
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Gel polymer electrolytes based on sulfonamide functional polymer nanoparticles for sodium metal batteries†
Pierre L.
Stigliano
 *ab,
Antonela
Gallastegui
b,
Thomas H.
Smith
a,
Luke
O’Dell
a,
David
Mecerreyes
bc,
Cristina
Pozo-Gonzalo
ad and
Maria
Forsyth
*ac
*ab,
Antonela
Gallastegui
b,
Thomas H.
Smith
a,
Luke
O’Dell
a,
David
Mecerreyes
bc,
Cristina
Pozo-Gonzalo
ad and
Maria
Forsyth
*ac
aInstitute for Frontier Materials, Deakin University, Geelong, Victoria 3216, Australia. E-mail: pstigliano@deakin.edu.au; maria.forsyth@deakin.edu.au
bPOLYMAT, University of the Basque Country UPV/EHU, 20018, Donostia-San Sebastian, Spain
cIkerbasque, Basque Foundation for Science, María Díaz de Haro 3, 48013, Bilbao, Spain
dInstituto de Carboquímica (ICB-CSIC), Miguel Luesma Castán, 4, 50018, Zaragoza, Spain
First published on 8th January 2025
Abstract
In this work, we investigate the development of polymer electrolytes for sodium batteries based on sulfonamide functional polymer nanoparticles (NaNPs). The synthesis of the polymer NaNPs is carried out by emulsion copolymerization of methyl methacrylate and sodium sulfonamide methacrylate in the presence of a crosslinker, resulting in particle sizes of 50 nm, as shown by electron microscopy. Then, gel polymer electrolytes are prepared by mixing polymer NPs and different organic plasticizers including carbonates, glymes, sulfolanes and ionic liquids. The chemical nature of the plasticizer resulted in different effects on the sodium coordination shell, which in turn impacted the properties of each membrane as investigated by FTIR. The transport properties were investigated by EIS and solid-state NMR. Among the organic gel polymer electrolytes (GPEs), the system comprising NaNPs and sulfolanes achieved the best ionic conductivity (1.1 × 10−4 S cm−1 at 50 °C) and sodium single-ion properties while for the ionogels, the best ionic conductivity was obtained by NaNPs mixed with pyrrolidinium-FSI IL (4.7 × 10−4 S cm−1 at 50 °C). From sodium metal symmetrical cell cycling, the use of ILs as plasticizers proved to be more beneficial for SEI formation and its evolution during cell cycling compared to the systems based on NPs and organic solvents. However, NPs + PC led to lower cell overvoltage than NPs + ILs (<0.4 V vs. >0.5 V). This study shows the potential of using Na-sulfonamide functional polymer nanoparticles to immobilize different plasticizers and thereby obtain soft-solid electrolytes for Na metal batteries.
Introduction
Sodium-based batteries are increasingly becoming a valid alternative to lithium-ion batteries for applications in which high energy density is not crucial. Their benefits include the high natural abundance of sodium, low production cost and lower environmental impact.1–4 Due to their high ionic conductivity and electrode wettability, liquid electrolytes are the most investigated materials for current sodium-based batteries. As much as liquid electrolytes played (and still do) a pivotal role in the advancement and commercialization of batteries, they feature intrinsic hazards, especially leaking and combustion, which could harm end-users.5–10 To avoid such problems, solid-state batteries are being widely investigated, which hold promise for enhanced safety, high-energy and high-power densities. Among solid-state technologies, polymer electrolytes are drawing attention as valid candidates to replace liquid electrolytes, owing to their thermal stability, wide electrochemical window, and limited risk of leakage.11,12 On the other hand, solid polymer electrolytes fail to achieve high ionic conductivity at room temperature and show poor interfacial compatibility with electrodes, hindering their practical application.9,13Gel polymer electrolytes (GPEs) are generally composed of a polymer framework and a liquid electrolyte component. GPEs are becoming widely investigated as they represent a good balance between liquid and solid electrolytes. The polymeric component provides mechanical strength and flexibility, while the liquid electrolyte component, often termed as plasticizer and/or solvent, supports high ionic conductivity and interfacial stability.14–16 GPEs were first studied back in 1975 when Feuillade et al.17 reported polyvinylpyrrolidone/polyacrylonitrile mixed with propylene carbonate (PC) as a gel polymer electrolyte for Li–CuS batteries, however, little interest was shown in investigating GPEs as ‘solid’ electrolytes until the 2000s when this class of materials was shown to have promising properties as quasi solid state devices, and since then GPEs have been drawing increasing attention.18 Nowadays, GPEs are being extensively investigated and the most common polymer frameworks studied are polyethylene oxide (PEO),19–22 polyvinylidene fluoride (PVDF),23–26 poly(vinylidene fluoride-hexafluoropropylene) (PVDF-HFP),27,28 polyacrylonitrile (PAN),29–31 and polymethyl methacrylate (PMMA).32–34 Regarding solvents/plasticizers, the most investigated are cyclic carbonates (EC, PC), glymes or other organic solvents such as acetonitrile, DMF or DMSO.14,18 Ionic liquids (ILs) in combination with polymers are also being investigated within a class of quasi-solid electrolytes defined as ionogels that have been drawing attention thanks to the high safety due to the thermal stability and low volatility and superior chemical and electrochemical properties these materials display.35–37
One main drawback of GPEs and ionogels is their limited mechanical strength. Nanofillers such as nanofibers or nanoparticles can be added to GPEs to reinforce their mechanical properties and thermal stability and improve their ionic conductivity and electrochemical properties.38–40 Inorganic nanoparticles for mechanical reinforcement and ionic conductivity boosting have been used in various systems; however, this kind of nanofiller can suffer from agglomeration and poor homogeneity.41–43 To overcome such problems, the functionalization of the nanoparticle surfaces with polymeric shells or ionic groups was investigated. Moreover, functionalizing the nanoparticles with immobilized anions or polyanions proved to be a viable method for obtaining single-ion conducting electrolytes.44,45 For instance, lithium conducting silica nanoparticles with a lithium sulfonamide polymer brush and investigated in PEO-based composite polymer electrolytes were reported by Bocharova et al.46 Recently, as an alternative to inorganic nanoparticles, functional polymer nanoparticles that are easier to synthesize by well-known emulsion polymerization47 have been investigated.
In a pioneering work, Porcarelli et al.48 reported the use of methacrylic polymer nanoparticles (100 nm size) endowed with lithium sulfonamide surface functionality as a new family of polymer electrolytes by mixing them with poly(ethylene oxide) or propylene carbonate, achieving improved mechanical stability (up to G′ = 106 Pa), promising ionic conductivity values (up to 10−4 S cm−1) and a high transference number (0.8 at 25 °C). More recently, Gallastegui et al.49,50 reported the optimized synthesis of small sulfonamide methacrylic nanoparticles (25 nm) which can be used to develop all-polymer nanocomposite electrolytes as well as 3D printable gel polymer electrolytes for lithium batteries. The application of such functionalised NPs in sodium devices has not been reported as yet and could provide a route to creating solid-state Na metal batteries.
Herein, sodium sulfonamide functionalized methacrylic polymer nanoparticles (NaNPs) were synthesized via simple batch emulsion polymerization of methyl methacrylate and sodium 1-(3-(methacryloyloxy)propylsulfonyl)-1-(trifluoromethylsulfonyl)imide (NaMTFSI). The small NaNPs were mixed with different plasticizers: three organic solvents (sulfolane, propylene carbonate, and diglyme), three ionic liquids ILs, N-butyl-N-methylpyrrolidinium bis(trifluoromethylsulfonyl)imide (C4mpyrTFSI), N-propyl-N-methylpyrrolidinium bis(fluorosulfonyl)imide (C3mpyrFSI), and trimethyl isobutyl phosphonium bis-fluorosulfonyl imide (P111i4FSI), and one hybrid plasticizer (diglyme + C4mpyrTFSI). The polymer electrolytes based on the combination of NaNPs and plasticizers were investigated with the goal of understanding how the sodium ions in the NaNPs interact with different plasticizers; the Na+ ion coordination environment was probed by FTIR while ion transport was investigated using ionic conductivity and NMR dynamic measurements. Interestingly, it was investigated how the chemical nature of the plasticizers changed radically the solvation shell of sodium and the competition of coordination strength between the Na-TFSI and Na-plasticizer. The electrochemical characteristics of the soft solid electrolytes were finally evaluated in sodium metal symmetric cells. To improve the electrochemical properties of organic solvent-based GPEs, especially the SEI formation, two systems including small amounts of sodium bis(fluoro sulfonyl) imide (NaFSI) were investigated, resulting in improved SEI formation and Na symmetrical cyclability.
Materials and characterization
Materials
Methyl methacrylate (MMA) and ethylene glycol dimethacrylate (EGDMA) were purchased from Sigma-Aldrich and vacuum distilled before use. Sodium 1-(3-(methacryloyloxy)-propylsulfonyl)-1-(trifluoromethylsulfonyl)imide (NaMTFSI) was purchased from Specific Polymers and used as is. Ascorbic acid, tert-butyl hydroperoxide (70% solution in water, TBHP) and sodium dodecyl sulfate (SDS) were purchased from Sigma-Aldrich and used as is. Sulfolane (SL), propylene carbonate (PC), and diglyme (DGME) were purchased from Sigma Aldrich and dried over molecular sieves for one week before use. C4mpyrTFSI (Solvionic, 99.9%), C3mpyrFSI (Solvionic, 99.9%), and P111i4FSI (boron molecular, 99.9%) were dried under vacuum at 50 °C overnight.Synthesis of sodium sulfonamide functional methacrylic polymer nanoparticles
Sodium-functionalized polymer NPs were synthesized through emulsion polymerization. This one-pot synthesis was carried out in water at 80 °C in 10 wt% solid content, over 6 h, employing methyl methacrylate and NaMTFSI as comonomers (in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 weight ratio), sodium dodecyl sulfate (SDS) as the surfactant, ascorbic acid and hydrogen peroxide as redox initiator systems, and EGDMA as a crosslinker. A 250 mL flask was filled with 180 g of Milli-Q water and SDS, heated to 80 °C, and purged with N2 for 20 min. In the meantime, three feeding solutions were prepared: one containing NaMTFSI, ascorbic acid, and Milli-Q water (18 g), one containing MMA and EGDMA, the third containing hydrogen peroxide and Milli-Q water. The three solutions were purged with N2 for 15 min at room temperature and subsequently injected into the flask containing water and SDS. The solution was left to stir vigorously for 6 h and then allowed to cool down to room temperature. Fig. 1a schematically illustrates the synthesis of polymer nanoparticles. After polymerization, the solution was passed through an 80 μm nylon mesh to filter coagulated nanoparticles and then dialyzed against Milli-Q water using Spectra-Por 4 membranes (MW cut-off 12000–14000 Da) to remove unpolymerized ionic species. After dialysis, the solution was freeze-dried for one week, until the complete removal of water in the system and a white powder of solid nanoparticles was obtained. The nanoparticles were further dried under vacuum at 50 °C overnight before storing them in an Argon-filled glove box.
