
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEvolution of silicate coordination in architected amorphous and crystalline magnesium silicates during carbon mineralization†
Xun
Gao
a,
Prince
Ochonma
 b,
Divya
Prasad
a,
Mahadeo A.
Mahadik
a,
Ivan
Kuzmenko
c,
Jan
Ilavsky
c and
Greeshma
Gadikota
*ab
b,
Divya
Prasad
a,
Mahadeo A.
Mahadik
a,
Ivan
Kuzmenko
c,
Jan
Ilavsky
c and
Greeshma
Gadikota
*ab
aSchool of Civil and Environmental Engineering, Cornell University, Ithaca, NY 14853, USA. E-mail: gg464@cornell.edu; Tel: +1 607-255-4796
bSmith School of Chemical and Biological Engineering, Cornell University, Ithaca, NY 14853, USA
cAdvanced Photon Source, Argonne National Laboratory, Lemont, Illinois 60439, USA
First published on 24th February 2025
Abstract
Advancing durable solutions for carbon storage and removal at the gigaton scale to produce solid carbonates via carbon mineralization requires harnessing earth abundant magnesium silicate resources. Calibrated insights linking the structural and morphological features of earth abundant amorphous and crystalline magnesium silicate phases to their reactivity are essential for scalable deployment but remain underdeveloped. To resolve the influence of silica coordination and mass transfer on carbon mineralization behavior, magnesium silicates bearing amorphous and crystalline phases (AC Mg-silicate) are synthesized. The structural and morphological transitions starting from colloidal precursors to their final synthesized form on heating are delineated using operando ultra small/small/wide angle X-ray scattering (USAXS/SAXS/WAXS) measurements. The evolution of the silicate phases on carbon mineralization of AC Mg-silicate is contrasted with that of highly crystalline Mg-silicate (HC Mg-silicate) when reacted at 200 °C and a CO2 partial pressure of 20 atm in water and 1 M NaHCO3 solution in stirred and unstirred environments. These experimental conditions are analogous to those of the water–gas-shift reaction for sustainable recovery of H2 with inherent carbon mineralization. Enhancement in the extent of carbon mineralization by 13.3–19.5% noted in the presence of NaHCO3 compared to water in AC and HC Mg-silicate with and without stirring, is attributed to the buffering effect which aids simultaneous silicate dissolution and carbon mineralization. Enhanced extents of carbon mineralization in the presence of NaHCO3 correspond to the formation of MgSiO3 and SiO2 phases from the starting Mg2SiO4 precursors in AC and HC Mg-silicate. Unlocking these silicate transformations during carbon mineralization by harnessing architected Mg-silicate precursors reveals the feasibility of integrating these chemical pathways with sustainable H2 conversion pathways with inherent carbon mineralization.
1. Introduction
The need to manage rising CO2 emissions has motivated advances in novel chemical pathways to capture and store these emissions in a sustainable, durable and usable manner, while meeting the ever-increasing demand for energy and resources. Geological CO2 storage, biological fixation, and carbon mineralization have been proposed to store CO2 emissions.1–4 Amongst these approaches, carbon mineralization is a thermodynamically favoured pathway that converts CO2 emissions into usable solid carbonate end products while reliably storing CO2 emissions at the scale of several gigatons.5,6 Furthermore, the integration of carbon mineralization with energy and resource conversions enhances the overall thermodynamic feasibility of these pathways and enables the co-production of multiple usable products essential for a sustainable resource future. For example, coupling carbon mineralization with H2 conversion pathways such as the water–gas shift reaction (CO + H2O → CO2 + H2) or biomass reforming reactions (CnH2yOn + nH2O → nCO2 + (y + n)H2) enhances the production of high purity hydrogen (H2) while capturing CO2 emissions in situ to produce solid carbonates.7–9 This approach is an alternative to conventional membrane or sorption-based approaches to separate CO2 from H2, and the need for subsequent compression, transport, and storage in subsurface geological environments.Additionally, the integration of CO2 capture and mineralization enables favourable thermodynamics and is an effective approach for both CO2 storage and resource conversion.10,11 The capture and conversion of CO2 in these systems draw inspiration from natural processes where CO2 is converted to calcium (Ca) or magnesium (Mg) carbonate by harnessing earth abundant Ca- and Mg-bearing silicate minerals in the subsurface environment.11,12 Notably, combining thermodynamically favourable CO2 mineralization processes with energy-intensive resource conversions can reduce the overall energy requirements, while simultaneously enabling durable CO2 storage. Table 1 presents examples of Ca- and Mg-bearing silicates for carbon mineralization and their associated free energies of reaction associated with carbon mineralization.13 The negative free energies of reaction indicate the spontaneity of the reaction. In addition to favourable thermodynamics, it is worth noting that the natural abundance of magnesium bearing silicates in ultramafic and mafic rocks enables large-scale, durable, and economical CO2 storage.14,15 Furthermore, the products of carbon mineralization such as silica, carbonates, or iron oxides have multiple industrial applications.
| Mineral | Formula | Carbonation reaction | ΔGr, kJ mol−1 |
|---|---|---|---|
| Forsterite | Mg2SiO4 | Mg2SiO4 + 2H2O + 2CO2 → 2MgCO3 + H4SiO4 | −66.80 |
| Larnite | Ca2SiO4 | Ca2SiO4 + 2H2O + 2CO2 → 2CaCO3 + H4SiO4 | −127.20 |
| Akermanite | Ca2MgSi2O7 | Ca2MgSi2O7 + 4H2O + 3CO2 → 2CaCO3 + MgCO3 + 2H4SiO4 | −127.90 |
| Diopside | CaMgSi2O6 | CaMgSi2O6 + 4H2O + 2CO2 → CaCO3 + MgCO3 + 2H4SiO4 | −26.20 |
Though promising, the acceleration of carbon mineralization during direct gaseous CO2-solid mineral or rock interactions often requires high temperatures (>300 °C) or pressures (>100 atm), and the kinetics of this process remains slow.16–18 To address this challenge, aqueous carbon mineralization has been proposed as a more efficient pathway. The presence of aqueous media in the coupled CO2-reaction fluid-mineral or rock environments enhances mineral dissolution and ion mobility, thus increasing the reaction rates, and decreasing energy requirements.18,19 In this integrated environment, several steps could influence the process, including the dissolution of silicate minerals and CO2, and the subsequent mineralization of the dissolved Mg2+ ions with dissolved CO2 species to form Mg-carbonates. The dissolution of silicate minerals is often reported as the rate limiting step of this process, and strategies such as increasing temperatures and CO2 partial pressure, tuning aqueous chemistries (acidic or alkaline), reducing the particle sizes, or adding chelating agents have been successfully used to accelerate the kinetics of dissolution and carbon mineralization of silicates.20,21 Prior studies have focused on investigating the carbon mineralization behavior of silicate-bearing materials, in the context of elucidating the reaction thermodynamics and kinetics. For instance, studies have shown that >80% conversion of olivine ((Mg,Fe)2SiO4) to Mg-bearing carbonate is achieved after 3 hours, at pCO2 of 139 atm and 185 °C, in the presence of NaCl and NaHCO3 solutions. In contrast, lower temperatures of 90 °C resulted in much lower conversion rates of ∼3% under similar conditions.12 Elevated temperatures during carbon mineralization accelerate the dissolution kinetics and reactivity of Ca- and Mg-bearing silicates, thereby increasing the extent of carbon mineralization.8,22 Furthermore, a carbon mineralization with a concurrent critical metal recovery approach has been reported to show >70% carbon mineralization efficiency with nickel recovery ∼80% in a single-step process driven by the dissolution of silicate minerals such as serpentinized peridotite under specific conditions (185 °C and 50 bar CO2 pressure) with EDTA. The dual-purpose process highlights the significance of silicate dissolution in both stable mineral carbonate precipitation and selective metal recovery.23 Additional research reports olivine carbon mineralization with 34.5 bar of pCO2, 175 °C, and 1.5 M NaHCO3, in which a fine particle size (≤80 μm) has been proved necessary for accelerated silicate dissolution and carbon mineralization. Notably, carbon mineralization efficiency enhancement ∼50% is reported when the particle size decreases from 80 μm to 25 μm, which can be attributed to the greater reactive surface area resulting from smaller grain sizes.12,24 This increased reactive surface area facilitates the release of divalent cations (Ca2+, Mg2+, Fe2+) and accelerates CO2 conversion to solid carbonates.18
