
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
How non-aqueous media direct the reaction of Ca(OH)2 with CO2 to different forms of CaCO3: operando mid-infrared and X-ray absorption spectroscopy studies†
Thokozile A.
Kathyola
 ab,
Sin-Yuen
Chang
b,
Elizabeth A.
Willneff
c,
Colin J.
Willis
d,
Giannantonio
Cibin
b,
Paul
Wilson
d,
Anna B.
Kroner
b,
Elizabeth J.
Shotton
b,
Peter J.
Dowding
d and
Sven L. M.
Schroeder
*ab
ab,
Sin-Yuen
Chang
b,
Elizabeth A.
Willneff
c,
Colin J.
Willis
d,
Giannantonio
Cibin
b,
Paul
Wilson
d,
Anna B.
Kroner
b,
Elizabeth J.
Shotton
b,
Peter J.
Dowding
d and
Sven L. M.
Schroeder
*ab
aSchool of Chemical and Process Engineering, University of Leeds, Leeds, LS2 9JT, UK. E-mail: s.l.m.schroeder@leeds.ac.uk
bDiamond Light Source, Didcot, Oxfordshire OX11 0DE, UK
cSchool of Design, University of Leeds, Leeds, LS2 9JT, UK
dInfineum UK Limited, Abingdon, Oxfordshire OX13 6BB, UK
First published on 22nd April 2025
Abstract
Time-resolved structural changes taking place during the reaction of Ca(OH)2 and CO2 forming different CaCO3 polymorphs, in aqueous and non-aqueous environments, were recorded operando using mid-infrared (mid-IR) and X-ray absorption near-edge structure (XANES) spectroscopy. Results show that Ca(OH)2 directly transforms into calcite in a pure water dispersion. In methanolic media with low water content, calcium di-methylcarbonate (Ca(OCOOCH3)2) is formed, which is hydrolysed to amorphous calcium carbonate (ACC) and vaterite in the presence of sufficient water. The addition of toluene shifts the equilibrium composition further from Ca(OH)2 to ACC and the crystalline forms of CaCO3, probably by affecting the activity of the methoxide intermediate. It can facilitate the formation of aragonite. No Ca(OH)2 conversion was detected in pure ethanol, isopropanol and toluene dispersions, except for nanoscale Ca(OH)2 in ethanolic dispersion, which formed calcium di-ethylcarbonate (Ca(OCOOCH2CH3)2). Our findings underline that vaterite formation is driven by the solution and solid state chemistry related to the reaction via alkoxides and carbonic acid esters of the alcohols, rather than the nucleation process in solution. The alcohol in these systems does not just act as a solvent but as a reactant.
Introduction
The molecular basis for modifying the polymorphic outcome of reactive calcium carbonate (CaCO3) crystallisation by choice of the solvent medium has attracted interest in various research fields, including nucleation science, biomineralisation, fuel additives, and conservation science.1–7 Short-chain alcohols such as methanol, ethanol, and isopropanol are well known additives that can influence the polymorphic outcome of CaCO3 formation.8 By tuning the reaction conditions in solvent systems with high alcohol content, any of the common CaCO3 forms can be obtained: amorphous CaCO3 (ACC), the metastable polymorphs vaterite and aragonite, and the thermodynamically stable polymorph calcite.5–22 A mechanistic study of CaCO3 formation by reaction of CO2 with methanolic calcium hydroxide (Ca(OH)2) dispersions has recently provided mechanistic insight into the action of alcohols at the molecular level.23 In an aqueous phase, the formation of calcite from Ca(OH)2 and CO2 is the dominant reaction pathway. For high methanol contents, it is kinetically outperformed by the formation of transient methoxide and carbonate ester salts formed with methanol. The alcohol thus acts both as a reactant and a solvent/dispersant, forming the key intermediates calcium hydroxide methoxide, Ca(OH)(OCH3), and calcium methoxide, Ca(OCH3)2, from Ca(OH)2. CO2 reacts with the methoxide ions to form calcium di-methylcarbonate, Ca(OCOOCH3)2, which transforms into an ACC sol–gel and vaterite when hydrolysed. It was confirmed that in methanolic (≥20 mol%) systems CaCO3 forms via the following reaction pathways:| Ca(OH)2 + CH3OH ⇌ Ca(OH)(OCH3) + H2O | (1) |
| Ca(OH)(OCH3) + CH3OH ⇌ Ca(OCH3)2 + H2O | (2) |
| Ca(OCH3)2 + 2CO2 → Ca(OCOOCH3)2 | (3) |
| Ca(OCOOCH3)2 + H2O → CaCO3 + 2CH3OH + CO2 | (4) |
The formation of the Ca(OCOOCH3)2 ester salt from methanol had occasionally been noted in the literature,24–26 but its crucial mechanistic role for directing CaCO3 formation in methanolic phase to its metastable forms appears to have been overlooked. The hydrolysis of Ca(OCOOCH3)2 followed by diffusion-limited ACC → vaterite → calcite transformations was observed after stopping the addition of CO2. Traces of aragonite were also detected. In the present paper, we will provide insight into the nature of transient species during the reactive formation of CaCO3 from Ca(OH)2 and CO2 in a ternary solvent system containing methanol, water and toluene (Fig. 1).