:1 weight ratio), sodium dodecyl sulfate (SDS) as the surfactant, ascorbic acid and hydrogen peroxide as redox initiator systems, and EGDMA as a crosslinker. A 250 mL flask was filled with 180 g of Milli-Q water and SDS, heated to 80 °C, and purged with N2 for 20 min. In the meantime, three feeding solutions were prepared: one containing NaMTFSI, ascorbic acid, and Milli-Q water (18 g), one containing MMA and EGDMA, the third containing hydrogen peroxide and Milli-Q water. The three solutions were purged with N2 for 15 min at room temperature and subsequently injected into the flask containing water and SDS. The solution was left to stir vigorously for 6 h and then allowed to cool down to room temperature. Fig. 1a schematically illustrates the synthesis of polymer nanoparticles. After polymerization, the solution was passed through an 80 μm nylon mesh to filter coagulated nanoparticles and then dialyzed against Milli-Q water using Spectra-Por 4 membranes (MW cut-off 12000–14000 Da) to remove unpolymerized ionic species. After dialysis, the solution was freeze-dried for one week, until the complete removal of water in the system and a white powder of solid nanoparticles was obtained. The nanoparticles were further dried under vacuum at 50 °C overnight before storing them in an Argon-filled glove box.
 | ||
| Fig. 1 (a) Schematic representation of the polymeric nanoparticle synthesis and structure, and (b) TEM images of the NaNPs dispersed in Milli-Q water; (c) DSC trace of neat NaNPs; (d) TGA trace of neat NaNPs. | ||
Characterization techniques
Dynamic light scattering (DLS) was used for the measurement of the nanoparticle size and PdI using a Malvern Zetasizer Nano ZS. The nanoparticles were dispersed in water after dialysis and were ri-diluted in 1 mL of Milli-Q water and transferred to disposable polystyrene cuvettes DTS0012. Differential scanning calorimetry (DSC) was employed to detect TG of the nanoparticles using a DSC Q2000 (TA Instruments), DSC scans were performed at heating and cooling rates of 10 °C min−1 from 0 °C to 200 °C. Thermal stability of the nanoparticles was measured via thermogravimetric analysis using a TGA Q500 (TA Instruments), heating 10 °C min−1 under N2 atmosphere from room temperature to 800 °C. Ionic conductivity was measured using electrochemical impedance spectroscopy with an MTZ-35 in the frequency range from 0.1 Hz to 10 MHz (amplitude of 0.01 V) in the temperature range of 30 and 90 °C. The gel polymer electrolyte membranes were cut into 12 mm diameter round discs and sandwiched between two stainless steel electrodes inside of a coin cell. The coin cell was then put in a custom-built barrel cell. All the spectra were fitted using the MT-lab software. 23Na NMR spectroscopy was done using a Bruker Avance III 500 MHz wide bore spectrometer. Samples were loaded in a 4 mm zirconia rotor. All the data were processed using the Topspin software. Full-width half-maximum (FWHM) values were obtained by fitting the peaks with the Gaussian/Lorentzian function. Fourier-transform infrared (FTIR) measurements were performed using a PerkinElmer instrument with a single diamond attenuated reflection unit. The spectra were measured in the region from 4000 to 650 cm−1 and fitted with Gaussian/Lorentzian curves.Electrochemical characterization
Electrochemical characterization was performed using coin cells with the electrolyte membrane (diameter 14 mm) sandwiched between Na metal disc (diameter 10 mm) and stainless-steel spacer. Cells were assembled inside an Ar-filled glovebox and connected to Biologic VMP3 potentiostat, and data were processed using the EC-Lab software. Cells were cycled at 0.02 mA cm−2 and 50 °C.Results and discussion
Synthesis and characterization of Na sulfonamide functionalised methacrylic crosslinked polymer NPs
Sodium-functionalized methacrylic nanoparticles (NaNPs) were synthesized by emulsion copolymerization of sodium sulfonamide methacrylate monomer (NaMTFSI) and methyl methacrylate (MMA), in the presence of a crosslinker (Fig. 1a). In our previous work, we reported the lithium analogue using a lithium-based surfactant. In this case, we introduced a modified sodium version of the monomer, NaMTFSI, and used sodium dodecyl sulfate as the surfactant. The copolymerization of MMA and NaMTFSI was carried out in water at 80 °C with a 10 wt% solid content for 6 h, employing ascorbic acid and hydrogen peroxide as the redox initiator system and EGDMA as the crosslinker. The composition of poly(MMA78-co-NaMTFSI22) is shown in the experimental section. After purification by dialysis, the latexes were freeze-dried and polymer NPs were obtained as fine powders in high weight conversion (>75 wt%). The final weight of NPs was used to calculate the conversion rate (75%). The Na content was investigated via ICP, resulting in 28.81 mg(Na) per g(NPs), while DLS was used to obtain the nanoparticle size (50 nm) and polydispersity index (PdI) (0.190).Nanoparticles were initially characterized by DLS, TEM, DSC, and TGA. The size of the nanoparticles was found to be of 50 nm and the sample showed a low PdI (Table S1, ESI†). From TEM imaging (Fig. 1b) the size of the polymer nanoparticles was confirmed, and low aggregation of the nanoparticles was observed. Regarding the thermal properties of the polymer nanoparticles, they are characterized by a single glass transition temperature (Tg) with onset at 145 °C, as shown by the DSC traces (Fig. 1b), and they are thermally stable up to 260 °C, where the TGA traces (Fig. 1c) shows the one-step degradation reaching a residual weight of 20 wt%.
The polymer nanoparticles were further characterized via FTIR to confirm the composition and the presence of the two comonomers in the NaNPs. In Fig. S1 and Table S1 (ESI†), the FTIR absorbance spectrum of the neat polymer nanoparticles is provided. The main peaks are highlighted in Fig. S1b and c (ESI†) for the two components NaMTFSI and MMA in blue and red, respectively. Analysing the FTIR spectrum, it was also possible to further understand the various interactions within the polymer nanoparticles, in particular the deconvolution of the spectral regions related to TFSI and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O suggested an unexpected coordination of the Na+ ion. In particular, whereas we might have expected all the anions attached to the NP to be coordinated with the Na+ cation, the IR spectra in Fig. S1 (ESI†) would indicate that some TFSI ions remain uncoordinated (‘free’) as observed from the vibrations at 740 and 760 cm−1, while additional coordination of the MMA CO with Na ions is visible from the 1714 cm−1 vibration.
O suggested an unexpected coordination of the Na+ ion. In particular, whereas we might have expected all the anions attached to the NP to be coordinated with the Na+ cation, the IR spectra in Fig. S1 (ESI†) would indicate that some TFSI ions remain uncoordinated (‘free’) as observed from the vibrations at 740 and 760 cm−1, while additional coordination of the MMA CO with Na ions is visible from the 1714 cm−1 vibration.
The coordination of Na+ ions in the polymer nanoparticles regulates how its transport will occur in polymer membranes when plasticizers, such as organic solvents or ILs, are mixed with the nanoparticles to obtain gel polymer electrolytes. This coordination mechanism could also help understand the physiochemical and electrochemical properties of the gel polymer electrolytes. The polymer nanoparticles were thus investigated in combination with different organic solvents (sulfolane, propylene carbonate, diglyme), an organic/IL hybrid solvent (diglyme/C4mpyrTFSI), and ILs (C4mpyrTFSI, C3mpyrFSI, P111i4FSI).
General procedure for preparation of gel polymer electrolytes based on sodium-functionalized polymer nanoparticles
Gel polymer electrolytes were simply prepared by mixing the NaNPs with a plasticizer in a given ratio. Fig. 2 shows the representative scheme of the polymer electrolyte preparation employing different plasticizers. As a result, flexible polymer films were obtained. The ratio between the polymer NPs and the plasticizer was 50 wt%, which was the optimized composition for having free standing flexible membranes. Polymer electrolytes were also prepared using carbonates commonly used in battery electrolytes as plasticizers such as SL, PC, DGME, one organic-IL hybrid electrolyte DGME and C4mpyrTFSI (molar ratio DGME:C4mpyrTFSI of 4:1) and different ILs (C4mpyrTFSI, C3mpyrFSI, and P111i4FSI) (Fig. 2). All membranes were prepared with 50 wt% NaNPs and 50 wt% plasticizer. It is worth noting that the only source of sodium in these gel polymer electrolytes comes from the sodium sulfonamide attached to the polymer NP without the addition of extra sodium salt.
