
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh-symmetry cage-like molecule N20(C2B2)30: computational insight into its bonding and reactivity†
Miaorun
Zhang
,
Lin
Zhang
 ,
Zexing
Cao
and
Yi
Zhao
*
,
Zexing
Cao
and
Yi
Zhao
*
State Key Laboratory of Physical Chemistry of Solid Surfaces, Collaborative Innovation Center of Chemistry for Energy Materials, Fujian Provincial Key Laboratory of Theoretical and Computational Chemistry, and Department of Chemistry, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, Fujian, People's Republic of China. E-mail: yizhao@xmu.edu.cn
First published on 28th January 2025
Abstract
On the basis of density functional theory (DFT) calculations and AIMD simulations, a novel Ih-symmetry cage-like molecule N20(C2B2)30 is constructed and characterized computationally. It is found that N20(C2B2)30 is structurally similar to fullerene C20, but it has high thermodynamic and kinetic stability. The designed N20(C2B2)30 exhibits strong chemical reactivities, including reactivities for the Diels–Alder reaction with butadiene (C4H6) and cyclopentadiene (C5H6), as well as for the [3+2] addition reaction with diazomethane (N2CH2). In addition, the presence of the boron site and the inverted C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond with the charge-shift (CS) bonding in N20(C2B2)30 make it quite active not only for cycloaddition reactions but also for capture of small molecules (e.g. H2, CO, NO, and NO2). Once N20(C2B2)30 complexes with a transition metal (TM) ion, the resultant complexes (TM)N20(C2B2)30+ (TM = Cu, Ag, and Au) can bind inactive CO2 and N2O at the TM site. Furthermore, AuN20(C2B2)30+ is able to effectively separate CO2 and N2O. Owing to its unique porous structure and reactivity as well as high stability, N20(C2B2)30 may further enrich the diversity of a highly symmetrical molecular family.
C bond with the charge-shift (CS) bonding in N20(C2B2)30 make it quite active not only for cycloaddition reactions but also for capture of small molecules (e.g. H2, CO, NO, and NO2). Once N20(C2B2)30 complexes with a transition metal (TM) ion, the resultant complexes (TM)N20(C2B2)30+ (TM = Cu, Ag, and Au) can bind inactive CO2 and N2O at the TM site. Furthermore, AuN20(C2B2)30+ is able to effectively separate CO2 and N2O. Owing to its unique porous structure and reactivity as well as high stability, N20(C2B2)30 may further enrich the diversity of a highly symmetrical molecular family.
1. Introduction
Highly symmetrical molecules with Ih symmetry can be traced back to closo-[B12H12]2−, as reported both experimentally and theoretically.1–3 Owing to its perfect structure and stable physicochemical properties in the closed borane family, this closo-[B12H12]2− has begun to play important roles in medicine, energetic materials, new energy, environmental management and optoelectronic materials.4 In 1985, another kind of soccer-like cage C60 with Ih symmetry was reported by Kroto et al.5 Henceforth, there has been long standing interest in using C60 in experimental and theoretical domains due to its perfect symmetry and applications in electrochemistry, organic chemistry, nanoscience, nanotechnology, and so on.6–9 Previous studies have shown that C60 can act as a dienophile to undergo a cycloaddition reaction because of its CC double bond character. Moreover, the Diels–Alder reactions of C60 with the derivatives of butadiene and cyclopentadiene, as well as the [3+2] cycloadditions of C60 with N2CR2 and ONCR are favorable to occur as usual across a 6,6-bond of C60.10–13 Recently, the aggregation of Ti atoms on the surface of C60 enables it as a potential hydrogen storage material.14 A C60-modified gold electrode is employed for the determination of dopamine in the excess of ascorbic acid using square-wave voltammetry.15 C60-buffered Cu/SiO2 can decrease the energy barriers to effectively synthesize ethylene glycol (EG) under ambient pressure conditions.16 Thus, C60 has been broadly applied in designing functional materials.