Despite these advances in the importance of silicate dissolution and the subsequent conditions that enable carbon mineralization, significant scientific gaps remain in understanding the underlying dissolution mechanisms. While the formation of a silica passivation layer has been reported during silicate dissolution in both acidic and alkaline solutions, its limiting impact on carbon mineralization remains less understood.25–27 To address this knowledge gap, this study is dedicated to the silicate coordination evolution during high-temperature aqueous carbon mineralization, and how coordination impacts the carbon mineralization efficiency, which will provide strategic guidance for tuning carbon mineralization across diverse operational scenarios (e.g., in situ and ex situ carbon mineralization, carbon mineralization with metal recovery, carbonate formation for use in construction materials). In the process of aqueous carbon mineralization, Mg2+ ions are released from the silicate lattice consumed by carbonate formation, which leads to the formation of a silica-rich layer on the surface of the reacting particles. This layer can inhibit further dissolution of the silica precursor, particularly Mg2+ ions, potentially limiting the formation of magnesite (MgCO3) as shown in Fig. 1(a). The possible forms of Si-bearing products resulting from silicate dissolution and carbon mineralization are shown in Fig. 1(b), where the Qn notation represents the number of bridging oxygens per Si tetrahedron in silicate-based materials and reflects the different corresponding phases from crystalline Mg2SiO4 (Q0) to amorphous SiO2 (Q4). Moreover, it has also been reported that silicates with different crystallinities exhibit varying solubilities at 30 °C following the order: MgSiO3 (Q2) > Mg2SiO4 (Q0) > SiO2 (Q4).28 These differences in solubility could potentially lead to different dissolution behaviours and varying carbon mineralization extents for Mg- and Ca-silicate bearing materials with varying Si coordination. For example, granular and amorphous SiO2 (Q4) particles are present alongside MgCO3 particles in Fig. 1(c), confirming the formation of amorphous silica after carbon mineralization. Despite the morphological evidence of silica formation, the abundance of various Si-bearing phases has not been extensively reported in the literature due to challenges associated with quantification. To address this challenge, the abundance of various silicate structures determined using XPS analyses is shown in Fig. 1(d).
 | ||
| Fig. 1 Schematic representation of (a) carbon mineralization, (b) possible structures of silicates, (c) image showing magnesium carbonate crystals formed alongside silica, and (d) relative abundance of various silicate phases present. | ||
The Si coordination evolves from 98% crystalline SiO4 (Q0) to 46% SiO3 (Q2) and 23% SiO2 (Q4) after carbon mineralization, indicating the release of Mg2+ ions from lattice during silicate dissolution and the formation of an amorphous Si-rich phase. The insights obtained from quantifying various Si-bearing phases can unlock new insights into the observed non-monotonic reactivity of Mg- and Ca-bearing silicates for carbon mineralization and the recovery and transport of energy relevant and critical metals such as iron and nickel.29 Additionally, while most studies on carbon mineralization have been conducted in aqueous media with stirring to overcome mass transfer limitations, there is a critical need to explore the influence of unstirred conditions to better understand the influence of diffusion limitations on the evolution of Si-bearing phases.11 This information can inform the development of novel reactor systems to enhance mass transfer without mechanical stirring elements and the natural weathering of silicates for carbon removal.
In this study, the influence of Si coordination in Mg-silicates on carbon mineralization behaviour is investigated given the natural abundance of Mg-silicates for carbon mineralization. The specific research questions addressed are: (i) How can we architect magnesium silicates with crystalline and amorphous phases to advance calibrated insights into carbon mineralization? (ii) How can we simultaneously capture the structural and morphological evolution in Mg-silicates as they are synthesized? (iii) What is the influence of highly crystalline and mixed amorphous and crystalline Mg-silicate phases on carbon mineralization behaviour in buffered and unbuffered environments, and stirred and unstirred environments? (iv) How do we quantify the relative abundance of silicate phases in the unreacted and reacted Mg-silicates?
To address these questions, amorphous and crystalline phases bearing Mg-silicates (AC-Mg-silicate) are synthesized. The structural and morphological evolution of these materials is characterized using operado ultra-small/small/wide angle X-ray scattering (USAXS/SAXS/WAXS) measurements. The reactivity of AC-Mg-silicate is contrasted with that of highly crystalline Mg-silicate (HC Mg-silicate) in water and 1 M NaHCO3 solution, at 200 °C and pCO2 of 20 atm in stirred and unstirred environments. In addition to determining the carbon mineralization behaviour of AC and HC Mg-silicates, the evolution in silica coordination with water and NaHCO3 at stirred and unstirred conditions is determined. Thus, these efforts are directed towards unlocking the influence of amorphous and crystalline phases of Mg-silicates on carbon mineralization.
2. Experimental methods
Magnesium nitrate hexahydrate, Mg(NO3)2·6H2O (Sigma-Aldrich, >98% purity) and tetraethyl orthosilicate (Sigma-Aldrich, 99.999% purity) are used to synthesize amorphous and crystalline phases bearing Mg-silicates (AC-Mg-silicate) for use in this study. Mg-silicate of relatively higher crystallinity is obtained from Xi'An Function Material Group Co. Ltd, and is referred to as highly-crystalline (HC) Mg-silicate in this study. The carbon mineralization behaviour of the AC and HC Mg-silicate are investigated using bone-dry CO2 gas (Airgas, 99.8% purity) via a high-temperature aqueous carbon mineralization process. Deionized water (18.2 MΩ cm, Millipore) was used in all the experiments as required.2.1. Synthesis of amorphous and crystalline phases bearing Mg-silicate (AC-Mg-silicate)
The amorphous Mg-silicate precursors are synthesized via a sol–gel method using magnesium nitrate hexahydrate and tetraethyl orthosilicate (TEOS) as the starting materials. The synthesized precursors are calcined in a muffle furnace to obtain the AC Mg-silicate samples, which are used as the alkaline source in the subsequent experiments.30,31 In this synthesis route, 17 g of tetraethyl orthosilicate is dissolved in 600 ml of 1 M nitric acid, and then 41 g of magnesium nitrate hexahydrate is added to the solution. The mixture is stirred for 4 hours at room temperature. A highly viscous gel is formed after drying the stirred solution at 80 °C for 24 hours. The gel is then calcined at 800 °C for 30 minutes in air, with a ramp rate of 10 °C min−1 to induce the transition from amorphous to crystalline phases (Fig. 2). During this process, magnesium nitrate hexahydrate is the Mg source and TEOS provides silica. The synthesis reaction can be expressed as reaction (R1),| 2Mg(NO3)2·6H2O + (CH3CH2O)4Si → Mg2SiO4 + 4CH3CH2OH + 8H2O + 4HNO3 | (R1) |
 | ||
| Fig. 2 Approach to synthesize the amorphous-crystalline (AC) Mg-silicate: 17 g of tetraethyl orthosilicate (TEOS) is dissolved in 600 ml of 1 M nitric acid, and then 41 g of magnesium nitrate hexahydrate is added to the solution. The mixture is stirred for 4 hours at room temperature and dried at 80 °C for 24 hours. The obtained gel is then calcined at 800 °C for 30 minutes in air, with a ramp rate of 10 °C min−1. | ||
2.2. Operando X-ray scattering to probe transformations of silicate gels to nanoparticles
As previously reported by Sanosh and co-workers, amorphous magnesium silicate samples undergo an agglomeration–aggregation process during calcination.30 In this process, the amorphous crystallites tend to aggregate as the temperature increases to reduce the surface energy. At a calcination temperature around 800 °C, these crystallites sinter and form larger secondary particles. To investigate the phase evolution and crystallite size changes during the amorphous to crystalline transition resulting in the formation of the AC Mg-silicate sample, cross-scale ultra-small/small/wide-angle X-ray scattering (WAXS) measurements are harnessed. These measurements are performed at sector 9-ID of the advanced photon source (APS), Argonne National Laboratory, Argonne, IL.32The gel precursor is placed in a quartz capillary and heated continuously under controlled conditions. The heating ramping rates are set as 10 °C min−1 from 25 °C to 300 °C and 5 °C min−1 from 300 °C to 800 °C. Each scan takes about 4 minutes and the data are collected sequentially during the thermal treatment. The total X-ray flux is set as 1013 photon mm−2 s−1, and the X-ray wavelength is 0.59 Å, corresponding to 21.0 keV energy.33,34 The collected USAXS/SAXS/WAXS data are reduced and analyzed using the Irena and Nika packages within the IgorPro software.35,36 The structural evolution obtained from the WAXS measurements uncovers the amorphous to crystalline transitions during thermal treatment. In addition, the fitted USAXS/SAXS data reveal the changes in the particle sizes as a function of temperature during calcination.