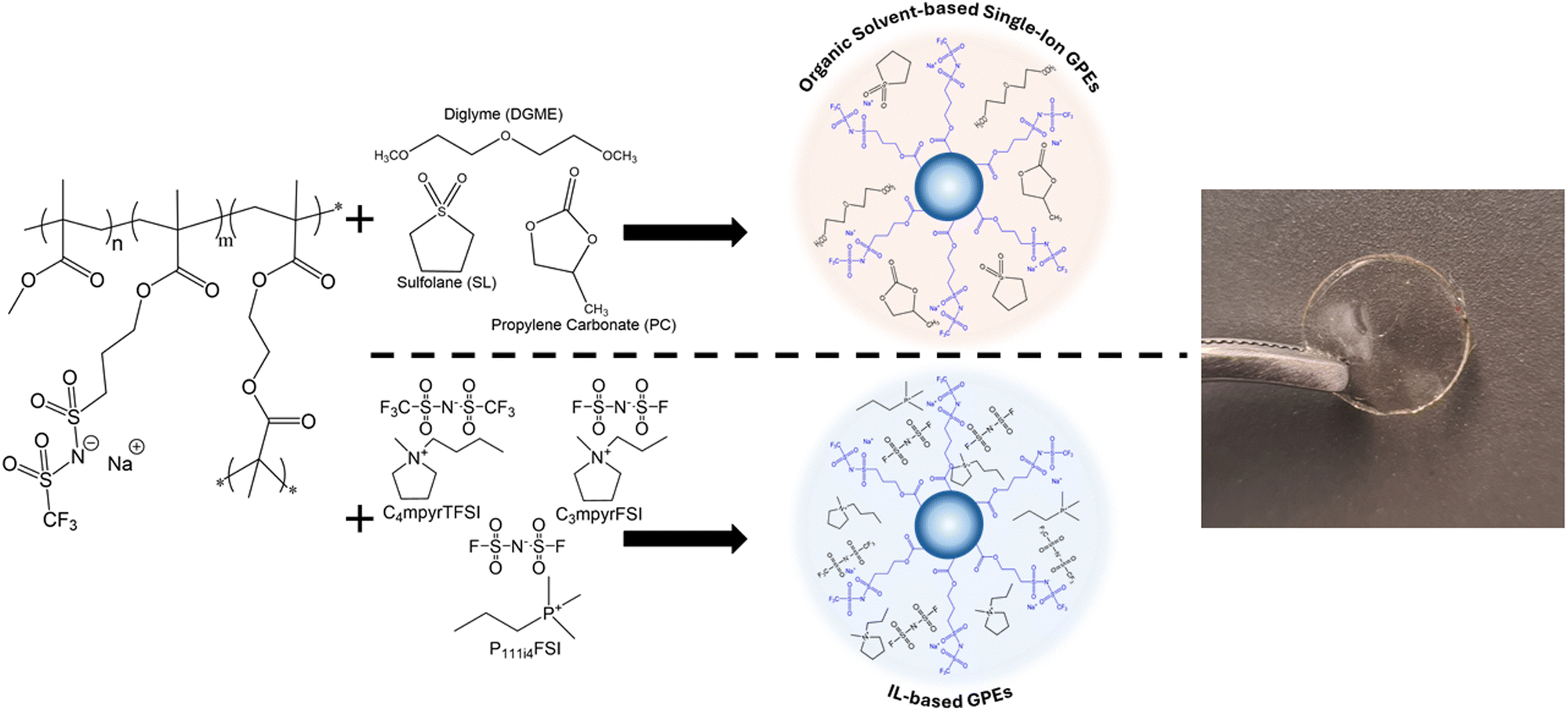 | ||
| Fig. 2 Preparation of gel polymer electrolytes based on sodium-functionalized polymer nanoparticles and different plasticizers. All membranes were prepared with 50 wt% of NaNPs and 50 wt% of plasticizer. | ||
Gel polymer electrolytes based on sodium-functionalized polymer nanoparticles and organic or hybrid organic/ionic liquids as plasticizers
First, we investigated the gel electrolytes prepared by mixing the NPs with organic solvents as plasticizers. The ionic conductivities of the polymer electrolytes are shown in Fig. 3. The highest conductivities were obtained for NPs + hybrid and NPs + SL, with values at 50 °C of 1.2 × 10−4 and 1.1 × 10−4 S cm−1, respectively. NPs + PC resulted in almost one order of magnitude lower values, with an ionic conductivity of 3.4 × 10−5 S cm−1. The lowest conductivity was obtained for NPs + DGME, having σ = 1.3 × 10−6 S cm−1. It must be remembered that in these systems, apart from NPs + hybrid, the anions are attached to the backbone of the polymer nanoparticles, meaning that the ionic conductivity only stems from the free Na ions making these systems single-ion conductors. Compared to similar systems based on single-ion Li-functionalized polymer nanoparticles mixed with PC,48 the systems investigated in this work are slightly less conductive. However, the coordination of the sodium ion, compared to lithium-ion, could be influencing the ion conduction in the systems. The conductivities were fit using the Arrhenius equation to calculate the activation energy (EA) of each system and the values obtained are summarized in Table S2 (ESI†). NPs + SL, NPs + PC, and NPs + hybrid obtain similar activation energies around 50 kJ mol−1, while EA for NPs + DGME was sensibly higher (72 kJ mol−1). By comparison with the similar work on Li single-ion polymer nanoparticles by Porcarelli,48 the NaNPs investigated here show comparable ionic conductivity at 50 °C but have higher activation energy (≈50 vs. ≈35 kJ mol−1). On the other hand, when comparing the results obtained here with a more recent work by Gallastegui et al.49, it appears that NPs + SL achieves slightly higher ionic conductivity than its lithium counterpart (1.1 × 10−4vs. 8.0 × 10−4 S cm−1). This result can be explained by the higher sulfonamide content in the present work compared to the cited reference. These results show how the coordination of the Na ion in the polymer nanoparticles influences the properties and conduction mechanism in the gel polymer membranes investigated. For this, a deeper look into the Na coordination and solvation shell was carried out and the results are shown below.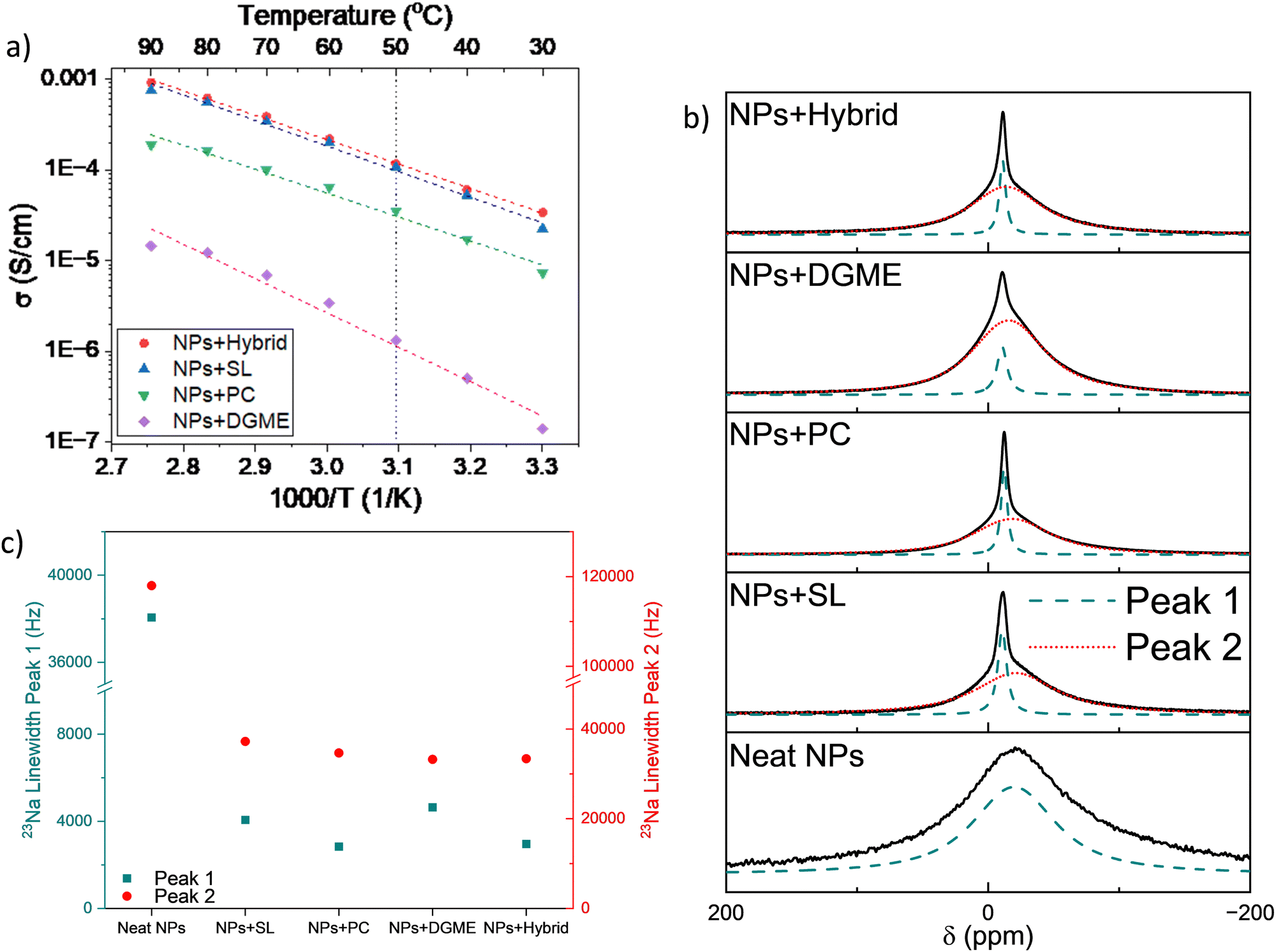 | ||
| Fig. 3 (a) Arrhenius plot for the gel polymer electrolytes based on sodium-functionalized polymer nanoparticles and organic or hybrid solvents; (b) 23Na NMR spectra for the gel polymer electrolytes based on sodium-functionalized polymer nanoparticles and organic or hybrid solvents and (c) full width half-maximum (FWHM) frequencies of 23Na. | ||
23Na NMR allows a better understanding of the solvation and local dynamics of the sodium ion in the systems. The 23Na NMR spectra of neat nanoparticles and organic/hybrid plasticizer-based GPEs at 60 °C are shown in Fig. 3b. The spectra of all membranes after deconvolution show two different peaks, one narrower positioned between −10 and −12 ppm (called Peak 1 from now on) and a broader one with a position ranging from −15 to −20 ppm (called Peak 2 from now on). The presence of multiple populations of Na ions with different mobility stemming from different environments has been already reported in polymer electrolytes.51,52
Erlich et al. showed that in 23Na NMR investigations, it is possible to relate the sodium chemical shift to the Gutmann donor number (DN) of the solvating ligands, with lower DN values a more upfield (negative) the shift is observed.53 In NaFSI/glyme electrolytes, Geysens et al. reported a dramatic upfield shift at higher salt concentrations given by the substitution of stronger coordinating glymes with weaker coordinating FSI anions.54 Thus, from the chemical shift of the peaks observed it is possible to have an idea of the coordinating strength in the different systems investigated. The chemical shifts for Peak 1 for NPs + SL, NPs + PC, NPs + DGME, and NPs + hybrid are −10.7, −12.2, −10.6, and −11.0 ppm, respectively. This could suggest a solvation shell in the NPs + PC system is characterized by weaker coordination, which could result in lower ionic conductivity.54 This hypothesis follows the ionic conductivity values shown in Fig. 3a, where NPs + PC has a lower ionic conductivity compared to NPs + SL and NPs + hybrid. However, the less upfield peak position of NPs + DGME compared to the other systems is in contrast with this interpretation. The lower upfield peak position of Peak 1 but the much lower ionic conductivity of NPs + DGME could be explained by the influence of two other factors as discussed further below.
The full width at half maximum (FWHM) is connected to the relaxation time NMR T2, which is in turn correlated to the mobility of a species. Generally, the narrower the peak width, the more mobile the species is, although this factor can be influenced by the exchange rate between different physical or chemical coordinating sites. In Fig. 3c the FWHM values in the different systems for both Peak 1 and Peak 2 are reported. NPs + DGME results have the widest Peak 1 FWHM, thus suggesting that the Na ions are less mobile compared to the other systems, especially NPs + PC and NPs + hybrid. Moreover, the population represented by Peak 2 in the systems generally shows FWHM values much higher than Peak 1, suggesting that this population is dramatically less mobile than the population connected to Peak 1. In the case of NPs + DGME, this Peak 2 population greatly contributes to the total sodium population, as the peak intensity is higher than Peak 1, while for the other systems Peak 1 is the most intense.
The FWHM of Peak 2 does not vary as much across the samples, while the chemical shift does. NPs + SL, NPs + PC, NPs + DGME, and NPs + hybrid show Peak 2 peak position at −20.9, −18.1, −15.7, and −13.6 ppm, respectively. Relating to the discussion above, generally, we can suggest that this population is more weakly coordinated to sodium, and NPs + SL appears to have the weakest coordination, while in NPs + DGME and NPS + hybrid, the “slow” sodium population is more strongly coordinated. This could explain the higher conductivity of NPs + SL compared to NPs + PC and NPs + DGME. The high ionic conductivity of NPs + hybrid is in contrast with the proposed discussion; however it must be remembered that in this system, the introduction of C4mpyrTFSI adds free charge carriers (as it will be discussed later in the FTIR spectra), thus the ionic conductivity of NPs + hybrid is not related only to the diffusion of Na ions.
These results already start shedding a light on the difference in coordination of sodium ions in the various systems, and a deeper understanding of the solvation shells is needed to better understand how the Na ions move in the different materials investigated.