Besides closo-[B12H12]2− and C60, some molecules and clusters with Ih symmetry are also reported experimentally or theoretically, such as B80,17 C60H60, C60F60, Si20H20, Si60H60, and so on.18,19 The edges of high symmetry molecules and clusters mentioned above are normal B–B, C–C, and Si–Si bonds. Inspired by the synthesis methods of covalent organic frameworks (COFs),20 we wonder whether high-symmetry cages with linkers on the edges can be obtained by the [C3+C2] topology. Herein, a type of high-symmetry cage N20(C2B2)30, mimicking fullerene C20 composed of C2 and C3 building blocks, is constructed using C2B2 linkers and N nodes, as illustrated in Fig. 1, in which C2B2 is taken from the molecule C2B2H2 reported experimentally in 2017.21
 | ||
| Fig. 1 The optimized structure of N20(C2B2)30, as well as selected bond lengths, bond angles, and calculated Wiberg bond indices (WBIs, in bracket) are presented. Color scheme: B atom (in pink), C atom (in green), and N atom (in blue). | ||
2. Computational methods
The geometry optimization of N20(C2B2)30 and small molecules (H2, CO, NO, NO2, CO2, N2O, C4H6, C5H6, and N2CH2), as well as the corresponding molecular complexes is performed by using the DFT/M06-2X method,22 in combination with the SDD basis set23 for Cu, Ag, and Au atoms and the 6-31G(d,p) basis set24 for the non-metallic atoms. Vibrational frequencies of the studied molecules are calculated at the same level of theory to confirm the true minima on the potential energy surfaces (PESs). The adsorption energies of small gas molecules are estimated by the difference in the total energies of their respective constituents, including the zero-point energy (ZPE) correction and the basis set superposition error (BSSE) correction.25 The Wiberg bond indices (WBIs) are derived via the natural bond orbital (NBO) analysis.26 The electron density (ρ) and Laplacian values (∇2ρ) of certain chemical bonds are calculated using the Multiwfn 3.8 program.27 Key molecular orbital analysis and the pore diameter of N20(C2B2)30 can be exported and visualized by using the Multiwfn 3.8 program27 and VMD 1.9.4 software,28 respectively. Nucleus independent chemical shifts (NICS) at the centers of the three-membered rings of CCB in N20(C2B2)30 are calculated using the gauge-independent atomic orbital (GIAO) method.29 All the above calculations are performed with the Gaussian16 program.30Ab initio molecular dynamics (AIMD) simulations with N20(C2B2)30 are carried out with the Vienna ab initio simulation package (VASP) program,31 where the core and valence electrons are represented by using the projector augmented wave (PAW)32 method and plane-wave basis functions with a kinetic energy cut-off of 520 eV. The generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhof (PBE)33 exchange–correlation functional is used in the AIMD calculations. The finite temperature simulations of the dynamic properties are performed at temperatures 298 and 600 K using the exact Hellmann–Feynman forces and applying the statistics of the canonical ensemble to the motion of atomic nuclei by means of a Nosé thermostat.34 Newton's equations of motion are integrated using the Verlet algorithm35 with a time step of 1 fs, and the total duration is 10 ps. N20(C2B2)30 is confined in a cubic box of 25 × 25 × 25 Å, and a vacuum distance of more than 10 Å is set to keep the interactions between the molecules in the adjacent boxes negligible. The Brillouin-zone sampling is restricted to the Γ-point. The geometry optimization is performed via a conjugate gradient algorithm until the residual forces acting on atoms are less than 0.1 eV Å−1.