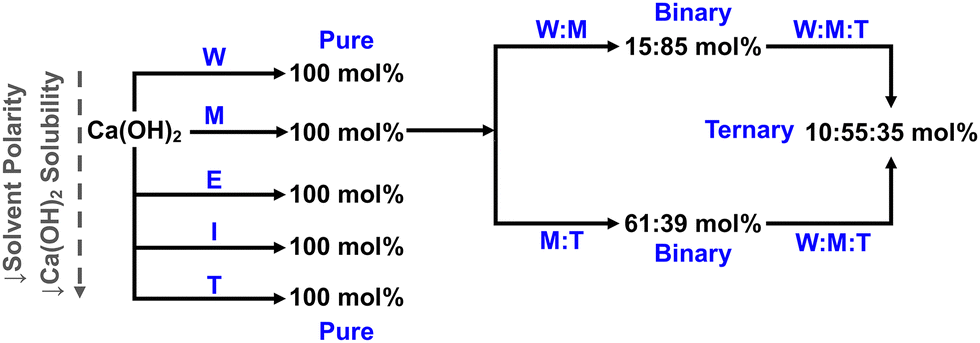 | ||
| Fig. 1 Roadmap of solvent systems examined in this paper: pure, binary, and ternary water (W), methanol (M), ethanol (E), isopropanol (I), and toluene (T) systems. | ||
We will also explore how higher alcohols, specifically ethanol and isopropanol, influence CaCO3 formation. Evidence for alkoxide formation in these solvents has previously been reported for nano-sized Ca(OH)2 particles (‘nanolimes’), which are used in art and stone conservation.5–7 These studies identified the formation of calcium ethoxide and isopropoxide, but the possibility of carbonic acid ester formation was not explored. We have therefore examined the reactions in ethanol and isopropanol systems using both micro- and nano-sized Ca(OH)2.
For all system we monitored the changes in Ca speciation in situ and/or operando by a combination of mid-infrared (mid-IR) and X-ray absorption spectroscopy (XAS). XAS was, in effect, integrated into a scaled down practical reaction platform as an in-line process analytical technology (PAT) tool.27,28In situ/operando XAS provides deep mechanistic insight because it provides quantitative information on the nature and concentrations of transient and stable Ca species during reactions. In-line mid-IR provides complementary information on the speciation. It discriminates better than XAS between the alkoxide and ester intermediates.23 As we will show, the combination of both analytical techniques leads to mechanistic scenarios explaining how alcohols determine the properties of CaCO3 products.
Experimental
Materials
CaCO3 was synthesised using Ca(OH)2 (95%; L’Hoist), ethanol, and isopropanol nanolimes (Nanorestore Plus; CSGI), CO2 (99.9%; BOC), methanol (99.9%; Fisher Scientific), and deionised water. Nitrogen (99.9%; BOC) and helium (99.9%; Air Products) were also used to control the environment during synthesis and X-ray absorption measurements respectively. Powdered samples of Ca(OCH3)2 (≥99%; Sigma Aldrich), Ca(OCOOCH3)2, ACC, vaterite, aragonite, and calcite (≥99%; Sigma Aldrich) were used as references. The Ca(OCOOCH3)2, ACC, vaterite, and aragonite were synthesised using methods previously described by Kathyola et al.,23 Koga et al.,29 Shivkumara et al.,30 and Kitamura et al.31 respectively.Reactive crystallisation process
Dispersions were prepared by mixing micro-sized Ca(OH)2 (∼30 μm; 4.2 g) with 750 ml of solvent. Solvent ratios of the binary and ternary solvent systems (Fig. 1) were determined based on the typical synthesis methods for sulphonate-stabilised CaCO3 particles.3,32 The commercial nanolimes were pre-mixed with 5 g of nano-sized Ca(OH)2 (∼170 nm) dispersed in 1 L of ethanol or isopropanol. Reactions were carried out in a 1 L glass baffled reactor using a conventional lab-scale setup (Fig. S1, ESI†) under constant N2 (30 ml min−1) flow. The temperature and stirrer speed were maintained at 28 °C and 400 rpm, respectively. Each dispersion was carbonated for a duration of 20 min using 95 wt% of the required stoichiometric amount of CO2. The post-carbonation dispersion was stirred for a further 10 min to allow for reaction completion and growth of carbonate particles.Mid-IR
Operando mid-IR spectra were collected with a Bruker Alpha FTIR spectrometer equipped with a DPR 210 ZnSe ATR probe (Hellma Analytics) was used (Fig. S1, ESI†). Spectra were acquired from 64 scans at a resolution of 4 cm−1 from 4000 to 650 cm−1. Measurements were controlled and data initially processed using the OPUS 7.0 software. The resulting spectra were analysed using Fityk 1.3.1 curve fitting software.33XAS
Operando fluorescence yield Ca K-edge X-ray absorption near-edge structure (XANES) spectra were collected in the photon energy range from 4000 eV to 4800 eV at beamline B18 of Diamond Light Source.34 Spectra were obtained every minute before (pre-), during and after (post-) the 20 min carbonation reactions, for a total duration of 40 min. To monitor real-time chemical state changes of Ca with XAS with minimal disruption to the synthesis, a sampling loop with a bespoke multi-modal liquid jet cell was attached to the baffled reactor (Fig. S1, ESI†).27,28 Aliquots of the polyphasic dispersions were continuously pumped from the reactor into the liquid jet cell and back at 320 ml min−1. Data were processed and analysed using Athena in the Demeter software package.35 Linear combination fitting (LCF) was performed on the operando spectra over a range of 4020 to 4090 eV. Ex situ total electron yield spectra of Ca(OH)2, Ca(OCH3)2, Ca(OCOOCH3)2, ACC, vaterite, aragonite, and calcite reference samples were used as standards for the LCF. All measurements were acquired at room temperature under a constant He environment.Results and discussion