Various studies have been undertaken to understand the coordination of metal ions (i.e. Li or Na) within different systems. First, the coordination of the different plasticizers with the Na ions is discussed, later the focus will be given to the change in coordination of the CO and TFSI anions in the nanoparticles. For each plasticizer, a specific spectral region was chosen to understand its behaviour in the systems investigated, as shown in Fig. 4. The chosen FTIR regions were picked to examine the predominant changes in vibration modes of the sodium-coordinating component in each organic/hybrid plasticizer. This allows for a better comprehension of the coordination type of sodium ions. The absorbance region chosen for SL is specifically associated with the scissoring vibration mode of the SO2 molecule.55 In the case of PC, the region between 1680 and 1860 cm−1 shows changes in the stretching vibration of the CO bond.56 As for DGME and hybrid, the selected FTIR region indicates the coordination of DGME with sodium ions through the OCCO moiety.57
 | ||
| Fig. 4 FTIR absorption spectra for the analysis of the coordination with Na+ ions in (a) NPs + SL; (b) NPs + PC; (c) NPs + DGME; (d) NPs + hybrid; the bands are related to the solvent vibrations. All membranes were prepared with 50 wt% of NaNPs and 50 wt% of plasticizer. | ||
In SL-based electrolytes using Li salts with weakly coordinated anions such as TFSI, the diffusion of metal ions appears to be dominated by ion hopping/exchange. Watanabe et al.58,59 reported that this mechanism is the result of a mix between monodentate configuration and an SL-bridged chainlike lithium-ion coordination, where two Li ions coordinate with two oxygen atoms from the SO2 moiety. The latter type of coordination was observed to be mainly present in the more concentrated SL-based electrolyte (SL/LiTFSA, x < 3). To understand the type of coordination of the Na with SL in this work, we investigated the same vibrational mode in the spectral region 540–600 cm−1 attributed to the scissoring vibration mode of SO2. As shown in Fig. 4a, a main peak at 568 cm−1 and a minor peak at 551 cm−1 appeared. Watanabe et al. observed that upon addition of Li salts, the peak around 550 cm−1 appeared, concomitant to the broadening and shift of the peak at 568 cm−1 to higher wavenumbers, the magnitude of the shift depending on the predominant Li-SL type of coordination between monodentate and bridging. From the position of the main peak observed in the NPs-SL membrane, we can infer that, in our system, the coordination of sodium with SL occurs mainly via monodentate interactions. However, due to the broadening observed for this peak, the bridging-type coordination cannot be completely excluded.
When mixed with metal ion (i.e. Li, Na) salts, it was previously shown that PC coordinates via the carbonyl oxygen (CO), and the coordination can be either with the metal ion or the anion from the salt. Upon addition of LiPF6, Yang et al.60 observed how different Li(PC)+ complexes form at different concentrations of Li salt. In more dilute solutions, the dominant complex is Li(PC)4+, while at higher concentrations, the anion from the salt enters the Li solvation shell and Li(PC)2(anion)2− becomes the dominant complex. The Li solvation shell comprising also the anion resulted in a lower number of coordinated PC molecules and lower conductivity. Lucht et al.56 observed the change in the IR spectrum of LiPF6/PC electrolytes in the spectral region of the CO, with a decrease of intensity of free PC bands at 1789 and 1800 cm−1 and the appearance of coordinated PC bands at 1770 and 1752 cm−1. The change in the IR spectrum was parallel to the change in the solvation shell of Li, shifting to anion-comprising complexes when the coordinated PC band was increasing. In the neat PC absorbance spectrum (Fig. S2, ESI†), we observe a peak at 1780 cm−1, which relates to the CO stretching vibration.61 Interestingly, in the NPs + PC membranes, differences with the literature are observed for the absorption bands. In NPs + PC (Fig. 4b), three peaks are observed at 1725, 1786, and 1812 cm−1. The band at 1725 cm−1 was previously assigned to the CO environment in the neat nanoparticles, thus related to MMA or MTFSI anion, and will be discussed later. The band at 1786 cm−1 is compatible with the presence of a free PC, as observed in the literature. The PC:Na mol ratio in this system is 8:1, and this could suggest that the solvation shell of Na in this system could include the TFSI anion from the polymer backbone. The NPs + PC gels had an ionic conductivity of 3.5 × 10−5 S cm−1 at 50 °C. This means that, although the anion (anchored to the polymer backbone) participates in the solvation of Na ions, the cation can still diffuse, although more understanding of the transport mechanism is needed.
Previous studies on the coordination of DGME Li- or Na-based electrolytes show that one or two organic solvent molecules can coordinate with the Na in ether crown complexes, leading to different solvated species for Na+.54,62 All the coordination shells for Na+–DGME stem from modification of the backbone conformation dependent on different torsional angles of the OCCO in the DGME molecule.57,63 It is possible to investigate the different conformers of the OCCO chain in the spectra region 800–900 cm−1. In pure diglyme, previous works observed the occurrence of three bands, which are 824 and 879 cm−1 (trans conformer) and 853 cm−1 (gauche conformer).64 When coordinating with Na+ ions, the gauche conformer band at 853 cm−1 splits into two bands at higher and lower wavelengths, and these two new bands can thus be related to coordinated DGME. By deconvoluting the absorbance spectrum of NPs + DGME (Fig. 4c) three main bands were found in the spectral region 800–900 cm−1, at 811, 840, and 864 cm−1. No presence of peaks related to free DGME was observed, suggesting that all the DGME molecules are coordinated. The DGME:Na mol ratio in the membrane is 6:1, this could suggest that, in this system, the preferential solvent shell for sodium is –[Na(diglyme)2]–, where two diglyme molecules form a cage surrounding the sodium ion. The Na–diglyme complex, being positively charged and thus forming a cation, could be still coordinated with the TFSI on the polymer backbone, resulting in significantly lower diffusion. However, the amount of diglyme present in the system is in excess for this type of coordination to sodium, suggesting that DGME also coordinates with the anion in the polymer backbone. As previously observed, NPs + DGME resulted in having the lowest conductivity and highest activation energy among all samples, thus the type of solvation and coordination of DGME in this system hinders the diffusion of Na.
In hybrid systems such as NaTFSI in DGME/C4mpyrTFSI, Garcia-Quintana et al.57 investigated the coordination of the Na ion at different concentrations of Na salt. From computational experiments, they investigated the solvation structure. The calculated solvation structure at lower concentrations is –[Na(DGME)2]+– while in more concentrated electrolytes the solvation structure becomes more complicated and diversified due to the introduction of the TFSI anion in the shell. To understand the coordination of Na ions in the hybrid system of this work, the peaks related to the torsional angle of OCCO in DGME were investigated in addition to the cis/trans conformations of TFSI anions. The latter, related to the TFSI anion, is already present in the polymer nanoparticles, thus making the analysis complicated as will be discussed later. In the OCCO-related spectral region of NPs + hybrid (Fig. 4d) more bands are observed compared to the NPs + DGME spectrum. Four peaks result from the deconvolution of the spectral region 800–900 cm−1 of NPs + hybrid. These include the splitting of the gauche conformer band (853 cm−1) into two bands at 865 and 839 cm−1 while the band at 817 cm−1 could be related to the presence of free uncoordinated DGME in the system. Note that the calculated mol ratio between DGME, IL, and Na is 3:1:1. The presence of free DGME in the FTIR peaks suggests that in this system the solvation shell of Na ions is composed of DGME and TFSI. This will be further discussed in the analysis of the spectral regions connected to the TFSI anion.
As shown in the FTIR analysis of the neat nanoparticles, the Na+ ion in the polymer nanoparticles is originally coordinating with the TFSI anion and the CO from either MTFSI or MMA. When mixing the polymer nanoparticles with organic or hybrid plasticizers, these coordinate as well with sodium, suggesting the change in the environment for TFSI and CO from the NPs. To investigate this change in the Na+–TFSI/CO coordination, the absorbance spectral regions assigned to these environments were investigated and shown in Fig. 5.
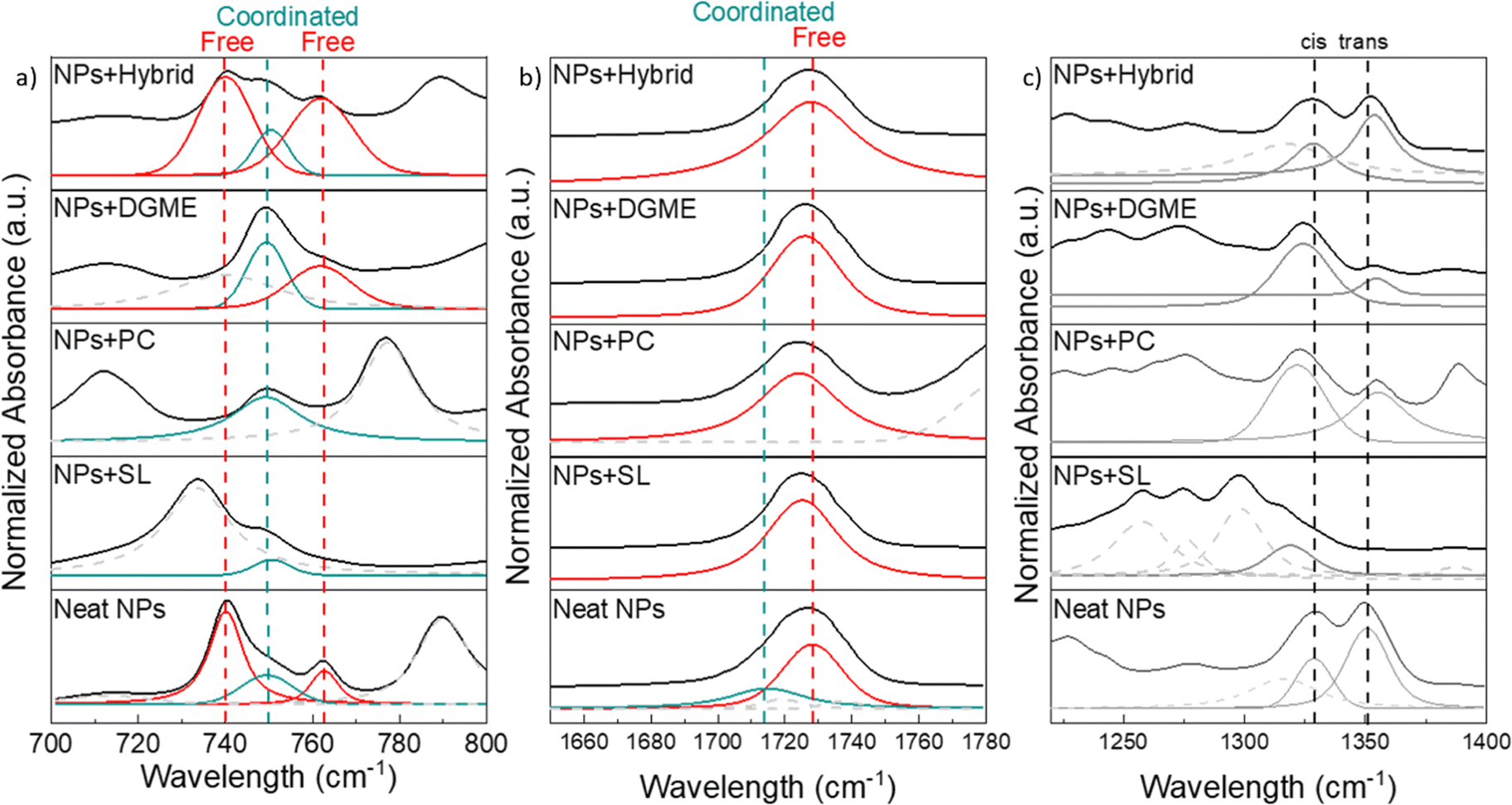 | ||
| Fig. 5 FTIR absorption spectra of neat nanoparticles and NPs + organic/hybrid membranes for the regions of TFSI anion (a), CO (b), and cis/trans conformers of TFSI (c). | ||
Fig. 5a shows the TFSI anion coordination for the organic/hybrid-based membranes against the neat nanoparticles. In all membranes but NPs + hybrid the peak at 740 cm−1, the main peak assigned to the free TFSI anion in the neat NPs, disappears, while the peak at 749 cm−1, related to the coordinated TFSI anion is still present. We suggest that the presence of the free TFSI anion peak at 740 cm−1 in the NPs + hybrid system stems from the introduction of free TFSI anions of the pyrrolidinium ionic liquid (C4mpyrTFSI).
