forming a negatively charged, protonated triazole ring. Additionally, compound 3 exhibited greater stability than compound 1 and underwent a multi-step decomposition process. The improved stability of compound 3 can be attributed to the hydrogen bonding and electron-withdrawing effects of its carboxymethyl group present in the compound.
Conclusions
The decomposition mechanisms of three triazole derivatives, namely 3-amino-1,2,4-triazole (compound 1), 2,4-dihydro-3H-1,2,4-triazol-3-ylidene-nitramide (compound 2), and 5-amino-1,2,4-triazol-3-yl-acetic acid (compound 3), were determined using a combination of thermal experiments, kinetic analysis, mass spectrometry, and TG-FTIR. The kinetic analysis of compound 1 indicated a single-step decomposition, while compounds 2 and 3 followed multistep decomposition kinetics, evident from the nonlinear relationship between the activation energy and the extent of conversion. GC/MS analysis of compound 1 (C2H4N4) showed the possible formation of 1A (CH3N3)+ by HCN elimination, and 1A (CH3N3)+ then underwent fragmentation to form 1B (CH3N2+). The degradation pattern obtained by Tandem mass spectrometry indicated that compound 2 led to the formation of 2B (C2HN5O)˙−via the elimination of hydroxyl radicals, and 2B (C2HN5O)˙− subsequently underwent a nitrogen extrusion reaction to yield 2C (C2HN3O)˙−. The degradation of compound 3 proceeded through the formation of 3B (C3H5N4)− by decarboxylation and 3B (C3H5N4)− then form 3C (C2N3)−via the elimination of methylamine (CH5N). The mass fragmentation pattern suggested the breaking of weak bonds, as confirmed by computational analysis. TG-FTIR analysis of compounds 1 and 3 suggested the possible elimination of H2O and CO2, respectively. Compound 2 showed the evolution of N2O, CO2, HN3, NO, and HCN. The average activation energy for the decomposition of 1 was found to be lower than that of 2. In compound 2, a triazole possessing a protonated imino nitrogen atom is resonance-stabilized by forming a negatively charged, protonated triazole ring. In addition, compound 1 decomposed endothermically, whereas compound 2 decomposed exothermically. These findings revealed that the substitution of amino groups with nitramino groups enhanced the thermal stability of the compound and altered its decomposition behaviour. Furthermore, the incorporation of a carboxymethyl group into amino triazole (compound 1) improved the thermal stability of the compound (compound 3). This understanding can be used to design novel 1,2,4-triazole-based HEMs with suitable explosophores to improve their performance and thermal stability.
Author contributions
The manuscript was written through the contributions of all authors.
Data availability
Most of the data supporting the findings of this study are included in the ESI.† Additional data are available from the corresponding author upon reasonable request.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors thank Defense Research and Development Organization [DRDO/ARMREB/HEM/2021/241], Science and Engineering Research Board [SERB/SRG/2021/001182], FRG-NIT Calicut, and Student Innovation Program (SIP)-NIT Calicut for the financial support. S. V. acknowledges Department of Science and Technology (DST) for INSPIRE Fellowship [IF190991]. The authors thank DST-FIST for HRMS facility at NIT Calicut, Centre for Materials Characterisation (CMC)-NIT Calicut for NMR facility, and Central Research Facility (CRF)-NIT Karnataka for GC-MS facility.
Notes and references
- M. Saqib, F. Izadi, L. U. Isierhienrhien, M. Ončák and S. Denifl, Decomposition of triazole and 3-nitrotriazole upon low-energy electron attachment, Phys. Chem. Chem. Phys., 2023, 25, 13892–13901 RSC
 .
.
- R. S. Mathpati, A. K. Yadav, V. D. Ghule and S. Dharavath, Potential energetic salts of 5,5′-methylenedi(4H-1,2,4-triazole-3,4-diamine) cation: synthesis, characterization and detonation performance, Energ. Mater. Front., 2022, 3, 90–96 CrossRef CAS .
- S. Seth and C. Pathak, 4,4′-Azobis(1,2,4-triazole): A Versatile Molecular Scaffold to Develop Tailor-Made Energetic Materials, Cryst. Growth Des., 2023, 23, 4669–4679 CrossRef CAS .
- T. Wang, H. Gao and J. M. Shreeve, Functionalized Tetrazole Energetics: A Route to Enhanced Performance, Z. Anorg. Allg. Chem., 2021, 647, 157–191 CrossRef CAS .
- A. A. Dippold and T. M. Klapötke, Nitrogen-Rich Bis-1,2,4-triazoles—A Comparative Study of Structural and Energetic Properties, Chem. – Eur. J., 2012, 18, 16742–16753 CrossRef CAS PubMed .
- K.-Y. Lee, C. B. Storm, M. A. Hiskey and M. D. Coburn, An improved synthesis of 5-amino-3-nitro-1H-1,2,4-triazole (ANTA), a useful intermediate for the preparation of insensitive high explosives, J. Energ. Mater., 1991, 9, 415–428 CrossRef CAS .
- K. Wang, D. A. Parrish and J. M. Shreeve, 3-Azido-N-nitro-1H-1,2,4-triazol-5-amine-Based Energetic Salts, Chem. – Eur. J., 2011, 17, 14485–14492 CrossRef CAS PubMed .
- V. Thottempudi, H. Gao and J. M. Shreeve, Trinitromethyl-Substituted 5-Nitro- or 3-Azo-1,2,4-triazoles: Synthesis, Characterization, and Energetic Properties, J. Am. Chem. Soc., 2011, 133, 6464–6471 CrossRef CAS PubMed .