Carbonation in pure water and in pure methanol
Operando mid-IR and XAS were simultaneously used to resolve the changes in Ca speciation during the 20 min carbonation of Ca(OH)2 dispersed in 100 mol% water and methanol. As expected from our previous study,23 the results (Fig. 2) indicated the formation of calcite and the monoester salt Ca(OCOOCH3)2 in the water and methanol systems, respectively. In aqueous phase, calcite crystallisation was evidenced by the distinctive mid-IR absorptions associated with out-of-plane bending (γCO3) at 871 cm−1, asymmetric stretching at 1402 cm−1, and in-plane bending (δCO3) at 710 cm−1 (Fig. 3a). These peaks are absent in the methanolic dispersion (Fig. 2b), which exhibits weak characteristic methoxycarbonyl IR absorptions arising from C
at 1402 cm−1, and in-plane bending (δCO3) at 710 cm−1 (Fig. 3a). These peaks are absent in the methanolic dispersion (Fig. 2b), which exhibits weak characteristic methoxycarbonyl IR absorptions arising from C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C(O)–O vibrational modes at 1641 (νCO), 1330
O and C(O)–O vibrational modes at 1641 (νCO), 1330  , 1095
, 1095 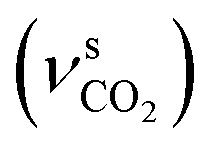 and 827 (δCO) cm−1. Ex situ mid-IR of the final product confirmed the formation of Ca(OCOOCH3)2 (reactions (1)–(3)), in line with our previous findings reported for systems with high methanol content.23
and 827 (δCO) cm−1. Ex situ mid-IR of the final product confirmed the formation of Ca(OCOOCH3)2 (reactions (1)–(3)), in line with our previous findings reported for systems with high methanol content.23
 | ||
| Fig. 2 Operando mid-IR and XANES of Ca(OH)2 carbonation in pure water (a) and (c) and methanol (b) and (d) – calcite and Ca(OCOCH3)2 (CDMC) are formed. The full line XA spectra in (c) and (d) were obtained by LCF of the experimental spectra (dots) with the reference spectra shown at the bottom and top of the stacked plots. | ||
 | ||
| Fig. 3 Operando mid-IR and XANES of Ca(OH)2 carbonation in binary solvent systems of (a) and (c) water–methanol and (b) and (d) methanol–toluene – Ca(OCOCH3)2 (CDMC) and ACC are formed. The full line XA spectra in (c) and (d) were obtained by LCF of the experimental spectra (dots) with the reference spectra shown at the bottom and top of the stacked plots. | ||
Inspection of the operando XANES spectra (Fig. 2c and d) for both systems revealed significant changes in the features arising from 1s → 3d (at ∼4040 eV) and 1s → 4p (from 4045 to 4060 eV) electronic transitions. The expected27 conversion of Ca(OH)2 to calcite was detected in the aqueous dispersion (Fig. 2c). Primary indicators for this were the growth of the post-edge peak at 4060 eV and a shift of about −2.5 eV in the edge position as a function of time. A similar shift was observed in the methanolic dispersion (Fig. 2d), but it was coupled with diminutions in the 1s → 4p features at about 4045 and 4060 eV, resulting in spectra comparable to the reference Ca(OCOOCH3)2 XANES. The post-carbonation product in methanol (at 35 min) was initially assumed to be ACC, based on the 1s → 4p features and the relatively prominent 1s → 3d peak at 4040 eV. However, this possibility was contradicted by the presence of the weak broad peak at 4060 eV in the post-carbonation and Ca(OCOOCH3)2 spectra, which is absent in the spectrum of ACC.23
The composition of the systems was further elucidated by linear combination fitting (LCF) of the XANES data. Initial fitting of the aqueous dispersion XANES (Fig. 2c) using only Ca(OH)2 and calcite references resulted in what appeared to be acceptable fits, but relatively high residuals indicated that the two reference spectra were not fully accounting for the chemical speciation in the system. Inclusion of a reference spectrum of hydrated Ca2+ cations (obtained from a calcium chloride solution) improved the fit significantly.36 It therefore appears that significant dissolution of Ca(OH)2 takes place. The effect of the dissolved Ca2+ ions on the XANES is less apparent in the initial 5 min of the reaction due to the low solubility of Ca(OH)2 in water. However, Ca(OH)2 dissolution becomes more noticeable as the reaction progresses, likely due to acidification associated with the dissolution of CO2.