The thermodynamic stability of a molecule can generally be measured by its cohesive energy. Generally, the greater the cohesive energy, the better the stability. The cohesive energy of N20(C2B2)30 is computed as follows
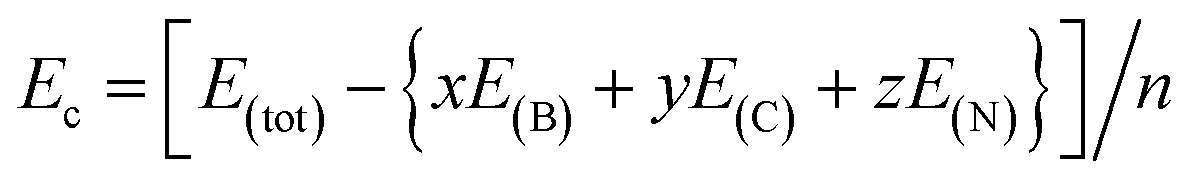
3. Results and discussion
3.1. Structure and stability of N20(C2B2)30
The optimized structure of the novel high-symmetry cage-like molecule N20(C2B2)30, as well as its key lengths and WBIs are shown in Fig. 1. Notably, this molecule is structurally similar to fullerene C20, i.e. this molecule contains twelve regular pentagons (see Fig. S1 in the ESI†), in which twenty vertexes are nitrogen atoms and thirty edges are rhombus C2B2 units. In contrast to fullerene C20,36–38 the optimized N20(C2B2)30 with Ih symmetry has the minimum on the PES without any imaginary frequency and Jahn–teller distortion. In addition, the lengths of B–N, B–C, and inverted CC bonds in N20(C2B2)30 are 1.43, 1.47, and 1.49 Å, respectively. The CC bonds in N20(C2B2)30 are inverted compared with the CC bond in ethylene, thus, we describe the CC bond to be the inverted CC bond. The corresponding WBIs of the B–N, B–C, and inverted CC bonds are 0.88, 1.07, and 1.38, respectively. Thus, the inverted CC bond has double bond character which is consistent with B-heterocyclic carbene (BHC), double B-heterocyclic carbene (DBHC), and Si, B-heterocyclic carbene (SiBHC),39–42 while the B–N and B–C are single bonds. Moreover, the key occupied molecular orbitals (MOs, HOMO−4 to HOMO) involving CC bonds of N20(C2B2)30 are evaluated in order to gain insights into its electronic properties, as shown in Fig. S2 (see the ESI†). It is found that these MOs are formed by the sp2 hybrid orbital of carbon atoms in the CC bonds, which is well consistent with the reported BHCs, DBHCs, and SiBHCs. Thus, the inverted CC in N20(C2B2)30 contains one π bond and one charge-shift (CS) bond.39–44
In order to compare the size of N20(C2B2)30 and C60, we optimize the structure of C60 using the M06-2X/6-31G(d,p) approach. Additionally, the pore diameters of the cavity of N20(C2B2)30 and C60 are calculated using the Multiwfn 3.8 program and are shown in Fig. S3 (see the ESI†). The computational results show that the pore diameter of N20(C2B2)30 is 11.29 Å, which is approximately three times as large as that of C60 (3.68 Å). Calculated cohesive energy of N20(C2B2)30 is −6.00 eV per atom, indicating that it has relatively high thermodynamic stability. To further confirm the chemical stability of N20(C2B2)30, AIMD simulations of N20(C2B2)30 are performed at T = 298 and 600 K and the specific details are shown in Fig. 2. The structure of N20(C2B2)30 has no significant changes at 298 and 600 K for 10 ps. According to the radial distribution of interatomic distances, it is suggested that during a 10 ps of AIMD simulation at both 298 and 600 K, the stability of the backbone of N20(C2B2)30 still remains unchanged. Additionally, all of the B–N, B–C, and C–C bonds of N20(C2B2)30 are not broken at T = 298 and 600 K during AIMD simulations for 10 ps. Thus, N20(C2B2)30 has high kinetic stability.
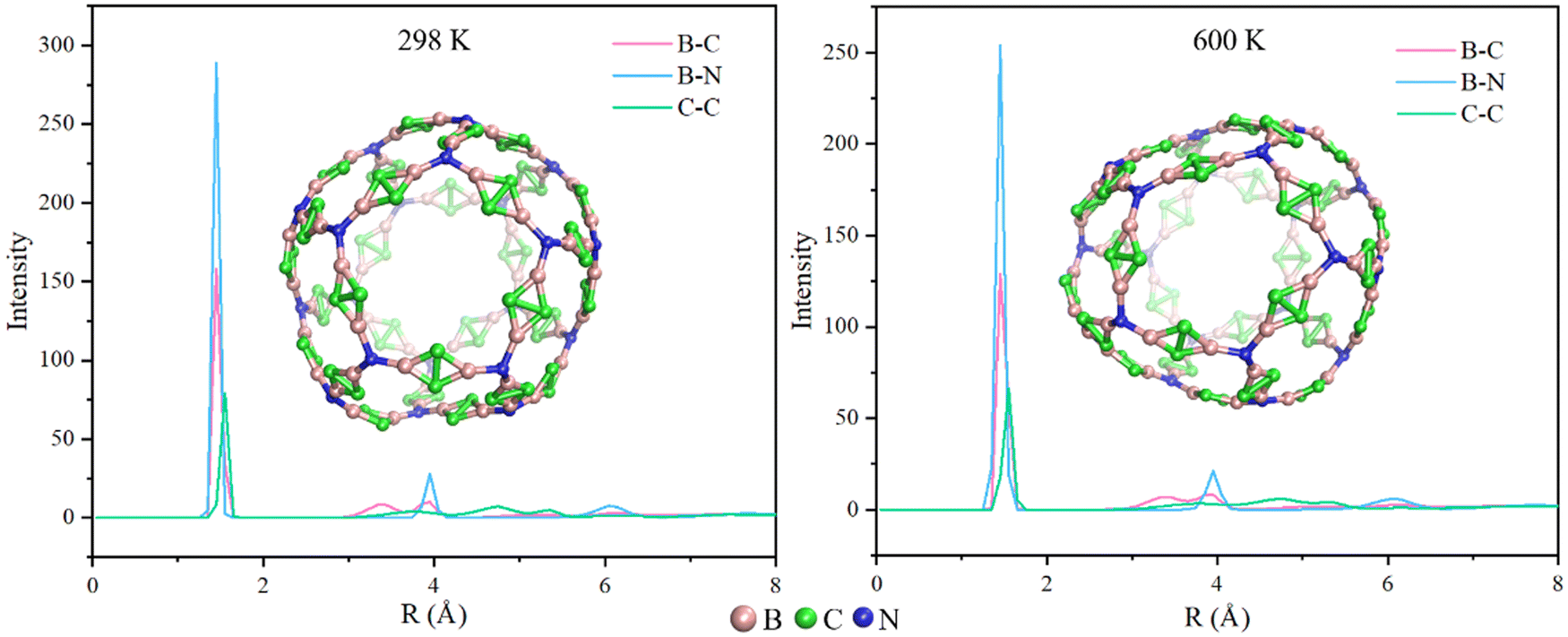 | ||
| Fig. 2 Radial distribution of interatomic distances at 298 and 600 K. Color scheme: B atom (in pink), C atom (in green), and N atom (in blue). | ||