2.3. Carbon mineralization experiments
To contrast the extents of carbon mineralization of the synthesized AC Mg-silicate and the procured HC Mg-silicate, high-temperature carbon mineralization experiments are conducted in a 50 mL high-pressure reactor equipped with a built-in 4-blade Rushton turbine stirrer (Micro Bench Top Reactor, Parr Instruments Co., USA). The reactions are carried out at 200 °C and pCO2 of 20 atm for 3 hours, both with and without stirring to assess the impact of mass transfer on carbon mineralization. A schematic representation of the experiment setup is shown in Fig. 3. In each experiment, a slurry mixture with 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :17 ratio of Mg-silicate and the fluid of interest such as deionized (DI) water or 1 M NaHCO3 solution is used. The reactor is first purged by injecting bone-dry CO2 into the headspace for 10 minutes to evacuate gaseous impurities. Afterwards, the outlet valve is closed, and the reactor is pressurized to 20 atm. Once the desired internal pressure is achieved, the inlet valve is closed and the heater is switched on to bring the reactor temperature to 200 °C, indicating the start of the experiment. For the stirring-mode experiments, the stirring rate is set as 300 rpm. After carbon mineralization, the reactor is allowed to cool to the ambient temperature and then depressurized. The products bearing solid carbonates are vacuum filtered, washed with deionized water three times, and dried in a heating oven at 80 °C for 24 hours. The dried samples are then prepared for further characterization.
:17 ratio of Mg-silicate and the fluid of interest such as deionized (DI) water or 1 M NaHCO3 solution is used. The reactor is first purged by injecting bone-dry CO2 into the headspace for 10 minutes to evacuate gaseous impurities. Afterwards, the outlet valve is closed, and the reactor is pressurized to 20 atm. Once the desired internal pressure is achieved, the inlet valve is closed and the heater is switched on to bring the reactor temperature to 200 °C, indicating the start of the experiment. For the stirring-mode experiments, the stirring rate is set as 300 rpm. After carbon mineralization, the reactor is allowed to cool to the ambient temperature and then depressurized. The products bearing solid carbonates are vacuum filtered, washed with deionized water three times, and dried in a heating oven at 80 °C for 24 hours. The dried samples are then prepared for further characterization.
 | ||
| Fig. 3 Schematic representation of the experimental setup for carbon mineralization studies. All the experiments were conducted at 200 °C and pCO2 of 20 atm in the presence of DI water and 1 M NaHCO3, respectively. | ||
2.4. Characterization of carbonate-bearing products
All unreacted Mg-silicate materials and carbonate-bearing products are analyzed using multiple instruments to characterize their structural and morphological evolution. The changes in structural arrangement of the samples are determined by X-ray diffractometry (XRD, Bruker D8 Advance ECO powder diffractometer, Bruker). The morphology and particle size distribution are analyzed with a field emission scanning electron microscope (FESEM, LEO 1550 FESEM, Bruker) and particle size analyzer (Anton Parr). The concentration of Mg2+ ions in the solution are determined by elemental analysis using inductive coupled plasma atomic emission spectroscopy (ICP-AES, Spectro Arcos FHE12) at a wavelength of 279.079 (Aqueous-Axial-iFR). The detection limit is specified to be 1 ppm. Ultra-high purity liquid argon from Airgas is used, and all solutions are prepared in 3% nitric acid from stock 70% nitric acid (TraceMetal Grade, Fisher Chemicals). Mg2+ ion standard solutions for ICP-AES calibration are prepared using 1 g L−1 Mg2+ ion stock solution (Sigma Aldrich) in 3% nitric acid. Furthermore, the extent of carbon mineralization is assessed using thermogravimetric analysis (TGA, SDT650, TGA Instrument). During TGA analysis, the samples are heated from room temperature to 1000 °C at a 2 °C min−1 ramp rate, using a nitrogen gas flow rate of 50 ml min−1. The extent of carbon mineralization, which indicates how far the sample is from its theoretical CO2 capacity is calculated using eqn (1): | (1) |
In the expression above, YCO2 is the extent of carbon mineralization, TGA is the weight loss associated with the dissociation of the carbonate-bearing materials, and RCO2 is the stoichiometric mass of the alkaline source to react with a specific amount of CO2.12
3. Result and discussion
3.1. Structural and morphological evolution of gels to Mg-silicate nanoparticles
At room temperature, the smooth curve indicates the amorphous nature of the Mg-silicate precursor with no long-range ordered crystalline structure (Fig. 4(a)). As the temperature increases, the precursor begins to exhibit characteristic peaks at 1.61 Å−1, 2.51 Å−1, 2.55 Å−1, and 4.20 Å−1 at 211 °C, corresponding to the (021), (131), (112), and (004) peaks of crystalline forsterite. These peaks indicate the structural rearrangement and the initiation of the amorphous-to-crystalline transition (PDF 01-078-1371). The appearance of these specific crystallographic planes is primarily due to their favoured surface energy under the given conditions. As the calcination temperature rises to 431 °C, peaks appear at 2.96 Å−1 and 4.23 Å−1 corresponding to the growth along the (112) and (260) crystallographic planes, signifying increased crystallinity of the sample (Fig. 4(a)). When the temperature is further raised to 665 °C, additional peaks emerge at 1.68 Å−1, 1.79 Å−1, 2.09 Å−1, 2.23 Å−1, 2.75 Å−1, and 3.57 Å−1, corresponding to the (101), (111), (031), (221) and (222) planes of forsterite. These new peaks indicate the nucleation and growth of specific crystals as the crystallization process progresses. Further increases in the temperature leads to an enhancement in the peak intensity, confirming the continued growth of the generated crystallographic planes at high temperatures. The exclusive crystalline phase is observed to be pure Mg2SiO4, a typical structure with Q0 Si coordination.39
 | ||
| Fig. 4 Phase evolution of amorphous-crystalline (AC) Mg-silicate gels on heating from (a) room temperature to 577 °C and (b) 577 °C to 756 °C, as determined by operando Wide Angle X-Ray Scattering (WAXS) measurements. | ||
| I(q) ≃ I(0)exp(−q2Rg2/3) | (2) |
 | ||
| Fig. 5 Evidence of morphological evolution of the amorphous-crystalline (AC) Mg-silicate gel at temperatures in the range of (a) 30–574 °C and (b) 574–745 °C using operando USAXS/SAXS measurements. The slope evolution demonstrates the aggregation and growth of particles during thermal treatment. | ||
In the expression above I(q) and I(0) represent the scattering intensity and forward scattering intensity, respectively. q is the scattering vector magnitude which equals (4p/l)sinq, and Rg is the radius of gyration. This approximation allows us to determine the radius of gyration from the scattering intensity slopes at specific scales of the detected samples. Since the particle sizes of the synthesized AC Mg-silicate are expected to range from 1 nm to 100 nm, we focus on the intensity slopes between the 10−3 to 10−1 Å−1q range. Also, since the emergence of characteristic peaks largely occurs at calcination temperatures above 550 °C, Rg is calculated for the data obtained above 550 °C. As shown in Fig. 6(b), during thermal treatment, the radius of gyration increases from 22.63 nm at 574 °C to 37.92 nm at 750 °C, demonstrating an increase in particle size. However, it is important to note that the radius of gyration only indicates the distance of the particles’ components from their center of mass, not the actual particle size. By assuming the synthesized forsterite particles are roughly spherical, the radius of gyration can be converted into the actual particle size using eqn (3):30,45
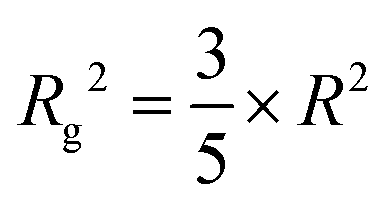 | (3) |
 | ||
| Fig. 6 A schematic representation of particle size evolution (a). The measured particle size of the amorphous-crystalline (AC) Mg-silicate characterized by (b) radius of gyration (Rg), (c) morphological analysis, and (d) particle size distribution. The particle size evolution highlights the aggregation-sintering mechanisms of the precursor during thermal treatment. | ||
Using this equation, the AC Mg-silicate sample has particles with a size of 29.22 nm at 574 °C, which grows to 48.96 nm at 750 °C. The increase in the particle size can be attributed to a sintering-aggregation mechanism. During thermal treatment, intermolecular forces such as van der Waals forces drive the nanocrystallites in the amorphous Mg-silicate to aggregate into clumps to minimize the surface free energy (Fig. 6(c)). Continued heating would cause these aggregated clumps to sinter and fuse into larger secondary particles at the micron scale. Fig. 6(d) shows the volume weighted particle size distribution of the transformed AC Mg-silicate sample, indicating the dominance of the micron-scale crystalline particles with a smaller fraction of nanoscale particles. The final AC Mg-silicate product is composed of fine particles with a mean particle size of approximately 13 μm, which provides a relatively higher surface-to-volume ratio and increased surface area for CO2 interaction compared to the HC Mg-silicate (25 μm), making it an ideal starting material for subsequent carbon mineralization.