- S. Dharavath, J. Zhang, G. H. Imler, D. A. Parrish and J. M. Shreeve, 5-(Dinitromethyl)-3-(trinitromethyl)-1,2,4-triazole and its derivatives: a new application of oxidative nitration towards gem-trinitro-based energetic materials, J. Mater. Chem. A, 2017, 5, 4785–4790 RSC .
- G. Zhao, D. Kumar, P. Yin, C. He, G. H. Imler, D. A. Parrish and J. M. Shreeve, Construction of Polynitro Compounds as High-Performance Oxidizers via a Two-Step Nitration of Various Functional Groups, Org. Lett., 2019, 21, 1073–1077 CrossRef CAS PubMed .
- T. S. Hermann, T. M. Klapötke, B. Krumm and J. Stierstorfer, The energetic 3-trinitromethyl-5-nitramino-1H-1,2,4-triazole and nitrogen-rich salts, New J. Chem., 2017, 41, 3068–3072 RSC .
- T. Liu, X. Qi, K. Wang, J. Zhang, W. Zhang and Q. Zhang, Green primary energetic materials based on N-(3-nitro-1-(trinitromethyl)-1H-1,2,4-triazol-5-yl)nitramide, New J. Chem., 2017, 41, 9070–9076 RSC .
- Y. Wang, L. Hu, S. Pang and J. M. Shreeve, Nitroimino as an energetic group in designing energetic materials for practical use, a tautomerism from nitroamino, J. Mater. Chem. A, 2023, 11, 13876–13888 RSC .
- W. Kowhakul, D. Inoue, Y. Nakagawa, H. Masamoto and M. Shigematsu, Thermal decomposition mechanisms of 1H-1,2,4-triazole derivatives: a theoretical study, J. Loss Prev. Process Ind., 2017, 50, 37–54 CrossRef CAS .
- W. Kowhakul, M. Kumasaki and M. Arai, Study on thermal behavior of 1H-1, 2, 4-triazole-copper complex with substituents, Sci. Technol. Energ. Mater., 2005, 66, 425–430 CAS .
- P. J. Sánchez-Soto, E. Morillo, J. L. Pérez-Rodríguez and C. Real, Thermoanalytical study of the pesticide 3-amino-1,2,4-triazole, J. Therm. Anal., 1995, 45, 1189–1197 CrossRef .
- V. L. Korolev, T. V. Petukhova, E. A. Bakhmatova, T. S. Pivina and A. B. Sheremetev, Molecular modeling of the mechanisms of thermolysis of nitramino-1,2,4-triazoles, Chem. Heterocycl. Compd., 2006, 42, 1267–1290 CrossRef CAS .
- N. Li, X. Yang, X. Li, Z. Peng, Z. Yin, C. Jiang, Z. Huang and Y. Li, Enhancing the detonation performance of azobistriazole energetic derivatives via inducing N-oxide groups, Phys. Chem. Chem. Phys., 2024, 26, 19863–19875 RSC .
- S. Venugopal and A. A. Vargeese, Decomposition Mechanism of Alkylidene Bridged Tetrazoles With Different Carbon Chain Lengths, ChemPhysChem, 2024, e202400943, DOI:10.1002/cphc.202400943 .
- S. Vyazovkin, Evaluation of activation energy of thermally stimulated solid-state reactions under arbitrary variation of temperature, J. Comput. Chem., 1997, 18, 393–402 CrossRef CAS .
- A. Benhammada and D. Trache, Thermal decomposition of energetic materials using TG-FTIR and TG-MS: a state-of-the-art review, Appl. Spectrosc. Rev., 2020, 55, 724–777 CrossRef CAS .
- M. A. Kumar, P. Ashutosh and A. A. Vargeese, Decomposition Mechanism of Hexanitrohexaazaisowurtzitane (CL-20) by Coupled Computational and Experimental Study, J. Phys. Chem. A, 2019, 123, 4014–4020 CrossRef CAS PubMed .
- S. Lal, R. J. Staples and J. M. Shreeve, Nitroiminotriazole (NIT) based potential solid propellants: synthesis, characterization, and applications, Dalton Trans., 2024, 53, 903–907 RSC .
- A. M. Abdel-Megeed, H. M. Abdel-Rahman, G.-E. S. Alkaramany and M. A. El-Gendy, Design, synthesis and molecular modeling study of acylated 1,2,4-triazole-3-acetates with potential anti-inflammatory activity, Eur. J. Med. Chem., 2009, 44, 117–123 CrossRef CAS PubMed .
- S. Saha, A. Chowdhury and N. Kumbhakarna, Effect of copper chromite on ammonium perchlorate decomposition-A TGA-FTIR-MS and FE-SEM study, J. Therm. Anal. Calorim., 2024, 149, 9401–9412 CrossRef CAS .
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li,M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega,G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi,J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision, Gaussian, Inc., Wallingford CT, 2016 Search PubMed .
- J. E. White, W. J. Catallo and B. L. Legendre, Biomass pyrolysis kinetics: a comparative critical review with relevant agricultural residue case studies, J. Anal. Appl. Pyrolysis, 2011, 91, 1–33 CrossRef CAS .
- M. Azam, A. Ashraf, S. S. Jahromy, W. Raza, H. Khalid, N. Raza and F. Winter, Isoconversional nonisothermal kinetic analysis of municipal solid waste, refuse-derived fuel, and coal, Energy Sci. Eng., 2020, 8, 3728–3739 CrossRef CAS .
|
| This journal is © the Owner Societies 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b and
Anuj A.
Vargeese
b and
Anuj A.
Vargeese