The coordination of the CO also changes when the NPs are mixed with solvents. In Fig. 5b the peak related to the coordinated CO disappears and only the free CO mode remains. These observations for the coordinating environments on the NPs suggest that the added solvents facilitate the dissociation of Na+ from both the MMA CO group as well as from the bound TFSI on the NP.
From the deconvolution of the spectral region related to νas (SO2) and the cis/trans conformations of the TFSI anion (1300–1400 cm−1, Fig. 5c), additional insights into the coordination of the anion and the change in the polymer backbone of the NPs can be found. In the sodium-functionalized polymer nanoparticles, the trans conformation of TFSI is more dominant than the cis. However, when the solvent is incorporated with the NPs, different behaviours are observed. In NPs + SL, the peaks seem to disappear or dramatically shift. From the deconvolution, only the peak related to the cis conformer could be seen at lower wavelengths, while the peak related to the trans conformer could not be identified. In NPs + PC and NPs + DGME, the cis conformer becomes more dominant. In NPs + hybrid the trans conformer is the dominant configuration for the TFSI anion. The cis conformer being dominant could suggest that, in the system where this behaviour is observed, Na clusters65,66 are formed incorporating both the organic solvent and the TFSI from the polymer nanoparticles, in accordance with previous observations. In NPs + hybrid the trans conformer appears to be the most present, suggesting that the IL anion introduced does not extensively interact with the sodium, thus remaining in the trans conformational state.
The analyses for the NPs + organic/hybrid membranes show that each solvent induces unique coordination states for the metal ion. This impacts the mobility of sodium, as seen from ionic conductivity and NMR, and can reflect on the electrochemical properties of the various systems, as will be shown later.
Gel polymer electrolytes based on sodium-functionalized polymer nanoparticles and ionic liquids
The three ILs mixed with the polymer NPs in 50 wt% ratios to prepare the ionogel membranes were C4mpyrTFSI, C3mpyrFSI, and P111i4FSI, as shown in Fig. 2.The ionic conductivity of the membranes was evaluated in the range of temperature 30–90 °C, and the results are shown in the Arrhenius format in Fig. 6. NPs + C3mpyrFSI and NPs + P111i4FSI obtained ionic conductivity values, reaching 4.7 × 10−4 and 1.2 × 10−4 S cm−1, respectively, at 50 °C while NPs + C4mpyrTFSI resulted in two orders of magnitude lower conductivity (4.8 × 10−6 S cm−1). Compared to the GPE membranes based on organic/hybrid solvents, the IL-based systems show in general higher conductivity. However, these systems are no longer single-ion conductors, as the ILs are also charge carriers. The higher conductivity obtained in the case of NPs + C3mpyrFSI and NPs + P111i4FSI could be related to the lower viscosity of C3mpyrFSI and P111i4FSI compared to C4mpyrTFSI. Nevertheless, as discussed in the case of the GPEs above, the coordination of the IL solvents with the Na and the polymer NPs deeply influences the transport mechanism. This aspect will be further investigated below.
 | ||
| Fig. 6 (a) Arrhenius plot for the gel polymer electrolytes based on sodium-functionalized polymer nanoparticles and ionic liquids; 23Na NMR spectra for the gel polymer electrolytes based on sodium-functionalized polymer nanoparticles and ionic liquids (b) and full width half-maximum (FWHM) frequencies of 23Na (c). | ||
Fig. 6b and c show the results of the 23Na NMR for the NPs + IL membranes. As observed for the NPs + organic/hybrid plasticizers, two different sodium populations are observed, one more mobile (narrower FWHM) at lower chemical shifts and one less mobile (broader FWHM) at higher chemical shifts. The two peaks in the NPs + ILs membranes are centered between −12 and −14 ppm (Peak 1) and between −18 and −23 ppm (Peak 2).
As expected, the position of the two peaks generally occurs at higher fields compared to the NPs + organic/hybrid systems as a result of the lower DN of ILs containing TFSI and FSI.54,67–69
Interestingly, NPs + C3mpyrFSI gave a lower 23Na chemical shift for Peak 1 at −12.6 ppm, followed by NPs + P111i4FSI (−13.5 ppm) and NPs + C4mpyrTFSI (−13.6 ppm), while for Peak 2 the chemical shift follows the trend NPs + C4mpyrTFSI (−18.2 ppm) < NPs + C3mpyrFSI (−22.4 ppm) < NPs + P111i4FSI (−23.8 ppm). From this analysis, it appears that the mobile Na population in the NPs + IL systems is more strongly coordinated in FSI systems than in TFSI systems, which should result in lower conductivity of the former. Indeed, as shown in Fig. 6a, NPs + C4mpyrTFSI shows ionic conductivity values two orders of magnitude lower than the other two systems. However, a discrepancy between NMR studies and ionic conductivity is observed from the analysis of FWHM of Peak 1. Surprisingly, NPs + C4mpyrTFSI appears to have the narrowest Peak 1 among the NPs + IL membranes, while NPs + P111i4FSI and NPs + C3mpyrFSI show higher FWHM around 40000 Hz, 4 times higher than NPs + C4mpyrTFSI. However, as observed previously for the NPs + organic/hybrid systems, the intensity of Peak 1 vs. Peak 2 follows the ionic conductivity trend. In the case of NPs + C4mpyrTFSI Peak 1 is less intense than Peak 2, while the other NPs + IL systems display opposite behaviour, thus explaining the higher conductivity of NPs + C3mpyrFSI and NPs + P111i4FSI.
As previously mentioned, the coordination of Na+ ions in the neat nanoparticles is shared between the TFSI anion and CO. FTIR absorbance spectra were obtained for the NPs + ILs membranes to analyze the influence of the ILs on sodium coordination as discussed for the GPEs above. Because of the presence of TFSI and FSI, the characteristic peaks of ILs overlap with those from the neat nanoparticles, complicating the analysis. Fig. 7 shows the spectral region for TFSI anion, CO, and the cis/trans conformers of TFSI anion for the neat nanoparticles and the ILs-based membranes.
 | ||
| Fig. 7 FTIR absorption spectra of neat nanoparticles and NPs + ILs membranes for the regions of TFSI anion (a), CO (b), and cis/trans conformers of TFSI (c). | ||
In the spectral region 700–800 cm−1 (Fig. 7a), the behaviour of the polymer-bound TFSI anion varies greatly depending on the nature of both the cation and anion of the IL used as a plasticizer in the system. In the case of pyrrolidinium ILs (C4mpyr and C3mpyr), the coordinated TFSI anion is the predominant contribution to the TFSI population in the system, in the case of C3mpyrFSI showing no free TFSI peaks while C4mpyrTFSI still shows the secondary uncoordinated TFSI peak at 762 cm−1. P111i4FSI on the other hand shows a significant contribution from the uncoordinated TFSI peaks of the polymer-bound anion, although the coordinated peak is also present.
In the 700–800 cm−1 spectral region peaks related to the FSI anion, coordination can be also found. In FSI-based ILs it was observed that the addition of metal salts such as LiFSI/LiTFSI would present a peak around 740 cm−1 while the ‘free’ FSI peak at 725 cm−1 lowers in intensity. In NPs + C3mpyrFSI and NPs + P111i4FSI we see different behaviours. In NPs + C3mpyrFSI, a peak at 725 cm−1 is observed, suggesting the presence of free FSI anion, which is in accord with the presence of only the broad coordinated TFSI at 749 cm−1 and no uncoordinated TFSI. In contrast, in the NPs + P111i4FSI system, the peak related to free FSI anion at 725 cm−1 could not be observed. The peak at 740 cm−1, which for the TFSI anion depicts an uncoordinated state, could actually be related to the coordinated FSI anion from the IL, which could also explain the larger width compared to the free TFSI peak observed for the neat NPs, as the FSI anion has higher dynamic disorder than TFSI.70,71 In this case, the coordination of the sodium in the two systems would be different, where in NPs + C3mpyrFSI sodium coordinates mainly with the polymer-bound TFSI anion, while the solvation shell of sodium in NPs + P111i4FSI is formed by a mix of TFSI and FSI. This is in accordance with the NMR observations, where the peak position of Peak 1 in NPs + C3mpyrFSI is at lower values than NPs + P111i4FSI, related to stronger coordination in the solvation shell of the mobile sodium population.66,71
In Fig. 7b, the systems with FSI-based ILs show the presence of coordinated CO, while NPs + C4mpyrTFSI does not show this. As in the FSI-based membranes, the coordinated CO peak at 1714 cm−1 undergoes a shift, which may also mean coordination changes in the presence of the IL.
In the spectral region related to the cis/trans configurations of the TFSI anion (Fig. 7c) the influence of the plasticizer varies greatly depending both on the nature of the cation and anion of the neat IL. In the NPs + C4mpyrTFSI system, the cis configuration is favoured compared to the trans conformer. This behaviour could suggest that in this system, the introduction of TFSI anions, and consequently the augmentation of TFSI:Na ratio, induces a change in the type of coordination and solvation shell for the metal ion. Based on other works,65,66,72,73 the new coordination of sodium could be associated with the formation of Na+ clusters.
In C3mpyrFSI/LiFSI systems it was observed that, upon addition of Li, the peaks of the IL assigned to νas(SO2) which are found at 1360 and 1377 cm−1 did not shift.74 In the NPs + C3mpyrFSI system we can also observe the presence of these same peaks without any shift, whereas the peaks assigned to the cis/trans conformers of the TFSI anion of the NPs indicate a shift of the cis peak to lower wavelengths. The trans peak could not be observed, possibly merging with νas(SO2) of FSI at 1360 cm−1. This observation still could suggest that in this system the cis configuration of the TFSI anion is the most thermodynamically stable, as observed in the NPs + C4mpyrTFSI system. In the NPs + P111i4FSI system, different from the C3mpyrFSI system, the cis/trans peaks do not appear to shift, but the cis conformer is still observed to be the most stable.
The different coordination behaviours within the NPs + IL membranes are connected to the Na cation mobility and the ionic conductivity of the membrane, and reflect on the electrochemical properties of each system, as presented below.