LCF analysis confirmed the presence of Ca(OCOOCH3)2 as the main product of the reaction with methanol. Attempts to include the aqueous Ca2+ ion spectrum in the LCF analysis of the methanolic dispersion XANES (Fig. 2d) indicated that its contribution was insignificant. Taken together, our results thus support the proposition that the post-carbonation Ca product in methanolic phase is an alkoxide and/or carbonic acid methyl ester salt.23–26 Carbonate formation does not occur in the absence of water because the ester formation between CO2 and methanol is kinetically favoured over the reaction of dissolved CO2 with the solid Ca(OH)2, which is limited by mass transport.
The accuracy of the LCF quantifications is illustrated by comparing with the expected stoichiometric amounts of calcite (Fig. 2c) and Ca(OCOCH3)2 (Fig. 2d) with respect to the reactants. The extents of Ca(OH)2 conversion were calculated based on the reported sequence of carbonation, methoxylation, and esterification (reactions (1)–(3)).23 The stoichiometry of these reactions predicts that an equimolar amount of CO2 added to the two systems should result in formation of about 95% calcite and 48% Ca(OCOOCH3)2. These values are very close to those observed through the LCF analysis of the operando XANES spectra, 96(4) and 41(3)%, respectively (Fig. 2). It is important to note that the absolute standard deviation values are mostly determined by the noise in the operando XAS data, which is caused by the mechanical instability of the liquid jet flow in the X-ray beam.27
Binary water–methanol and methanol–toluene dispersions
As for the pure methanol system, Ca(OCOOCH3)2 was found to be the main product in the mid-IR and XANES spectra for both binary water–methanol (15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :85 mol%) and methanol–toluene (61:39 mol%) systems (Fig. 3). Significant ACC was detected in the water–methanol dispersion (γCO3 at 866 cm−1 in Fig. 3a) as the 15 mol% water hydrolyses Ca(OCOOCH3)2 to CaCO3 (reaction (4)).23 LCF analysis of the complementary XANES (Fig. 3c) showed that the final post-carbonation product consisted of 32(7)% Ca(OCOOCH3)2 and 30(7)% ACC. Post-synthesis ex situ mid-IR (Fig. S2, ESI†) revealed that the mixed Ca(OCOOCH3)2/ACC product converted into vaterite and calcite upon ageing. This is similar to the reaction pathway previously observed in 100 and 90 mol% methanol systems, where the sequence Ca(OCOOCH3)2 + water → ACC → vaterite → calcite was found.23
:85 mol%) and methanol–toluene (61:39 mol%) systems (Fig. 3). Significant ACC was detected in the water–methanol dispersion (γCO3 at 866 cm−1 in Fig. 3a) as the 15 mol% water hydrolyses Ca(OCOOCH3)2 to CaCO3 (reaction (4)).23 LCF analysis of the complementary XANES (Fig. 3c) showed that the final post-carbonation product consisted of 32(7)% Ca(OCOOCH3)2 and 30(7)% ACC. Post-synthesis ex situ mid-IR (Fig. S2, ESI†) revealed that the mixed Ca(OCOOCH3)2/ACC product converted into vaterite and calcite upon ageing. This is similar to the reaction pathway previously observed in 100 and 90 mol% methanol systems, where the sequence Ca(OCOOCH3)2 + water → ACC → vaterite → calcite was found.23
In pure toluene, no significant Ca(OH)2 conversion takes place at all (Fig. S3, ESI†), presumably because of the low solubilities of both CO2 and Ca(OH)2 in toluene, while the solvent also acts as a barrier to mass transport of CO2 to Ca(OH)2. The LCF XANES analysis for the methanol–toluene dispersion indicated the presence of Ca(OCOOCH3)2 only, with a final composition of 60(3)% (Fig. 4d). As for pure methanol phase, no carbonate formation can take place due to the absence of water. It is sometimes assumed that no reaction at all takes place in methanol–toluene systems,37 but this is clearly not supported by our data. Interestingly, both mid-IR and XANES data suggest that more Ca(OH)2 is converted to Ca(OCOOCH3)2 (∼60% vs. ∼40% ester salt content) in the methanol–toluene system (Fig. 3b and d) compared to the pure methanol dispersion (Fig. 2b and d). It appears that the reaction equilibrium is shifted somewhat towards the ester salt in the presence of toluene, perhaps because the methoxide ion is destabilised in less polar media,38,39 or because dissolution or solubilisation of the ester salt takes place in the resulting ternary system.