3.2. Carbon mineralization of Mg-silicates
Carbon mineralization of Mg-silicate sorbent is a multiphase process that can be broken down into three key steps: (i) the hydration of gaseous CO2; (ii) the dissolution of Mg-silicate; and (iii) the precipitation of the carbonate products. To elucidate, CO2 hydration leads to the formation of dissolved CO2 species such as carbonic acid (H2CO3), bicarbonate (HCO3−) and carbonate (CO32−) ions (reaction (R2)–(R4)). Simultaneously, Mg-silicate dissolves and continuously releases Mg2+ ions into the solution, which may also lead to the formation of MgSiO3 and silicic acid (H4SiO4) (reaction (R5) and (R6)). H4SiO4 can also undergo dehydration to reprecipitate SiO2 (reaction (R7)). Finally, Mg-carbonate precipitate occurs when Mg2+ ions combine with the carbonate species (reaction (R8)). The overall carbon mineralization pathway in this multiphase environment can be expressed by reaction (R9).| CO2(g) + H2O(l) ↔ H2CO3(aq) | (R2) |
| H2CO3(aq) ↔ H+(aq) + HCO3−(aq) | (R3) |
| HCO3−(aq) ↔ H+(aq) + CO32−(aq) | (R4) |
| Mg2SiO4(s) + 2H+(aq) ↔ MgSiO3(s) + Mg2+(aq) + H2O(aq) | (R5) |
| MgSiO3(s) + 2H+(aq) + H2O(aq) ↔ Mg2+(aq) + H4SiO4(aq) | (R6) |
| H4SiO4(aq) ↔ 4H+(aq) + SiO2(s) | (R7) |
| Mg2+(aq) + CO32−(aq) ↔ MgCO3(s) | (R8) |
| Mg2SiO4(s) + 2CO2(g) ↔ 2MgCO3(s) + SiO2(s) | (R9) |
Thus, the rate and extent of carbon mineralization depend on the multiphase chemical interactions shown in reactions (R2)–(R9). In this study, an initial CO2 partial pressure of 20 atm for the theoretical capacity of Mg-silicate (1/RCO2) of 0.6256 ensures excessive supply of CO2, as predicted by Henry's law.46 Hence, the differences in carbon mineralization extents are likely primarily driven by the distinct dissolution mechanisms of different alkaline Mg-silicates used (AC and HC Mg-silicate), the stirring conditions (300 rpm and no stirring), and the aqueous compositions (H2O and 1 M NaHCO3).
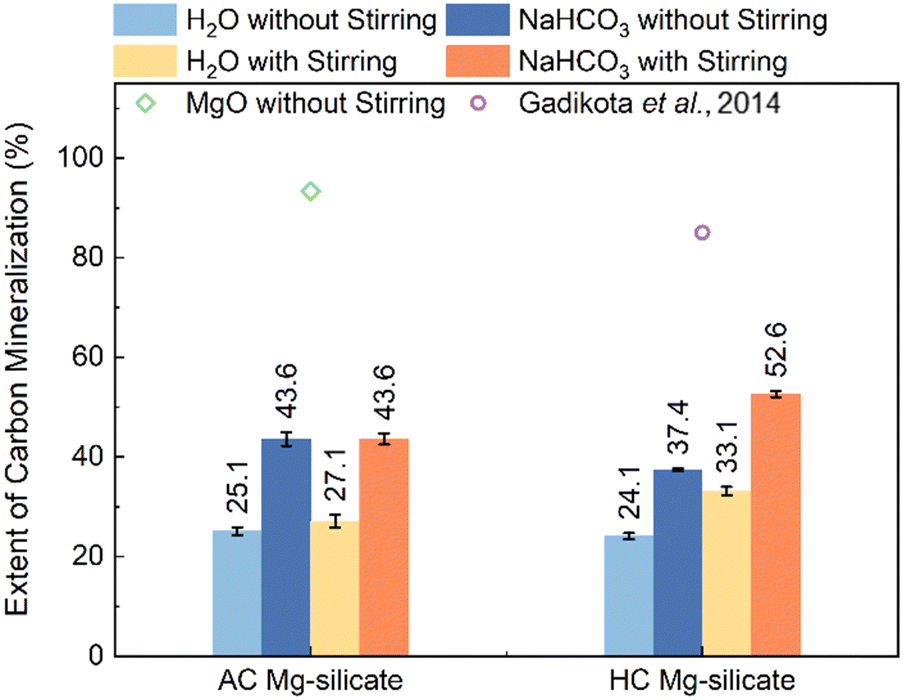 | ||
| Fig. 7 Extents of carbon mineralization of AC (amorphous-crystalline) Mg-silicate and HC (highly crystalline) Mg-silicates reacted at CO2 partial pressure of 20 atm and 200 °C in DI-water and 1 M NaHCO3 solutions, with and without stirring. Additional data corresponding to the extent of carbon mineralization of MgO reacted in DI water without stirring, and olivine reacted at 185 °C, CO2 partial pressure of 139 atm, 1 M NaHCO3 solution are shown.12 The addition of NaHCO3 enhances the extent of carbon mineralization, while the carbon mineralization of HC Mg-silicate is influenced by stirring. | ||
Moreover, as shown in Fig. 7, the addition of NaHCO3 significantly enhances the carbon mineralization extent of AC Mg-silicate, both with and without stirring, by 16.5% and 18.5%, respectively. For HC Mg-silicate, the NaHCO3 enhancement is 19.5% under stirring, and 13.3% without stirring. NaHCO3 solution enhances CO2 uptake and likely acts as a buffer to maintain a relatively stable pH during mineralization which promotes dissolution of Mg-silicate and carbon mineralization to produce magnesite.