Electrochemical properties of gel polymer electrolytes based on sodium-functionalized polymer nanoparticles
From the physiochemical characterization of the GPE membranes, it was shown how each plasticizer influences the sodium coordination uniquely. In NPs + organic solvents, NMR and FTIR experiments suggested that the Na solvation shell is formed by both the organic solvent and TFSI from the nanoparticles' polymeric backbone, resulting in anionic super structures that coordinate with sodium. In the case of NPs + SL and NPs + PC, these super-structures are still able to diffuse and conduct the cation thanks to the presence of free organic solvent, hence the good ionic conductivity. The superior ionic conductivity of NPs + SL compared to NPs + PC could be given by the different transport mechanisms, where in NPs + SL a mix of diffusion and hopping could happen, while in NPs + PC only diffusion would regulate sodium transport. In NPs + DGME, the absence of free diglyme could hinder sodium diffusion. In NPs + ILs the competition between the TFSI from the polymeric backbone and the anion of the IL regulates the possibility of sodium diffusion in the systems. In NPs + C4mpyrTFSI, the low ionic conductivity suggests that TFSI from the IL does not coordinate as strongly with the sodium to promote the cation diffusion, while in the systems using FSI-based ILs, the FSI is able to enter the Na coordination shell and promote sodium transport. These observations were used to investigate the electrochemical properties of the GPE membranes.To evaluate the potential application of the NaNPs-based gel polymer electrolytes in sodium-metal batteries, the behaviour in sodium symmetrical cells at 50 °C was investigated. The membranes were sandwiched between two sodium metal electrodes and the impedance and cycling stability were tested. The only system that was not evaluated electrochemically is NPs + DGME, as the membrane was too brittle and with too low ionic conductivity to be interesting for its application.
Fig. 8 presents the electrochemical characterization of the NPs + SL, NPs + PC, and NPs + hybrid gel membrane while the NPs + IL are given in Fig. S3 (ESI†). The evolution of the impedance spectra as a function of time was used to characterize the stability of the various samples against sodium metal.75 In general terms, there are four main processes that comprise impedance spectra: bulk electrolyte, surface reaction layer, charge transfer at the Na interface, and diffusion.76 Surface layer and charge transfer have similar time constants, in the impedance spectra their impedance responses fall in the same region forming a depressed semicircle, making it difficult to discern between the two contributions.75 For this reason, surface layer and charge transfer are together referred to as interfacial impedance (or resistance), which can be used to evaluate how the sodium–electrolyte interface layer forms and evolves. This sodium–electrolyte interface layer can be called solid electrolyte interphase (SEI) and has a pivotal role in the battery functioning. The interfacial resistance, in particular the charge transfer component, also reflects the activation energy of the electrolyte membranes. In the case of the GPEs based on organic or hybrid solvents, all membranes obtained similar activation energy values, with the exception of NPs + DGME, thus the differences between these materials in the impedance spectra can be attributed only to the SEI. The EIS spectra in Fig. 8a, d and g reveal how different organic solvents contribute to SEI formation and growth differently. This difference can be linked to the sodium-solvent coordination. NPs + SL show a high interfacial resistance and over time the impedance spectrum shows an initial decrease, which can be attributed to the reduction of the membrane thickness and higher adhesion of the electrolyte to the sodium metal, followed by an increase. This shows how NPs + SL forms an unstable and resistive SEI when in contact with sodium metal. On the other hand, NPs + PC and NPs + hybrid show better interfacial resistance and the SEI layer forming upon contact with sodium metal is stable over time (Fig. 8d and g). The EIS evolution shows interfacial resistances around 9000 Ω and 15000 Ω for NPs + PC and NPs + hybrid, respectively, and remain stable over time. To further investigate the evolution of the SEI layer, the sodium symmetrical cells were cycled at 50 °C and 0.02 mA cm−2 for 5 cycles. NPs + SL (Fig. 8b) shows a high overvoltage, starting 1.5 V on the 1st cycle and increasing up to 3 V in the 5th cycle. The high and deteriorating overvoltage profile of NPs + SL corroborates the formation of a resistive and unstable SEI layer between sodium metal and the membrane, as it can also be observed from the dramatically higher interfacial resistance in the impedance spectrum after cycling (Fig. 8c).
 | ||
| Fig. 8 EIS of initial 24 h, symmetrical cycling at 0.02 mA cm−2, and EIS spectra before and after cycling at 50 °C of NPs + SL (a)–(c), NPs + PC (d)–(f), and NPs + hybrid (g)–(i). | ||
Conversely, the SEI layer between sodium metal and NPs + PC or NPs + hybrid is confirmed to be more conductive and stable, as the overvoltage is lower (<1 V) and the cycling is stable as seen in Fig. 8e and h. Furthermore, in the impedance spectra after cycling in Fig. 8f and i, the interfacial resistance does not vary much before and after cycling. During cycling, both NPs + PC and NPs + hybrid encounter problems with soft short-circuits, as can be seen by the drops in cell voltage during the cycles. The occurrence of a soft short-circuit can be linked to the softer nature of the membrane compared to the NPs + SL membrane.
In the NPs + ILs system, the presence of ILs as plasticizers greatly improves the interfacial stability in the symmetrical cells (Fig. S3, ESI†); this can be observed by the lower interfacial resistance of these membranes compared to NPs + organic solvents, and further explains the results observed for NPs + hybrid. As it has been observed, fluorinated species such as FSI and TFSI are beneficial for the formation of a stable SEI layer.77 While for NPs + C4mpyrTFSI (Fig. S3a, ESI†) an increase in the impedance is observed over time, in NPs + C3mpyrFSI and NPs + P111i4FSI the interfacial layer formed between sodium metal and electrolyte appears to remain stable (Fig. S3d and g, ESI†). However, upon cycling (Fig. S3b, e and h, ESI†), all systems show similar overpotential and after cycling, the SEI layer shows even better stability, as the impedance spectra show a pronounced decrease in the interfacial resistance, even for NPs + C4mpyrTFSI (Fig. 3g, h and i). The most improved system after cycling is NPs + P111i4FSI, suggesting the better compatibility with the NPs observed from the IR and NMR spectroscopy and the subsequent solvation of Na ions contributes to a better SEI formation.
The influence of the compatibility between the gel polymer electrolytes and the sodium metal on the SEI formation and electrochemical performance was also observed when we attempted to evaluate the sodium transference number for the systems investigated, measured using the Bruce–Vincent method. To use this method, one requires that the SEI is stable and does not influence the transport number. However, even with numerous attempts to determine TNa+ from the polarisation and EIS data, numbers obtained were non-sensical, sometimes close to zero or negative. Despite this, the polarization curves obtained during this measurement (Fig. S4, ESI†) show an initial current I0 that then stabilises to a measurable steady state current Iss, However, the EIS semi-circle (and hence the interphase/charge transfer resistances) both before and after polarisation is applied indicate a very large resistive interface. Moreover, the EIS spectra indicate that this interface is not stable and actually increases post polarisation. We therefore believe that it is not appropriate to apply the Bruce–Vincent method to determine TNa+ as the interface will interfere with the measurement which is essentially assumed to be a bulk electrolyte property. Nevertheless, we can qualitatively see that some electrolytes support Na electrochemistry more than others. In NPs + PC, which achieved the most promising Na/Na cycling performance, I0 and Iss which followed after some time indicated that there is indeed a current supported by the Na+ transport and this is consistent with a lower interfacial resistance. Conversely, the NPs + SL system exhibits significant current decline during polarization, accompanied by a larger resistance which could be reflecting the poor SEI stability and electrochemical performance seen in the symmetrical cycling. Similar trends are observed in IL-containing systems, where larger differences between initial (I0) and steady-state (Iss) currents, coupled with lower ISS values, correlate with inferior electrochemical performance.
The electrochemical results presented above, particularly for the solvent-based GPEs, were surprisingly poor given the conductivities and materials properties of these materials and appear to be due to a highly resistive SEI layer formation. One possible explanation for this is that the SEI requires a surface-active species to breakdown and react with the Na+ ions to form a suitable SEI. In traditional electrolytes in which the anion (e.g. TFSI or FSI) is also mobile, this can react on the metal surface to form a suitable SEI.78 However, in our case the anion is tethered to the polymer, not present in high concentrations and thus the probability of it reaching and reacting with the metal interface is reduced. Thus, despite the good ionic conductivity an alternative method is required to improve the SEI. To investigate this hypothesis, membranes formed by sodium-functionalized nanoparticles, organic solvents, and a small amount of NaFSI were tested, where the FSI anion was added to help the formation of a stable SEI layer. 10 mol% of NaFSI, calculated on the organic solvent, was added. Interestingly the sodium salt addition resulted in more fragile membranes and thus the load of NaNPs had to be increased to 55 wt%, calculated on the amount of liquid electrolyte (NaFSI/organic solvent), to obtain a self-standing membrane. The addition of sodium salt indeed did dramatically improve the interfacial stability with the impedance spectra over time of both NPs + SL + NaFSI and NPs + PC + NaFSI (Fig. S5a and d, ESI†) showing lower interfacial resistance compared to the systems without sodium salt. Upon cycling at 0.02 mA cm−2, both membranes show stable overpotential lower than 200 mV (Fig. S5b and e, ESI†), while when the current density is increased to 0.05 mA cm−2 the plating/stripping behaviour becomes unstable and both membranes show signs of soft short-circuit. This is likely due to the non-homogeneity of the membranes which has a significant role in achieving stable cycling. The EIS spectra in Fig. S5c and f (ESI†) present the behaviour before cycling, after 5 cycles at 0.02 mA cm−2, and after 5 cycles at 0.05 mA cm−2. Here both systems show stable SEI formation and evolution during cycling. The addition of NaFSI improved the electrochemical performance of these NP-based GPEs, which shows the potential of the NPs for the gelation of various systems, from neat organic solvents or ILs, to liquid electrolytes, although more engineering of the NPs is needed to fully optimize these systems.
Conclusions
In this work, we reported the synthesis, characterization and application of sodium-sulfonamide functionalized methacrylic polymer nanoparticles for sodium metal batteries. The nanoparticles were mixed with various plasticizers to obtain gel polymer electrolytes and the sodium coordination was analyzed via EIS, NMR and FTIR. The nature of the plasticizer deeply influenced the sodium coordination shell, impacting the properties of each membrane. Using organic plasticizers, the highest conductivity was achieved by NPs + SL, showing predominantly a monodentate type of sodium coordination. However, sulfolane as a plasticizer resulted in poor SEI layer formation before and after symmetrical cycling. NPs + PC, although showing a weaker sodium coordination and lower ionic conductivity than NPs + SL, results in a more stable and conductive SEI layer than NPs + SL, as shown by an overvoltage lower than 300 mV at a current density of 0.02 mA cm−2. The NPs + ILs gel systems, due to the presence of free fluorinated species, show good ionic conductivity and a stable SEI layer. The stabilizing effect of fluorinated species on the SEI layer was also proved from NPs + SL/PC + NaFSI, which show more conductive and stable SEI layers compared to the system without sodium salt. Interestingly, when attempts were made to measure TNa+ using the Bruce–Vincent method, values could not be determined which we attribute to a highly resistive (in particular to Na+) and unstable SEI. Finally, although the data show the need for improvement in the engineering of the polymer nanoparticles and GPEs to enhance the interface compatibility, the potential application of NaNPs to create sodium conductiving polymer membranes was demonstrated, showing promising results.Author contributions
Pierre L. Stigliano: materials synthesis, sample preparation, physiochemical characterization, NMR characterization, electrochemical characterization, data analysis, and manuscript preparation; Antonela Gallastegui: materials synthesis, physiochemical characterization, results discussion, and revised manuscript; Thomas H. Smith: NMR characterization and results discussion; Luke O’Dell: NMR characterization, results discussion; David Mecerreyes: project conception, supervision, results discussion, and revised manuscript; Cristina Pozo-Gonzalo: supervision, results, discussion, and revised manuscript; Maria Forsyth: supervision, project conception, results discussion, and revised manuscript.Data availability
The data that support the findings of this study are included within the article and its ESI.†Conflicts of interest
All authors declare that they have no conflicts of interest.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement no. 860403. The authors acknowledge the Australian Research Council (ARC) for funding through Discovery Programme DP160101178 and the ARC Industry Transformation Training Centre for Future Energy Technologies (storEnergy) for funding under grant agreement no. IC180100049.References
- C. Zhao, L. Liu, X. Qi, Y. Lu, F. Wu and J. Zhao, et al., Solid-State Sodium Batteries, Adv. Energy Mater., 2018, 8(17), 1703012, DOI:10.1002/aenm.201703012.