 | ||
| Fig. 4 Operando mid-IR and XANES of Ca(OH)2 carbonation in a ternary solvent system of water–methanol–toluene (a) and (c) – Ca(OCOCH3)2 (CDMC) and ACC are formed; and time-resolved changes in the composition of carbonated species based on the (b) mid-IR and (d) XANES. The full line XA spectra in (c) were obtained by LCF of the experimental spectra (dots) with the reference spectra shown at the bottom and top of the stacked plots. Error bars for the XAS speciation analysis were provided from the correlation matrix of the multiparameter LCF analysis. Increased noise in the data towards the end of the XAS analysis reflect increasing jet instability due to the formation of Ca(OCOCH3)2 and ACC particles. | ||
Ternary water–methanol–toluene dispersion
An even higher conversion of Ca(OH)2, forming both Ca(OCOOCH3)2 and ACC, was indicated by the mid-IR for the water–methanol–toluene (10:55:35 mol%) system (Fig. 4). Analysis of the γCO3 deformation peak area (Fig. 4b) shows that ACC formed at a faster rate in the ternary dispersant than in the binary water–methanol system. This effect may again be related to the destabilisation of methoxide ions by the presence of the non-polar toluene molecules, which results in a higher rate of ester salt formation and subsequent hydrolysis by the water in the system (reaction (3) and (4)). The toluene also decreases the viscosity of the dispersions40 and may promote diffusive mass transport of CO2 to both the methanol and Ca(OH)2 components, increasing the rates further. The δHOH peak at 1642 cm−1 (Fig. 4a) revealed that both reactions also resulted in a significant increase in water content, above the initial 10 mol%, pushing the equilibrium further towards the hydrolysis product ACC (Fig. 3a). These conclusions are also confirmed by the LCF of the XANES (Fig. 4d) that shows about 50% less Ca(OCOOCH3)2 in the ternary system for the majority of the reaction.