Additionally, carbon mineralization studies with MgO are conducted at the same conditions for comparison. Due to its significantly higher reactivity and faster dissolution kinetics compared to silicate samples,47 MgO reached a carbon mineralization extent of 93.3% in H2O even without stirring, and the addition of NaHCO3 had minimal enhancement (Fig. S1, ESI†). However, unlike Mg-silicate minerals, MgO is not as abundantly available on earth. The earth abundance of Mg-silicates and the associated extensive CO2 storage capacity are well-suited for scalable carbon mineralization efforts. Therefore, extensive prior studies were focused on harnessing Mg-silicate rich olivine minerals for carbon mineralization. Extents of carbon mineralization of olivine as high as 85% were achieved on reacting olivine at 185 °C, with a CO2 partial pressure of 139 atm in 1 M NaHCO3 solution for 3 hours.12 These studies show that higher carbon mineralization extents are achieved at elevated CO2 partial pressures. Nevertheless, the observed enhancement in reactivity in the presence of NaHCO3 compared to water is consistent with prior studies.12
3.2.2.1. Si coordination evolution. During silicate dissolution, the release of the metal ions decreases the metal-to-silicon (metal/Si) ratio, which often leads to the formation of a Si-rich layer on the surface of the particles and suppresses further dissolution.48 The formation and dissolution of this Si-rich passivation layer are crucial factors in controlling the dissolution of Mg-silicates, which in turn influences their carbon mineralization behavior. Specifically, HC and AC Mg-silicates exhibit distinct Si coordination, which plays an important role in the formation of these silica-rich layers. To investigate how differences in Si coordination between unreacted HC and AC Mg-silicates impact the phases and compositions of carbonate-bearing products, XPS analysis is employed. The XPS spectra are calibrated using the C 1s peak (284.8 eV).49 Fig. S2 (ESI†) shows the Si 2p deconvolution results for the unreacted Mg-silicate materials and the carbonate-bearing solids under different conditions with stirring. The unreacted AC Mg-silicate sample shows a SiO4 peak at around 102.7 eV, along with an intensified SiO3 peak located at 103.7 eV (Fig. S2(a-1), ESI†). This is likely due to the incomplete crystallization of MgSiO3 and MgO during thermal treatment, which results in the presence of non-crystalline phases like MgSiO3.50 In contrast, the unreacted HC Mg-silicate sample shows a major SiO4 tetrahedra peak at 102.7 eV, with a minor shoulder at approximately 105 eV, indicating the presence of minor quantities of amorphous SiO2 (Fig. S2(b-1), ESI†).51,52
After carbon mineralization, the Si 2p spectra shifts, reflecting phase evolution that influences carbon mineralization. This phase evolution to create Si coordination with more bridging oxygens is observed due to the decreasing Mg/Si ratio and the reprecipitation of dissolved phases on the surface of the Mg-silicate particles, resulting in the formation of –SiO3 and SiO2, respectively. For AC Mg-silicate reacted in water with stirring, the SiO4 peak at 102.7 eV significantly decreases, while the SiO3 peak at 103.7 eV intensifies, and a minor SiO2 peak emerges (Fig. S2(a-2), ESI†). These results indicate that the continuous dissolution of Mg-silicates promotes the formation of Si coordination with more bridging oxygens and the reprecipitation of the amorphous SiO2 layer. The buffering effect of NaHCO3 enables dissolution of Mg-silicate and the precipitation of Mg-carbonate through the availability of carbonate species (Fig. S4, ESI†). Consequently, a higher SiO2 composition and decreased SiO4 content are observed (Fig. S2(a-3), ESI†). Similarly, for HC Mg-silicate reacted in water, a reduced SiO4 peak at 102.7 eV is observed, along with a newly emerged SiO3 peak at 103.9 eV and an enhanced SiO2 peak around 105 eV (Fig. S2(b-2), ESI†).50–52 This suggests a similar Mg-silicate dissolution pathway during the mineralization of both the AC and HC Mg-silicate. The addition of NaHCO3 also leads to a higher SiO2 composition with less SiO4 residue, further indicating enhanced dissolution and carbonate formation (Fig. S2(b-3), ESI†).
In non-stirring conditions, the SiO2 peak around 105 eV is more pronounced, indicating the accumulation of an amorphous Si layer on the particle surface (Fig. S3, ESI†). This Si-rich layer hinders the diffusion of Mg2+ ions and limits the exposure of the unreacted particles to the aqueous phase, thereby restricting further carbonate formation.29 In the absence of stirring, the re-dissolution of this amorphous SiO2 layer becomes slower due to diminished mass transfer and lack of particle collision, making the SiO2 layer dissolution the rate-limiting step in the overall process. The relatively low concentrations of Mg2+ ions in the aqueous phase collected from non-stirring cases provide further evidence of limited dissolution caused by the SiO2 layer (Fig. S4, ESI†).
The compositional changes in different Si coordination states before and after carbon mineralization were also quantified from high-resolution XPS spectra. For AC Mg-silicate, 75% of the Si in the unreacted sorbent is present as SiO3 rather than SiO4 phases based on the quantitative XPS (Fig. 8(a)). The presence of crystalline silica phases bearing SiO4 is confirmed using XRD (Fig. 9(a-1)). After carbon mineralization with stirring, the SiO3 composition increases to 95% in water and 84% in NaHCO3 solution reflecting enhanced Mg2+ dissolution. Notably, in the NaHCO3 case, 16% SiO2 is reported compared to 2% observed with water, highlighting the facilitated dissolution of Mg2SiO4 and SiO2 reprecipitation driven by NaHCO3 (Fig. 8(a)). For HC Mg-silicate, 98% of the Si in the unreacted material is in the SiO4 phase. However, after carbon mineralization in water with stirring, 85% of the Si goes into the SiO3 phase with a minor fraction (5%) in the SiO2 form. In the presence of NaHCO3, SiO2 composition increases to 17%, further confirming the role of NaHCO3 in promoting the dissolution of Mg2SiO4 to release Mg2+ ions and facilitate subsequent carbonate formation (Fig. 8(b)).
 | ||
| Fig. 8 Silicate coordination in unreacted and carbonate-bearing materials where (a), (b), (c), and (d) represent AC Mg-silicate reacted in a stirring mode, HC Mg-silicate reacted in a stirring mode, AC Mg-silicate reacted in a non-stirring mode, and HC Mg-silicate reacted in a non- stirring mode, respectively. Silicate coordination with fewer non-bridging oxygen species emerge after carbon mineralization, indicating the continuous consumption of Mg2+ ions for dissolution or to produce solid carbonates. | ||
 | ||
| Fig. 9 Crystalline phases present in (a-1) unreacted AC Mg-silicate; (a-2) carbonate-bearing AC Mg-silicate reacted in DI-water; (a-3) carbonate-bearing AC Mg-silicate reacted in 1.0 M NaHCO3; (b-1) unreacted HC Mg-silicate; (b-2) carbonate-bearing HC Mg-silicate reacted in DI-water; (b-3) carbonate-bearing HC Mg-silicate reacted in 1.0 M NaHCO3. Experiments were conducted in a stirring mode at 200 °C and CO2 partial pressure of 20 atm. Crystalline phases were determined by X-Ray Diffraction (XRD) measurements. The exclusive carbonate-bearing product is magnesite (MgCO3). | ||
In the non-stirring mode, the SiO4 content remains relatively high in the reacted and carbonate-bearing HC Mg-silicate (32% and 28% with H2O and NaHCO3 solution respectively), indicating that the Si-rich passivation layer limits the release of Mg2+ ions (Fig. 8(d)). In contrast, the transition from –SiO4 to –SiO3 in the carbonate-bearing AC Mg-silicate is still enhanced by 25% in the NaHCO3 case, even without stirring (Fig. 8(c)). These data suggest that Mg2SiO4 dissolves continuously to release Mg2+ ions in the case of AC Mg-silicate despite the accumulation of the Si-rich layer. The enhanced release of Mg2+ ions due to lower crystallinity in AC Mg-silicate is associated with the near complete transformation of SiO4 to SiO3 and SiO2 phases, unlike in HC Mg-silicate.