- C. Delmas, Sodium and Sodium-Ion Batteries: 50 Years of Research, Adv. Energy Mater., 2018, 8(17), 1703137, DOI:10.1002/aenm.201703137.
- A. Eftekhari and D. W. Kim, Sodium-ion batteries: New opportunities beyond energy storage by lithium, J. Power Sources, 2018, 395, 336 CrossRef CAS.
- H. S. Hirsh, Y. Li, D. H. S. Tan, M. Zhang, E. Zhao and Y. S. Meng, Sodium-Ion Batteries Paving the Way for Grid Energy Storage, Adv. Energy Mater., 2020, 10(32), 2001274, DOI:10.1002/aenm.202001274.
- Z. Y. Li, Z. Li, J. L. Fu and X. Guo, Sodium-ion conducting polymer electrolytes, Rare Met., 2023, 42, 1–16 CrossRef.
- M. Forsyth, L. Porcarelli, X. Wang, N. Goujon and D. Mecerreyes, Innovative Electrolytes Based on Ionic Liquids and Polymers for Next-Generation Solid-State Batteries, Acc. Chem. Res., 2019, 52(3), 686, DOI:10.1021/acs.accounts.8b00566.
- F. Gebert, J. Knott, R. Gorkin, S. L. Chou and S. X. Dou, Polymer electrolytes for sodium-ion batteries, Energy Storage Mater., 2021, 36, 10–30 CrossRef.
- G. Rollo-Walker, N. Malic, X. Wang, J. Chiefari, M. Forsyth and F. Lufrano, et al., Development and Progression of Polymer Electrolytes for Batteries: Influence of Structure and Chemistry, Polymers, 2021, 13, 4127 CrossRef CAS PubMed.
- P. Ding, Z. Lin, X. Guo, L. Wu, Y. Wang and H. Guo, et al., Polymer electrolytes and interfaces in solid-state lithium metal batteries, Mater. Today, 2021, 51, 449 CrossRef CAS.
- Y. An, X. Han, Y. Liu, A. Azhar, J. Na and A. K. Nanjundan, et al., Progress in Solid Polymer Electrolytes for Lithium-Ion Batteries and Beyond, Small, 2022, 18, 2103617, DOI:10.1002/smll.202103617.
- L. Qiao, X. Judez, T. Rojo, M. Armand and H. Zhang, Review—Polymer Electrolytes for Sodium Batteries, J. Electrochem. Soc., 2020, 167(7), 070534, DOI:10.1149/1945-7111/ab7aa0.
- J. Yang, H. Zhang, Q. Zhou, H. Qu, T. Dong and M. Zhang, et al., Safety-Enhanced Polymer Electrolytes for Sodium Batteries: Recent Progress and Perspectives, ACS Appl. Mater. Interfaces, 2019, 11(19), 17109, DOI:10.1021/acsami.9b01239.
- F. Wu, K. Zhang, Y. Liu, H. Gao, Y. Bai and X. Wang, et al., Polymer electrolytes and interfaces toward solid-state batteries: Recent advances and prospects, Energy Storage Mater., 2020, 33, 26–54 CrossRef.
- J. Pan, N. Wang, H. J. Fan, J. Pan, H. J. Fan and N. Wang, Gel Polymer Electrolytes Design for Na-Ion Batteries, Small Methods, 2022, 2201032, DOI:10.1002/smtd.202201032.
- S. Alipoori, S. Mazinani, S. H. Aboutalebi and F. Sharif, Review of PVA-based gel polymer electrolytes in flexible solid-state supercapacitors: Opportunities and challenges, J. Energy Storage, 2020, 27, 101072 CrossRef.
- R. Bhandary and M. Schönhoff, Polymer effect on lithium ion dynamics in gel polymer electrolytes: Cationic versus acrylate polymer, Electrochim. Acta, 2015, 174, 753 CrossRef CAS.
- G. Feuillade and P. Perche, Ion-conductive macromolecular gels and membranes for solid lithium cells, J. Appl. Electrochem., 1975, 5, 63–69 CrossRef CAS.
- J. Zheng, W. Li, X. Liu, J. Zhang, X. Feng and W. Chen, Progress in Gel Polymer Electrolytes for Sodium-Ion Batteries, Energy Environ. Mater., 2023, 6, e12422, DOI:10.1002/eem2.12422.
- V. Jabbari, V. Yurkiv, M. G. Rasul, M. T. Saray, R. Rojaee and F. Mashayek, et al., An efficient gel polymer electrolyte for dendrite-free and long cycle life lithium metal batteries, Energy Storage Mater., 2022, 46, 352 CrossRef.
- W. Li, Y. Pang, J. Liu, G. Liu, Y. Wang and Y. Xia, A PEO-based gel polymer electrolyte for lithium ion batteries, RSC Adv., 2017, 7(38), 23494 RSC.
- M. Dotter, J. L. Storck, M. Surjawidjaja, S. Adabra and T. Grothe, Investigation of the long-term stability of different polymers and their blends with peo to produce gel polymer electrolytes for non-toxic dye-sensitized solar cells, Appl. Sci., 2021, 11(13), 5834, DOI:10.3390/app11135834.
- J. L. Storck, M. Dotter, B. Brockhagen and T. Grothe, Evaluation of novel glycerol/peo gel polymer electrolytes for non-toxic dye-sensitized solar cells with natural dyes regarding long-term stability and reproducibility, Crystals, 2020, 10(12), 1–15 CrossRef.
- Y. Wu, Y. Li, Y. Wang, Q. Liu, Q. Chen and M. Chen, Advances and prospects of PVDF based polymer electrolytes, J. Energy Chem., 2022, 64, 62–84 CrossRef CAS.
- X. Pei, Y. Li, T. Ou, X. Liang, Y. Yang and E. Jia, et al., Li−N Interaction Induced Deep Eutectic Gel Polymer Electrolyte for High Performance Lithium-Metal Batteries, Angew. Chem., Int. Ed., 2022, 61(31), e202205075 CrossRef CAS PubMed.
- Y. Q. Shen, F. L. Zeng, X. Y. Zhou, A. B. Wang, W. K. Wang, N. Y. Yuan and J. N. Ding, A novel permselective organo-polysulfides/PVDF gel polymer electrolyte enables stable lithium anode for lithium–sulfur batteries, J. Energy Chem., 2020, 48, 267 CrossRef.
- T. Shi, S. Kang, K. Zhang, F. Xue and W. Lu, High-performance lithium-ion batteries with gel polymer electrolyte based on ultra-thin PVDF film, Ionics, 2022, 28(7), 3269 CrossRef CAS.
- R. Huang, R. Xu, J. Zhang, J. Wang, T. Zhou and M. Liu, et al., PVDF-HFP-SN-based gel polymer electrolyte for high-performance lithium-ion batteries, Nano Res., 2023, 16(7), 9480 CrossRef CAS.
- T. Shi, S. Kang, K. Zhang, F. Xue and W. Lu, High-performance lithium-ion batteries with gel polymer electrolyte based on ultra-thin PVDF film, Ionics, 2022, 28(7), 3269 CrossRef CAS.
- Z. Shen, J. Zhong, S. Jiang, W. Xie, S. Zhan and K. Lin, et al., Polyacrylonitrile Porous Membrane-Based Gel Polymer Electrolyte by in Situ Free-Radical Polymerization for Stable Li Metal Batteries, ACS Appl. Mater. Interfaces, 2022, 14(36), 41022 CrossRef CAS PubMed.
- R. Singh, S. Janakiraman, M. Khalifa, S. Anandhan, S. Ghosh and A. Venimadhav, et al., A high thermally stable polyacrylonitrile (PAN)-based gel polymer electrolyte for rechargeable Mg-ion battery, J. Mater. Sci.: Mater. Electron., 2020, 31(24), 22912 CrossRef CAS.
- X. Yuan, A. A. Razzaq, Y. Chen, Y. Lian, X. Zhao and Y. Peng, et al., Polyacrylonitrile-based gel polymer electrolyte filled with Prussian blue forhigh-performance lithium polymer batteries, Chin. Chem. Lett., 2021, 32(2), 890 CrossRef CAS.
- A. Hosseinioun and E. Paillard, In situ crosslinked PMMA gel electrolyte from a low viscosity precursor solution for cost-effective, long lasting and sustainable lithium-ion batteries, J. Membr. Sci., 2020, 594 Search PubMed.
- X. Liu, X. Xin, L. Shen, Z. Gu, J. Wu and X. Yao, Poly(methyl methacrylate)-Based Gel Polymer Electrolyte for High-Performance Solid State Li-O2Battery with Enhanced Cycling Stability, ACS Appl. Energy Mater., 2021, 4(4), 3975 CrossRef CAS.
- D. Kumar, K. Gohel, D. K. Kanchan and K. Mishra, Dielectrics and battery studies on flexible nanocomposite gel polymer electrolyte membranes for sodium batteries, J. Mater. Sci.: Mater. Electron., 2020, 31(16), 13249 CrossRef CAS.
- W. J. Hyun, C. M. Thomas and M. C. Hersam, Nanocomposite Ionogel Electrolytes for Solid-State Rechargeable Batteries, Adv. Energy Mater., 2020, 10, 2002135 CrossRef CAS.
- S. Wang, Y. Jiang and X. Hu, Ionogel-Based Membranes for Safe Lithium/Sodium Batteries, Adv. Mater., 2022, 34, 2200945 CrossRef CAS PubMed.
- N. Chen, H. Zhang, L. Li, R. Chen and S. Guo, Ionogel Electrolytes for High-Performance Lithium Batteries: A Review, Adv. Energy Mater., 2018, 8, 1702675 CrossRef.
- D. Zhou, D. Shanmukaraj, A. Tkacheva, M. Armand and G. Wang, Polymer Electrolytes for Lithium-Based Batteries: Advances and Prospects, Chem, 2019, 5, 2326 CAS.
- Mishra K. Harshlata and D. K. Studies Rai, on ionic liquid based nanocomposite gel polymer electrolyte and its application in sodium battery, Mater. Sci. Eng., B, 2021, 267 Search PubMed.
- S. A. Hashmi, M. Y. Bhat, M. K. Singh, N. T. K. Sundaram, B. P. C. Raghupathy and H. Tanaka, Ionic liquid-based sodium ion-conducting composite gel polymer electrolytes: effect of active and passive fillers, J. Solid State Electrochem., 2016, 20(10), 2817 CrossRef CAS.