The time-dependent conversions in water, water–methanol, and water–methanol–toluene are summarised in Fig. 4b and d. All of the Ca(OH)2 in the pure water system converted into CaCO3, whereas partial conversions (≤52%) were achieved in the presence of non-aqueous solvents. According to the XANES (Fig. 4d), the final post-carbonation product from the ternary system consists of about 44% less CaCO3 than for the dispersion in pure water (Fig. 3c), reflecting the mechanistic shift to the ester hydrolysis forming ACC (reaction (4)). The Ca(OH)2 conversion may also be affected by diffusion-limited precipitation due to the presence of methanol and the associated densification of the solvent media.41Ex situ X-ray diffraction (XRD) analysis (Fig. S4, ESI†) of the post-carbonation product from a ternary solvent dispersion with 14 times more Ca(OH)2 revealed that the toluene reduced the stability of the amorphous sol–gel formed. Peaks due to calcite and vaterite were present in the initial XRD pattern of the gel (15 min post-carbonation). This is unlike the water–methanol (10:90 mol%) sol–gel from our previous study,23 which was stable in its amorphous form up to 90 min. Interestingly, both XRD and mid-IR (Fig. S4 and S5, ESI†) for the ternary system showed that upon aging (60 hours) the Ca(OCOOCH3)2 and ACC converted mainly into aragonite. Traces of aragonite were detected previously in the mid-IR of the aged pure methanol product, but in the presence of toluene it is the predominant polymorph. The formation of aragonite and vaterite may be attributed to kinetic stabilisation induced by solvation/adsorption of toluene. Kinetic stabilisation due to adsorption of organic compounds, such as ethanol, has been reported for these two polymorphs.6,8,42 Additionally, toluene has been shown to affect the surface structure of Ca(OH)2.43 Toluene may perhaps be behaving like Mg2+ ions in restricting crystal growth and preventing the formation of calcite whilst promoting that of aragonite.42,44
Pure ethanol and isopropanol dispersions
Carbonation in the presence of ethanol and isopropanol was associated with no significant changes in the mid-IR (Fig. S6, ESI†) and XANES (Fig. 5) when standard coarse grained (∼30 μm particle size) Ca(OH)2 was carbonated in the presence of pure ethanol and isopropanol. Compared to methanol, Ca(OH)2 has lower solubility in the ethyl and isopropyl alcohols, reducing the rate of carbonation reaction.45,46 Nonetheless, the formation of calcium ethoxide and isopropoxide has been reported in alcoholic dispersions of nano-sized (∼170 nm) Ca(OH)2.5–7 In analogy to the methanolic systems discussed above, these alkoxide salts were identified as precursors for ACC and vaterite. Analysis of the mid-IR (Fig. 5a) for ethanol solutions of nanolimes shows evidence for the conversion of the nano-sized Ca(OH)2 to calcium di-ethylcarbonate, Ca(OCOOCH2CH3)2. This was determined from the peak assignments for the previously discussed Ca(OCOOCH3)2 ester and literature data on the ethoxycarbonyl (–COOCH2CH3) group.23,47 Some minor changes were observed in the isopropanol mid-IR (Fig. 5b), which could be due to the formation of calcium di-isopropylcarbonate, Ca(OCOOCH2CH2CH3)2. | ||
| Fig. 5 Operando mid-IR and XANES of the carbonation of micro- and nano-sized Ca(OH)2 in pure ethanol (a) and (c) and isopropanol (b) and (d) – Ca(OCOOCH2CH3)2 (CDEC) and traces of Ca(OCOOCH2CH2CH3)2 (in mid-IR only) are formed. | ||
LCF analysis of the XANES (Fig. 5c and d) for the ethanol and isopropanol systems was not possible due to the lack of reference spectra for the respective ester intermediates. However, a qualitative comparison of the pre- and post-carbonation spectra clearly shows that conversion from Ca(OH)2 only occurred in the ethanol nanolimes system. Ultimately, these results show that increasing the alkyl chain length decreases the reactivity of the alcohol and that Ca(OH)2 particle size has a significant influence on the reaction rate. Formation of carbonated Ca species in the ethanol system is comparable to methanol only when the Ca(OH)2 particle size is sufficiently reduced to overcome the mass transport limitations associated with the lower ester concentration in the mixtures.
Conclusions