3.2.2.2. Structural arrangement and morphological analysis. To gain further insight into the dissolution mechanisms and confirm the formation of carbonates, the structural evolution of various Mg-silicates before and after carbon mineralization is determined using X-ray diffraction (XRD) analyses. Fig. 9(a-1) and (b-1) show that the unreacted AC and HC Mg-silicates exhibit characteristic peaks corresponding to crystalline Mg2SiO4 with a minor amount of MgO, possibly caused by surface segregation of Mg2+ cations and the decoupling of MgO and amorphous MgSiO3. The presence of a small quantity of SiO2 in the HC Mg-silicate is also consistent with the XPS results (Fig. S2(a-1), ESI†). The relatively low peak intensities observed for AC Mg-silicate indicate lower crystallinity and the likely co-existence of amorphous phases.
After carbon mineralization with stirring, the carbonate-bearing products exhibit XRD peaks at 32.3°, 42.8°, 46.7°, and 53.9°, which correspond to the (104), (113), (202), and (116) planes of MgCO3, respectively (Fig. 9(a-2), (a-3), (b-2) and (b-3)). Based on the XRD pattern, MgCO3 or magnesite is the only carbonate phase observed, which is consistent with the favourable high temperature and pressure conditions that enable the formation of stable and anhydrous Mg-carbonate phases.53 In addition to the residual Mg2SiO4, all the carbonate-bearing samples (AC Mg-silicate and HC Mg-silicate) show an XRD peak at 20° corresponding to the (003) plane of MgSiO3 and expanded shoulders of amorphous SiO2 at 34.5° and 60.5°. The changes reflect a decrease in the Mg/Si ratio and the formation of SiO2 passivation layers. Notably, the XRD patterns in Fig. 9 reveal that the carbonate-bearing HC Mg-silicate exhibits more pronounced MgCO3 peaks compared to AC Mg-silicate under stirring conditions.
This observation aligns with the higher carbon mineralization extents of HC Mg-silicate indicated by the TGA analysis in Fig. 7. Carbonate-bearing Mg-silicates under no-stirring conditions exhibit similar phase compositions to those under stirring conditions, but with lower MgCO3 peak intensity, indicating a lower content of carbonate (Fig. S5, ESI†). Moreover, additional MgSiO3 peaks have been detected in both carbonate-bearing AC and HC Mg-silicate, indicating Si coordination rearrangement caused by Mg2+ release, which is consistent with the XPS analysis (Fig. 8).
Previous studies have shown that the structural and morphological properties of carbonate-bearing products are strongly influenced by a balance between nucleation and growth during the carbonate formation process.54 Specifically, homogeneous and heterogeneous nucleation dominate under different supersaturation levels of metal cations and carbonate species, impacting the product morphology and extent of carbon mineralization.55 Homogeneous nucleation occurs preferentially when uniform nuclei form in the solution, while heterogeneous nucleation is favoured on existing secondary nucleating surfaces.56 Given the abundance of surfaces available for nucleation in the material systems of interest in this work, carbonate formation due to heterogeneous nucleation is dominant.
To further explore the morphological changes during carbon mineralization, FE-SEM and particle size analyses (PSA) are used to investigate the mechanisms underlying carbonate formation. As shown in Fig. 10(a-1) and (b-1), both unreacted AC and HC Mg-silicates initially exhibit spherical particles. Despite the continuous aggregation and growth of AC Mg-silicate during thermal treatment, its mean particle diameter is 13.05 μm while that of HC Mg-silicate is 25.04 μm (Table S1, ESI† and Fig. 11(a) and (b)).
 | ||
| Fig. 10 Morphological features determined using Scanning Electron Microscopy (SEM) of: (a-1) unreacted AC Mg-silicate; (a-2) carbonate-bearing AC Mg-silicate reacted in DI-water; (a-3) carbonate-bearing AC Mg-silicate reacted in NaHCO3 (b-1) unreacted HC Mg-silicate; (b-2) carbonate-bearing HC Mg-silicate reacted in DI-water; (b-3) carbonate-bearing HC Mg-silicate reacted in NaHCO3. The Mg-silicate materials are reacted at 200 °C and CO2 partial pressure of 20 atm in a stirring mode. Cubic MgCO3 particles are detected in addition to the spherical Mg-silicate residues. | ||
 | ||
| Fig. 11 Particle size distributions of the unreacted and carbonate-bearing silicate materials obtained after carbon mineralization at 200 °C and CO2 partial pressure of 20 atm where (a), (b), (c), and (d) represent samples corresponding to AC Mg-silicate obtained on stirring, HC Mg-silicate obtained on stirring, AC Mg-silicate obtained without stirring and HC Mg-silicate obtained without stirring, respectively. The stirring mode creates uniform carbonate-bearing particles, while the non-stirring mode leads to particles with broader size distribution. | ||
On carbon mineralization with stirring, cubic particles of MgCO3 or magnesite are observed (Fig. 10(a-2), (a-3), (b-2) and (b-3)).57,58 With stirring, the particles smaller than 5 μm in AC Mg-silicate dissolve and Mg-carbonate precipitation occurs which increases the average mean particle size in the presence of water and NaHCO3 (Table S1, ESI† and Fig. 11). In the no-stirring case and in the presence of NaHCO3, the particles larger than 30 μm dissolve and smaller carbonate crystals form resulting in a mean particle size of 9.61 μm compared to the mean particle diameter of 13.05 μm of unreacted AC Mg-silicate. In the presence of water, however, a smaller increase in the mean particle diameter to 14.03 μm is noted in AC Mg-silicate. Particles in the range of 20–40 μm dissolve and the number of particles above 40 μm increase. These results show that varying particle size distributions are obtained with and without stirring. In contrast, the mean particle sizes of the reacted HC Mg-silicate are significantly lower compared to the unreacted material which is 25.04 μm. A significant reduction in the particle sizes above 20 μm due to dissolution is noted when HC Mg-silicate is reacted in all cases. The formation of smaller sized magnesium carbonate particles predominantly occurs below 20 μm in HC Mg-silicate. These results indicate that the dissolution and carbonate formation behaviour differ in HC and AC Mg-silicates.
The observations from the particle size distributions suggest that in the non-stirring mode, heterogeneous nucleation is more likely to occur due to the lower Mg2+ supersaturation level caused by the lack of shear force and mass transfer. Therefore, particles with broader size distribution are expected, as non-uniform nucleation and growth lead to less efficient carbon mineralization.59
As shown in Fig. S6 (ESI†), the cubic particles formed under non-stirring conditions tend to cluster together, further suppressing the carbon mineralization. Notably, as can be seen in Fig. S6(a) and (b) (ESI†), the carbonate-bearing HC Mg-silicate without stirring possesses granular particles on cubic MgCO3, likely due to the presence of residual Mg2SiO4 and reprecipitated amorphous SiO2. The carbonate particle agglomeration observed in the non-stirring cases emphasize the significance of stirring in enhancing particle collision, which reduces the amorphous SiO2 layer and breaks aggregated particles into smaller parts. As shown in Fig. 11(c) and (d) for non-stirring cases, the widespread particle size distribution indicates heterogeneous carbonate formation, especially for the HC Mg-silicate, aligned with the congregated particles observed in Fig. S6 (ESI†). Consequently, compared with the cases with stirring, the no-stirring mode generally results in a lower extent of carbon mineralization due to the limited mass transfer.