- L. Fan, S. Wei, S. Li, Q. Li and Y. Lu, Recent Progress of the Solid-State Electrolytes for High-Energy Metal-Based Batteries, Adv. Energy Mater., 2018, 8, 1702657 CrossRef.
- H. Che, S. Chen, Y. Xie, H. Wang, K. Amine and X. Z. Liao, et al., Electrolyte design strategies and research progress for room-temperature sodium-ion batteries, Energy Environ. Sci., 2017, 10, 1075 RSC.
- S. Srivastava, J. L. Schaefer, Z. Yang, Z. Tu and L. A. Archer, 25th Anniversary article: Polymer-particle composites: Phase stability and applications in electrochemical energy storage, Adv. Mater., 2014, 26, 201 CrossRef CAS PubMed.
- K. M. Diederichsen, E. J. McShane and B. D. McCloskey, Promising Routes to a High Li+ Transference Number Electrolyte for Lithium Ion Batteries, ACS Energy Lett., 2017, 2(11), 2563 CrossRef CAS.
- I. Villaluenga, S. Inceoglu, X. Jiang, X. C. Chen, M. Chintapalli and D. R. Wang, et al., Nanostructured Single-Ion-Conducting Hybrid Electrolytes Based on Salty Nanoparticles and Block Copolymers, Macromolecules, 2017, 50(5), 1998–2005 CrossRef CAS.
- V. Bocharova, X. C. Chen, S. P. Jeong, Z. Zhou, R. L. Sacci and J. K. Keum, et al., Single Ion Conducting Hairy Nanoparticle Additive to Improve Cycling Stability of Solid Polymer Electrolytes, ACS Appl. Energy Mater., 2023, 6(15), 8042 CrossRef CAS.
- J. M. Asua, Emulsion Polymerization: From Fundamental Mechanisms to Process Developments, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 1025 CrossRef CAS.
- L. Porcarelli, P. Sutton, V. Bocharova, R. H. Aguirresarobe, H. Zhu and N. Goujon, et al., Single-ion conducting polymer nanoparticles as functional fillers for solid electrolytes in lithium metal batteries, ACS Appl. Mater Interfaces, 2021, 13(45), 54354, DOI:10.1021/acsami.1c15771.
- A. Gallastegui, R. Del Olmo, M. Criado-Gonzalez, J. R. Leiza, M. Forsyth and D. Mecerreyes, Printable Single-Ion Polymer Nanoparticle Electrolytes for Lithium Batteries, Small Science, 2024, 4(3), 2300235 CrossRef CAS.
- J. L. Olmedo-Martínez, R. Del Olmo, A. Gallastegui, I. Villaluenga, M. Forsyth and A. J. Müller, et al., All-Polymer Nanocomposite as Salt-Free Solid Electrolyte for Lithium Metal Batteries, ACS Polym. Au, 2024, 4(1), 77–85, DOI:10.1021/acspolymersau.3c00035.
- Y. S. Pak, K. J. Adamic, S. G. Greenbaum, M. C. Wintersgill, J. J. Fontanella and C. S. Coughlin, Complex impedance and multifrequency 23Na NMR study of poly(propylene oxide) complexed with NaB (C6H5)4, Solid State Ionics, 1991, 45, 277–284 CrossRef CAS.
- K. J. Adamić, S. G. Greenbaum, M. C. Wintersgill and J. J. Fontanella, Ionic conductivity in solid, crosslinked dimethylsiloxane-ethylene oxide copolymer networks containing sodium, J. Appl. Phys., 1986, 60(4), 1342 CrossRef.
- R. H. Erlich and A. I. Popov, Spectroscopic Studies of Ionic Solvation. X. A Study of the Solvation of Sodium Ions in Nonaqueous Solvents by 23Na Nuclear Magnetic Resonance, J. Am. Chem. Soc., 1971, 93(22), 5620, DOI:10.1021/ja00751a005.
- P. Geysens, V. S. Rangasamy, S. Thayumanasundaram, K. Robeyns, L. Van Meervelt and J. P. Locquet, et al., Solvation Structure of Sodium Bis(fluorosulfonyl)imide-Glyme Solvate Ionic Liquids and Its Influence on Cycling of Na-MNC Cathodes, J. Phys. Chem. B, 2018, 122(1), 275 CrossRef CAS PubMed.
- J. E. Katon and W. R. Feairheller, The vibrational spectra and molecular configuration of sulfolane, Spectrochim. Acta, 1965, 21(1), 199–201 CrossRef CAS.
- M. Nie, D. P. Abraham, D. M. Seo, Y. Chen, A. Bose and B. L. Lucht, Role of solution structure in solid electrolyte interphase formation on graphite with LiPF6 in propylene carbonate, J. Phys. Chem. C, 2013, 117(48), 25381 CrossRef CAS.
- L. Garcia-Quintana, F. Chen, N. Ortiz-Vitoriano, Y. Zhang, L. A. O’Dell and D. R. MacFarlane, et al., Unravelling the Role of Speciation in Glyme:Ionic Liquid Hybrid Electrolytes for Na−O2 Batteries, Batteries Supercaps, 2021, 4(3), 513, DOI:10.1002/batt.202000261.
- K. Dokko, D. Watanabe, Y. Ugata, M. L. Thomas, S. Tsuzuki and W. Shinoda, et al., Direct Evidence for Li Ion Hopping Conduction in Highly Concentrated Sulfolane-Based Liquid Electrolytes, J. Phys. Chem. B, 2018, 122(47), 10736, DOI:10.1021/acs.jpcb.8b09439.
- A. Nakanishi, K. Ueno, D. Watanabe, Y. Ugata, Y. Matsumae and J. Liu, et al., Sulfolane-Based Highly Concentrated Electrolytes of Lithium Bis(trifluoromethanesulfonyl)amide: Ionic Transport, Li-Ion Coordination, and Li-S Battery Performance, J. Phys. Chem. C, 2019, 123(23), 14229 CrossRef CAS.
- H. Jiang, Q. Zhang, Y. Zhang, L. Sui, G. Wu and K. Yuan, et al., Li-Ion solvation in propylene carbonate electrolytes determined by molecular rotational measurements, Phys. Chem. Chem. Phys., 2019, 21(20), 10417 RSC.
- R. A. Pethrick and A. D. Wilson, Infrared and Raman spectroscopic studies of cyclic carbonates, Spectrochim. Acta, Part A, 1974, 30(5), 1073 CrossRef.
- J. Wahlers, K. D. Fulfer, D. P. Harding, D. G. Kuroda, R. Kumar and R. Jorn, Solvation Structure and Concentration in Glyme-Based Sodium Electrolytes: A Combined Spectroscopic and Computational Study, J. Phys. Chem. C, 2016, 120(32), 17949 CrossRef CAS.
- K. Ueno, R. Tatara, S. Tsuzuki, S. Saito, H. Doi and K. Yoshida, et al., Li+ solvation in glyme-Li salt solvate ionic liquids, Phys. Chem. Chem. Phys., 2015, 17(12), 8248 RSC.
- P. Johansson, J. Grondin and J. C. Lassègues, Structural and vibrational properties of diglyme and longer glymes, J. Phys. Chem. A, 2010, 114(39), 10700 CrossRef CAS PubMed.
- M. Beran, J. Příhoda, Z. Žák and M. Černík, A new route to the syntheses of alkali metal bis(fluorosulfuryl)imides: Crystal structure of LiN(SO2F)2, Polyhedron, 2006, 25(6), 1292 CrossRef CAS.
- J. B. Haskins, W. R. Bennett, J. J. Wu, D. M. Hernández, O. Borodin and J. D. Monk, et al., Computational and experimental investigation of li-doped ionic liquid electrolytes: [pyr14][TFSI], [pyr13][FSI], and [EMIM][BF4], J. Phys. Chem. B, 2014, 118(38), 11295 CrossRef CAS PubMed.
- M. Schmeisser, P. Illner, R. Puchta, A. Zahl and R. Van Eldik, Gutmann donor and acceptor numbers for ionic liquids, Chem. - Eur. J., 2012, 18(35), 10969 CrossRef CAS PubMed.
- K. Shigenobu, T. Sudoh, J. Murai, K. Dokko, M. Watanabe and K. Ueno, Ion Transport in Glyme- and Sulfolane-Based Highly Concentrated Electrolytes, Chem. Rec., 2023, 23(8), e202200301 CrossRef CAS PubMed.
- V. Chaudoy, J. Jacquemin, F. Tran-Van, M. Deschamps and F. Ghamouss, Effect of mixed anions on the transport properties and performance of an ionic liquid-based electrolyte for lithium-ion batteries, Pure Appl. Chem., 2019, 1361 CrossRef CAS.
- J. C. Lassègues, J. Grondin, C. Aupetit and P. Johansson, Spectroscopic identification of the lithium ion transporting species in LiTFSI-doped ionic liquids, J. Phys. Chem. A, 2009, 113(1), 305, DOI:10.1021/jp806124w.
- M. Kerner, N. Plylahan, J. Scheers and P. Johansson, Ionic liquid based lithium battery electrolytes: fundamental benefits of utilising both TFSI and FSI anions?, Phys. Chem. Chem. Phys., 2015, 17(29), 19569 RSC.
- J. L. Nowinski, P. Lightfoot and P. G. Bruce, Structure of LiN(CF3SO2)2, a novel salt for electrochemistry, J. Mater. Chem., 1994, 4(10), 1579 RSC.
- L. Xue, C. W. Padgett, D. D. DesMarteau and W. T. Pennington, Synthesis and structures of alkali metal salts of bis[(trifluoromethyl)sulfonyl]imide, Solid State Sci., 2002, 4(11–12), 1535 CrossRef CAS.
- J. Huang and A. F. Hollenkamp, Thermal behavior of ionic liquids containing the FSI anion and the Li+ cation, J. Phys. Chem. C, 2010, 114(49), 21840 CrossRef CAS.
- P. Vadhva, J. Hu, M. J. Johnson, R. Stocker, M. Braglia and D. J. L. Brett, et al., Electrochemical Impedance Spectroscopy for All-Solid-State Batteries: Theory, Methods and Future Outlook, ChemElectroChem, 2021, 8, 1930 CrossRef CAS.
- W. Choi, H. C. Shin, J. M. Kim, J. Y. Choi and W. S. Yoon, Modeling and applications of electrochemical impedance spectroscopy (Eis) for lithium-ion batteries, J. Electrochem. Sci. Technol., 2020, 11, 1–13 CrossRef CAS.
- G. G. Eshetu, M. Martinez-Ibañez, E. Sánchez-Diez, I. Gracia, C. Li and L. M. Rodriguez-Martinez, et al., Electrolyte Additives for Room-Temperature, Sodium-Based, Rechargeable Batteries, Chem. – Asian J., 2018, 13, 2770 CrossRef CAS PubMed.
- J. Ma, X. Feng, Y. Wu, Y. Wang, P. Liu and K. Shang, et al., Stable sodium anodes for sodium metal batteries (SMBs) enabled by in situ formed quasi solid-state polymer electrolyte, J. Energy Chem., 2023, 77, 290 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp04703f |
| This journal is © the Owner Societies 2025 |