Combined operando mid-IR and XAS studies of multiphase multicomponent CaCO3 crystallisation from Ca(OH)2 and CO2 were carried out. The rates of Ca(OH)2 conversion to different CaCO3 forms were monitored in real-time in various solvent systems, including pure and mixed systems of water, methanol, ethanol, isopropanol, and toluene. Calcite was confirmed to be the final product in pure water (100 mol% water). In methanolic systems with low water content the formation of the carbonate ester salt Ca(OCOOCH3)2 dominates. It hydrolyses to ACC and vaterite in the presence of water. The addition of toluene to methanol/water systems increased reaction rates of ester formation, indicating destabilisation of the methoxide intermediate by the non-polar solvent, and facilitated the crystallisation of aragonite. No conversion of crystalline Ca(OH)2 to carbonic acid alkyl ester salts was observed in pure ethanol and isopropanol dispersions, except when highly dispersed (‘nanolime’) Ca(OH)2 was contacted with the alcohol. For this case, the formation of Ca(OCOOCH2CH3)2 and traces of Ca(OCOOCH2CH2CH3)2 were detected. Overall, the results show that depending on solvent composition the carbonated ester intermediate can transform into various forms of CaCO3. The most prevalent polymorphs are ACC, vaterite and calcite, but aragonite can be accessed by the addition of toluene. The examination of the nanolimes indicated that ethanol is probably a viable replacement for methanol only when the Ca(OH)2 particle size is reduced to the nano-scale.Author contributions
All authors contributed to the work presented in this paper. T. A. K., S.-Y. C., E. A. W., C. W., G. C., A. B. K., P. J. D. and S. L. M. S. contributed to the conception and design of the experiments. C. W., G. C., P. W. and A. B. K. provided technical support. T. A. K., S.-Y. C., E. A. W., C. W., P. W., E. J. S., P. J. D. and S. L. M. S. performed the experiments. T. A. K. analysed the data and wrote the manuscript, which was edited by E. A. W. and S. L. M. S.Data availability
All data supporting this study are provided either in the Results section of this paper or in the ESI,† accompanying it.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by Infineum UK Ltd., the Royal Academy of Engineering, Diamond Light Source (DLS; Beamtime Awards – SP14673 and SP21040), the EPSRC Centre for Doctoral Training in Complex Particulate Products and Processes, cP3 (Grant Number – EP/L015285/1) and EPSRC Future Continuous Manufacturing and Advanced Crystallisation (CMAC) Research Hub (Grant Number – EP/P006965/1). T. A. K. gratefully acknowledges financial support from cP3, University of Leeds (UoL), Infineum UK Ltd., and the CMAC Research Hub. S. L. M. S. acknowledges the support of the Bragg Centenary Chair by the Royal Academy of Engineering, Infineum UK Ltd., and DLS. Thanks to Matthew Guindy (UoL), Arturs Pugejs (UoL), Robert W. M. Hooley (UoL) and Samuel G. Booth for their aid with some of the data collection; and Muling Zeng (UoL) for the ACC sample.References
- F. C. Meldrum and H. Colfen, Chem. Rev., 2008, 108, 4332–4432 CrossRef CAS.
- J. F. Marsh, Chem. Ind., 1987, 14, 470–473 Search PubMed.
- J. P. Roman, P. Hoornaert, D. Faure, C. Biver, F. Jacquet and J. M. Martin, J. Colloid Interface Sci., 1991, 144, 324–339 CrossRef CAS.
- C. Belle, R. Gallo, F. Jacquet, P. Hoornaert and J. P. Roman, Lubr. Sci., 1992, 5, 11–31 CrossRef CAS.
- C. Rodriguez-Navarro, A. Suzuki and E. Ruiz-Agudo, Langmuir, 2013, 29, 11457–11470 CrossRef CAS PubMed.
- C. Rodriguez-Navarro, K. Elert and R. Sevcik, CrystEngComm, 2016, 18, 6594–6607 RSC.
- C. Rodriguez-Navarro, I. Vettori and E. Ruiz-Agudo, Langmuir, 2016, 32, 5183–5194 CrossRef CAS PubMed.
- K. K. Sand, J. D. Rodriguez-Blanco, E. Makovicky, L. G. Benning and S. L. S. Stipp, Cryst. Growth Des., 2012, 12, 842–853 CrossRef CAS.
- T. Yasue, A. Mamiya, Y. Takahashi, R. Tsukisaka and Y. Arai, Nippon Kagaku Kaishi, 1984, 1984, 1107–1113 CrossRef.
- T. Yasue, A. Mamiya, T. Fukushima and Y. Arai, Gypsum Lime, 1985, 1985, 245–252 Search PubMed.
- Y. Ueda, K. Komatu, S. Shimizu, H. Nishioka, M. Hanazaki and S. Minayoshi, Gypsum Lime, 1994, 1994, 105–114 Search PubMed.
- F. Manoli and E. Dalas, J. Cryst. Grow., 2000, 218, 359–364 CrossRef CAS.
- S. R. Dickinson and K. M. McGrath, J. Mater. Chem., 2003, 13, 928–933 RSC.
- J.-K. Park, J.-W. Ahn, Y.-S. Park and C. Han, Geosyst. Eng., 2004, 7, 89–94 CrossRef CAS.
- K. S. Seo, C. Han, J. H. Wee, J. K. Park and J. W. Ahn, J. Cryst. Grow., 2005, 276, 680–687 CrossRef CAS.
- H. S. Lee, T. H. Ha and K. Kim, Mater. Chem. Phys., 2005, 93, 376–382 CrossRef CAS.
- S. F. Chen, S. H. Yu, J. Jiang, F. Q. Li and Y. K. Liu, Chem. Mater., 2006, 18, 115–122 CrossRef CAS.
- S. F. Chen, H. Colfen, M. Antonietti and S. H. Yu, Chem. Commun., 2013, 49, 9564–9566 RSC.
- Y. D. Hu, Y. H. Zhou, X. R. Xu and R. K. Tang, Cryst. Res. Technol., 2015, 50, 312–318 CrossRef CAS.
- G. Magnabosco, I. Polishchuk, B. Pokroy, R. Rosenberg, H. Colfen and G. Falini, Chem. Commun., 2017, 53, 4811–4814 RSC.