In summary, the non-uniform morphology and lower extents of carbon mineralization observed in the non-stirring mode can be attributed to two main factors: (i) the possible accumulation of the SiO2 passivation layer, which reduces the exposed surface area of the unreacted material; and (ii) the prevalence of heterogeneous nucleation, which leads to the formation of congregated carbonate particles that may suppress further carbon mineralization by limiting mass transfer. In contrast, the enhanced carbon mineralization observed in the stirring mode is linked to enhanced mass transfer and the formation of smaller uniform MgCO3 particles. The narrowest size distribution is also associated with the highest carbon mineralization extent observed in HC Mg-silicate with NaHCO3, which is consistent with prior studies.12
3.3. Insights on the Mg-silicate dissolution and carbon mineralization mechanisms
Carbon mineralization is a multiphase process, where the dissolution of alkaline silicate sources is often the rate limiting step due to the relatively slow release rates of metal cations and the formation of a SiO2 passivation layer. Hence, understanding the mechanisms underlying silicate dissolution is essential for tuning carbon mineralization. This study focuses on the effect of particle size and crystallinity on dissolution and carbon mineralization behaviour of HC and AC Mg-silicates. In the presence of stirring, mass transfer is enhanced to promote both dissolution and carbonate formation. Stirring facilitates particle collisions, which help break congregated particles and limit the extensive growth of amorphous SiO2 passivation layers. As a result, the key factors contributing to the release of Mg2+ ions into the aqueous phase are the Mg/Si ratio of the alkaline sources and the localized concentration of Mg2+ ions in addition to the silica passivation layer. Consistent with the carbon mineralization results in Fig. 7, the HC Mg-silicate, with a 3.31% higher Mg content and 0.36 higher Mg/Si ratio, results in a higher Mg2+ supersaturation level, leading to a higher extent of carbon mineralization (Table 2).| Atom% | Mg (%) | Si (%) | O (%) | C (%) | Mg/Si |
|---|---|---|---|---|---|
| AC Mg-silicate | 26.04 | 17.12 | 49.94 | 6.9 | 1.52 |
| HC Mg-silicate | 29.35 | 15.64 | 50.04 | 4.97 | 1.88 |
In contrast, without stirring, the carbonate-bearing particles tend to congregate due to heterogeneous nucleation and limited mass transfer. The reprecipitated SiO2 on the surface of the carbonate-bearing particles limits further release of Mg2+ ions. As a result, the dissolution of Mg2+ ions and the extent of carbon mineralization are controlled by the available reactive surface area, which is influenced by the dissolution rate of the SiO2 layer and the exposure of the Mg-rich layer to the solution.60,61 Specifically, the smaller mean particle size of the unreacted AC Mg-silicate ensures a larger contact area between the external SiO2 layer and the aqueous phase, which facilitates the dissolution of amorphous SiO2 and accelerates the exposure of the Mg-rich core, promoting the release of Mg2+ ions. Moreover, with a higher content of amorphous phases, the AC Mg-silicate lacks long-range order and a rigid lattice structure, favouring Mg2+ release from the alkaline silicate source which leads to elevated Mg2+ concentrations in the aqueous phase, especially for the cases with no stirring (Fig. S4, ESI†). The higher extents of carbon mineralization observed with AC Mg-silicate without stirring are consistent with the enhanced Si and Mg dissolution despite the lower Mg/Si ratio compared to HC Mg-silicate.
4. Conclusions
Unlocking fundamental insights into silica transformations in amorphous and crystalline Mg-silicates is crucial for advancing durable CO2 storage and removal via carbon mineralization. To this end, approaches to architect amorphous and crystalline phases (AC) bearing Mg-silicates and determine the dynamic evolution in the structural and morphological evolution of Mg-silicates on heating as determined using operando USAXS/SAXS/WAXS measurements, are developed. The limited long-range order and less rigid structure in AC Mg-silicate favour the release of Mg2+ ions and enhance carbon mineralization, in non-stirred environments, compared to highly crystalline (HC) Mg-silicate. The extents of carbon mineralization with AC Mg-silicate are 1.0% and 6.2% higher compared to HC Mg-silicate in water and NaHCO3 solutions, respectively. These experiments are conducted at 200 °C and CO2 partial pressure of 20 atm in water and 1 M NaHCO3 solution. Although stirring does not further improve the extents of carbon mineralization of AC Mg-silicate due to its lower Mg/Si ratio, it significantly enhances the carbon mineralization of HC Mg-silicate in both water and NaHCO3 solutions, with improvements of 9% and 15.2%, respectively. These increases in the extents of carbon mineralization are attributed to the enhanced mass transfer and particle collisions caused by stirring, which break silicate particles into smaller pieces and limit diffusion limitations arising from the formation of silica-rich passivation layers.The addition of NaHCO3 is shown to effectively balance the concentrations of Mg2+ and carbonate species, promoting carbonate precipitation and further increasing the mineralization extents. Specifically, NaHCO3 increases the carbon mineralization extents by 18.5%, 16.5%, 13.3%, and 19.5% for AC Mg-silicate without stirring, AC Mg-silicate with stirring, HC Mg-silicate without stirring, and HC Mg-silicate with stirring, respectively. Overall, stirring and addition of NaHCO3 significantly enhance Mg2+ release and carbon mineralization efficiency. The formation of magnesite (MgCO3) at 200 °C, pCO2 of 20 atm in water and 1 M NaHCO3 solution starting from Mg-silicate precursors is consistent with the need for elevated temperature and pressure to facilitate magnesite growth. Narrower particle size distributions of the carbonate-bearing products are associated with higher extents of carbon mineralization. The near complete conversion of SiO4 to SiO3 and SiO2 phases in AC Mg-silicate in the presence of NaHCO3 and in well-stirred environments occurred. These fundamental insights into silicate transformations during carbon mineralization of Mg-silicate inform advances in integrating these methods and processes for scalable industrial carbon management including sustainable production of H2 or the recovery of energy critical metals.
Author contributions
Greeshma Gadikota – conceptualization, methodology, data curation, formal analysis, investigation, project administration, resources, validation, visualization, supervision, funding acquisition, writing – original draft, writing – review & editing; Xun Gao – conceptualization, methodology, data curation, formal analysis, investigation, resources, validation, visualization, supervision, writing – original draft, writing – review & editing; Prince Ochonma – methodology, investigation, data curation, validation, visualization, writing – review & editing. Divya Prasad – investigation, validation, visualization, writing – review & editing; Mahadeo A. Mahadik – investigation, validation, visualization, writing – review & editing; Ivan Kuzmenko – investigation, validation; Jan Ilavsky – investigation, validation.Data availability
The data supporting this article have been included in the main manuscript and as part of the ESI.†Conflicts of interest
Greeshma Gadikota is the co-founder of Carbon to Stone which is commercializing technologies for industrial decarbonization and carbon management. The other authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This material is based upon the work supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Geosciences Program under the Award Number: DE-SC0020263. Shared instruments available through the Cornell Center for Materials Research (CCMR) were used. The authors also gratefully acknowledge the support of Dr Malcolm Thomas and Dr Alicia Tripp at CCMR and Peilong Lu at Cornell University for assisting in this effort. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science user facility at Argonne National Laboratory and is based on research supported by the U.S. DOE Office of Science-Basic Energy Sciences, under Contract No. DE-AC02-06CH11357.References
- S. Bachu, Energy Convers. Manage., 2000, 41, 953–970 CrossRef.
- P. G. Brewer, E. T. Peltzer and F. M. Orr, SPE Annual Technical Conference and Exhibition, SPE, 2001 Search PubMed.
- S. Zhang and Z. Liu, J. Chem. Technol. Biotechnol., 2021, 96, 1475–1495 CrossRef.
- A. Raza, G. Glatz, R. Gholami, M. Mahmoud and S. Alafnan, Earth-Sci. Rev., 2022, 229, 104036 CrossRef.
- G. Gadikota, Nat. Rev. Chem., 2020, 4, 78–89 CrossRef PubMed.
- P. Ochonma, X. Gao and G. Gadikota, Acc. Chem. Res., 2024, 57, 267–274 CrossRef PubMed.
- X. Gao, D. Prasad, M. Mahadik and G. Gadikota, React. Chem. Eng., 2025, 10, 576–592 RSC.
- G. Gadikota, Commun. Chem., 2021, 4, 23 CrossRef PubMed.
- P. Ochonma, C. Blaudeau, R. Krasnoff and G. Gadikota, Front. Energy Res., 2021, 9, 742323 CrossRef.
- T. Yin, S. Yin, A. Srivastava and G. Gadikota, Resour., Conserv. Recycl., 2022, 180, 106209 CrossRef.
- M. Liu and G. Gadikota, Fuel, 2020, 275, 117887 CrossRef.
- G. Gadikota, J. Matter, P. Kelemen and A. H. A. Park, Phys. Chem. Chem. Phys., 2014, 16, 4679–4693 RSC.
- P. Renforth, C.-L. Washbourne, J. Taylder and D. A. C. Manning, Environ. Sci. Technol., 2011, 45, 2035–2041 CrossRef PubMed.
- O. Parlak, M. Delaloye and E. Bíngöl, Geol. Rundsch., 1996, 85, 647 CrossRef.
- A. Sanna, M. R. Hall and M. Maroto-Valer, Energy Environ. Sci., 2012, 5, 7781 RSC.
- C. Wang, L. Jia, Y. Tan and E. J. Anthony, Fuel, 2008, 87, 1108–1114 CrossRef CAS.
- G. H. Wolf, A. V. G. Chizmeshya, J. Diefenbacher and M. J. McKelvy, Environ. Sci. Technol., 2004, 38, 932–936 CrossRef PubMed.
- R. Zevenhoven, S. Teir and S. Eloneva, Energy, 2008, 33, 362–370 CrossRef.
- W. K. O’Connor, D. C. Dahlin, G. E. Rush, S. J. Gerdemann, L. R. Penner and D. N. Nilsen, Aqueous mineral carbonation: mineral availability, pretreatment, reaction parametrics, and process studies. Technical Report, National Energy Technology Laboratory, US DOE, DOE/ARC-TR-04-002 ( 2005).
- K. S. Lackner, C. H. Wendt, D. P. Butt, E. L. Joyce and D. H. Sharp, Energy, 1995, 20, 1153–1170 CrossRef.
- K. S. Lackner, Annu. Rev. Energy Environ., 2002, 27, 193–232 CrossRef.
- M. Liu and G. Gadikota, Fuel, 2018, 227, 379–385 CrossRef.
- S. Katre, P. Ochonma, H. Asgar, A. M. Nair, K. Ravi and G. Gadikota, Phys. Chem. Chem. Phys., 2024, 26, 9264–9283 RSC.
- F. Wang, D. Dreisinger, M. Jarvis and T. Hitchins, Chem. Eng. J., 2021, 404, 126522 CrossRef.
- J. S. Loring, C. J. Thompson, C. Zhang, Z. Wang, H. T. Schaef and K. M. Rosso, J. Phys. Chem. A, 2012, 116, 4768–4777 CrossRef.
- F. K. Crundwell, Hydrometallurgy, 2014, 149, 265–275 CrossRef.
- W. J. J. Huijgen, G.-J. Witkamp and R. N. J. Comans, Environ. Sci. Technol., 2005, 39, 9676–9682 CrossRef PubMed.
- G. Rim, A. K. Marchese, P. Stallworth, S. G. Greenbaum and A.-H. A. Park, Chem. Eng. J., 2020, 396, 125204 CrossRef.
- S. Katre, P. Ochonma, H. Asgar, A. M. Nair, K. Ravi and G. Gadikota, Phys. Chem. Chem. Phys., 2024, 26, 9264–9283 RSC.
- K. P. Sanosh, A. Balakrishnan, L. Francis and T. N. Kim, J. Alloys Compd., 2010, 495, 113–115 CrossRef.
- X. Gao, H. Asgar, I. Kuzmenko and G. Gadikota, Microporous Mesoporous Mater., 2021, 327, 111381 CrossRef.
- J. Ilavsky, F. Zhang, A. J. Allen, L. E. Levine, P. R. Jemian and G. G. Long, Metall. Mater. Trans. A, 2013, 44, 68–76 CrossRef.
- G. Gadikota, Minerals, 2017, 7, 169 CrossRef.
- G. Gadikota, F. Zhang and A. J. Allen, Fuel, 2017, 196, 195–209 CrossRef PubMed.
- J. Ilavsky and P. R. Jemian, J. Appl. Crystallogr., 2009, 42, 347–353 CrossRef.
- J. Ilavsky, J. Appl. Crystallogr., 2012, 45, 324–328 CrossRef.
- S. J. Day, S. P. Thompson, A. Evans, J. E. Parker, L. D. Connor and C. C. Tang, Meteorit. Planet. Sci., 2013, 48, 1459–1471 CrossRef.
- R. Zheng, Z. Ren, H. Gao, A. Zhang and Z. Bian, J. Alloys Compd., 2018, 757, 364–371 CrossRef.
- J. R. Smyth and R. M. Hazen, Am. Mineral., 1973, 58, 588–593 Search PubMed.
- K. P. Sanosh, M. C. Chu, A. Balakrishnan, T. N. Kim and S. J. Cho, Bull. Mater. Sci., 2009, 32, 465–470 CrossRef.
- J. Gomez-Morales, J. Torrent-Burgues and R. Rodriguez-Clemente, Cryst. Res. Technol., 2001, 36, 1065–1074 CrossRef.
- R. T. Chancey, P. Stutzman, M. C. G. Juenger and D. W. Fowler, Cem. Concr. Res., 2010, 40, 146–156 CrossRef.
- A. Guinier and G. Fournet, Small-Angle Scattering of X-Rays, Wiley, 1955 Search PubMed.
- C. D. Putnam, J. Appl. Crystallogr., 2016, 49, 1412–1419 CrossRef PubMed.
- M. Y. Lobanov, N. S. Bogatyreva and O. V. Galzitskaya, Mol. Biol., 2008, 42, 623–628 CrossRef.
- S. M. Klara, Chem. Eng. Commun., 1990, 90, 23–33 CrossRef.
- A. I. Fernández, J. M. Chimenos, M. Segarra, M. A. Fernández and F. Espiell, Hydrometallurgy, 1999, 53, 155–167 CrossRef.
- D. Daval, O. Sissmann, N. Menguy, G. D. Saldi, F. Guyot, I. Martinez, J. Corvisier, B. Garcia, I. Machouk, K. G. Knauss and R. Hellmann, Chem. Geol., 2011, 284, 193–209 CrossRef.
- D. Fang, F. He, J. Xie and L. Xue, J. Wuhan Univ. Technol., Mater. Sci. Ed., 2020, 35, 711–718 CrossRef.
- A. Douy, J. Sol-Gel Sci. Technol., 2002, 24, 221–228 CrossRef.
- I. Yanase, K. Sato, H. Kobayashi, T. Doe and T. Naka, Chem. Eng. J., 2019, 356, 81–90 CrossRef.
- A. Kaur, P. Chahal and T. Hogan, IEEE Electron Device Lett., 2016, 37, 142–145 Search PubMed.
- N. V. Skorodumova, A. B. Belonoshko, L. Huang, R. Ahuja and B. Johansson, Am. Mineral., 2005, 90, 1008–1011 CrossRef.
- D. Wang, C. Xiong, W. Li and J. Chang, ACS Sustainable Chem. Eng., 2020, 8, 14718–14731 CrossRef.
- F. Liendo, M. Arduino, F. A. Deorsola and S. Bensaid, J. Cryst. Growth, 2022, 578, 126406 CrossRef.
- N. T. K. Thanh, N. Maclean and S. Mahiddine, Chem. Rev., 2014, 114, 7610–7630 CrossRef PubMed.
- S. B. Choi, N. W. Kim, D. K. Lee and H. Yu, J. Nanosci. Nanotechnol., 2013, 13, 7577–7580 CrossRef CAS PubMed.
- F. M. Hossain, B. Z. Dlugogorski, E. M. Kennedy, I. V. Belova and G. E. Murch, Solid State Commun., 2010, 150, 848–851 Search PubMed.
- A. M. Bremen, T. Ploch, A. Mhamdi and A. Mitsos, Chem. Eng. J., 2021, 404, 126480 CrossRef CAS.
- H. Béarat, M. J. Mckelvy, A. V. G. Chizmeshya, D. Gormley, R. Nunez, R. W. Carpenter, K. Squires and G. H. Wolf, Environ. Sci. Technol., 2006, 40, 4802–4808 Search PubMed.
- K. Jarvis, R. W. Carpenter, T. Windman, Y. Kim, R. Nunez and F. Alawneh, Environ. Sci. Technol., 2009, 43, 6314–6319 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp04762a |
| This journal is © the Owner Societies 2025 |