- M. Farhadi-Khouzani, D. M. Chevrier, P. Zhang, N. Hedin and D. Gebauer, Angew. Chem., Int. Ed., 2016, 55, 8117–8120 CrossRef CAS.
- Y. Nakashima, C. Takai, H. Razavi-Khosroshahi, W. Suthabanditpong and M. Fuji, Adv. Powder Technol., 2018, 29, 904–908 CrossRef CAS.
- T. A. Kathyola, E. A. Willneff, C. J. Willis, P. J. Dowding and S. L. M. Schroeder, ACS Phys. Chem. Au, 2024, 4, 555–567 CrossRef CAS.
- A. Buzágh, Kolloidn. Zh., 1926, 38, 222–226 Search PubMed.
- J. Plank, H. Hoffmann, J. Schölkopf, W. Seidl, I. Zeitler and Z. Zhang, Res. Lett. Mater. Sci., 2009, 2009, 1–3 Search PubMed.
- G. J. Witkamp, S. A. P. Escobar and R. S. Gaertner, US Pat., 20150344319A1, 2015 Search PubMed.
- S. Y. Chang, T. A. Kathyola, E. A. Willneff, C. J. Willis, P. Wilson, P. J. Dowding, G. Cibin, A. B. Kroner, E. J. Shotton and S. L. M. Schroeder, React. Chem. Eng., 2019, 4, 679–687 RSC.
- T. A. Kathyola, S.-Y. Chang, E. A. Willneff, C. J. Willis, G. Cibin, P. Wilson, A. B. Kroner, E. J. Shotton, P. J. Dowding and S. L. M. Schroeder, Ind. Eng. Chem. Res., 2023, 62, 16198–16206 CrossRef CAS.
- N. Koga, Y. Z. Nakagoe and H. Tanaka, Thermochim. Acta, 1998, 318, 239–244 CrossRef CAS.
- C. Shivkumara, P. Singh, A. Gupta and M. S. Hegde, Mater. Res. Bull., 2006, 41, 1455–1460 CrossRef CAS.
- M. Kitamura, H. Konno, A. Yasui and H. Masuoka, J. Cryst. Grow., 2002, 236, 323–332 CrossRef CAS.
- I. Markovic, R. H. Ottewill, D. J. Cebula, I. Field and J. F. Marsh, Colloid Polym. Sci., 1984, 262, 648–656 CrossRef CAS.
- M. Wojdyr, J. Appl. Crystallogr., 2010, 43, 1126–1128 CrossRef CAS.
- A. J. Dent, G. Cibin, S. Ramos, A. D. Smith, S. M. Scott, L. Varandas, M. R. Pearson, N. A. Krumpa, C. P. Jones and P. E. Robbins, J. Phys. Conf. Ser., 2009, 190, 012039 CrossRef.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- J. L. Fulton, S. M. Heald, Y. S. Badyal and J. M. Simonson, J. Phys. Chem. A, 2003, 107, 4688–4696 CrossRef CAS.
- M. L. Robbins, F. Leder, E. Chludzinski and G. R. Chludzinski, US Pat., 3429811A, 1969 Search PubMed.
- J. I. Brauman and L. K. Blair, J. Am. Chem. Soc., 1968, 90, 6561–6562 CrossRef CAS.
- J. I. Brauman and L. K. Blair, J. Am. Chem. Soc., 1970, 92, 5986–5992 CrossRef CAS.
- U. B. Bray, C. R. Dickey and V. Voorhees, Ind. Eng. Chem. Prod. Res. Dev., 1975, 14, 295–298 CrossRef CAS.
- Y. Oaki and H. Imai, Cryst. Growth Des., 2003, 3, 711–716 CrossRef CAS.
- F. C. Meldrum, Int. Mater. Rev., 2003, 48, 187–224 CrossRef CAS.
- I. Pashalidis and C. R. Theocharis, J. Chem. Technol. Biotechnol., 1998, 71, 223–226 CrossRef CAS.
- Y. Kitano, D. W. Hood and K. Park, J. Geophys. Res., 1962, 67, 4873 CrossRef CAS.
- A. Seidell, Solubilities of Inorganic and Organic Compounds: A Compilation of Solubility Data from the Periodical Literature, D. Van Nostrand Company, New York, 1919 Search PubMed.
- G. Just, Z. Phys. Chem., 1901, 37, 342–367 CrossRef.
- N. P. G. Roeges, in Guide to the complete interpretation of infrared spectra of organic structures, ed. A. N. Lobachev, John Wiley & Sons Ltd., Chichester, England, 1994, pp. 263–269 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Figures of the experimental setup and complementary XAS, XRD and mid-IR plots. See DOI: https://doi.org/10.1039/d4cp04774e |
| This journal is © the Owner Societies 2025 |