A previous study shows that C2B2H2 has the aromatic character according to the NICS(0) and NICS(1) values at the ring center of the three-membered CCB moiety in C2B2H2.21 The NICS(0) and NICS(1) values (inside and outside of the cage) of N20(C2B2)30 are calculated and shown in Fig. S4 (see the ESI†). It is shown that the NICS(0) value of N20(C2B2)30 is −17.49, and the NICS(1) values of inside and outside of the cage of N20(C2B2)30 are −21.32 and −15.30, respectively. Note that the NICS(1) value inside the cage is close to that of C2B2H2 molecules,21 indicating that N20(C2B2)30 also possesses aromatic character.
3.2. Cycloaddition reaction of N20(C2B2)30
BHCs, DBHCs, and SiBHCs can behave as dienophiles to undergo Diels–Alder reactions with dienes due to their CC bond character.39,40,42 Herein, in order to investigate the reactivity of the CC bond, the mechanism for the Diels–Alder reactions of N20(C2B2)30 with butadiene and cyclopentadiene is investigated. The predicted relative free energy profiles of the two Diels–Alder reactions, along with the optimized structures of transition states and products for analyzing the formation of chemical bonds, are shown in Fig. 3. Note that Diels–Alder reaction of N20(C2B2)30 with butadiene is exothermic and yields the product (PC1) via the transition state (TS1) with a free energy barrier of 20.9 kcal mol−1 and an energy release of 48.0 kcal mol−1, which is similar to the addition reaction of BHCs with butadiene.40 In the Diels–Alder reaction of N20(C2B2)30 with cyclopentadiene, a loose intermolecular complex (RC) is formed at first, and then the reaction undergoes the transition state (TS2) to generate the product (PC2), which is consistent with the Diels–Alder reaction of fullerene C60 with cyclopentadiene.45 The free energy barrier for the Diels–Alder reaction of N20(C2B2)30 with cyclopentadiene is 20.6 kcal mol−1, and the Gibbs free energies of the reaction ΔG is −15.5 kcal mol−1. Clearly, the Diels–Alder reactions of N20(C2B2)30 with butadiene and cyclopentadiene are feasible, both thermodynamically and kinetically.
 | ||
| Fig. 3 The predicted relative free energy profiles (in kcal mol−1) for the Diels–Alder reactions of N20(C2B2)30 with butadiene and cyclopentadiene are shown. Optimized structures, key bond lengths (Å, in plain), and WBIs (in italic). Color scheme: H atom (in white), B atom (in pink), C atom (in green), and N atom (in blue). | ||
The [3+2] cycloaddition reaction of C60 as well as its derivatives towards N2CH2 undergo either thermal or photochemical loss of dinitrogen to give C60CH2.13 Here, the mechanism for the [3+2] cycloaddition reaction of N20(C2B2)30 with N2CH2, including loss of dinitrogen, is illustrated in Fig. 4. It is revealed that the [3+2] cycloaddition proceeds to an intermediate (IM) through the transition state (TS3) with an energy barrier of 14.6 kcal mol−1 and an energy release of 25.8 kcal mol−1. Thus, the [3+2] cycloaddition is feasible both thermodynamically and dynamically. Followed by the release of N2, i.e., N20(C2B2)30N2CH2 (IM) → N20(C2B2)30CH2 (PC3) + N2, the final product N20(C2B2)30CH2 (PC3) of the [3+2] cycloaddition reaction, containing a 3, 4-boratricyclo[1.1.1]pentane, is formed, and the overall Gibbs free energies of the reaction ΔG is −47.6 kcal mol−1 relative to the initial reactants, showing the [3+2] cycloaddition is quite feasible, both thermodynamically and kinetically.
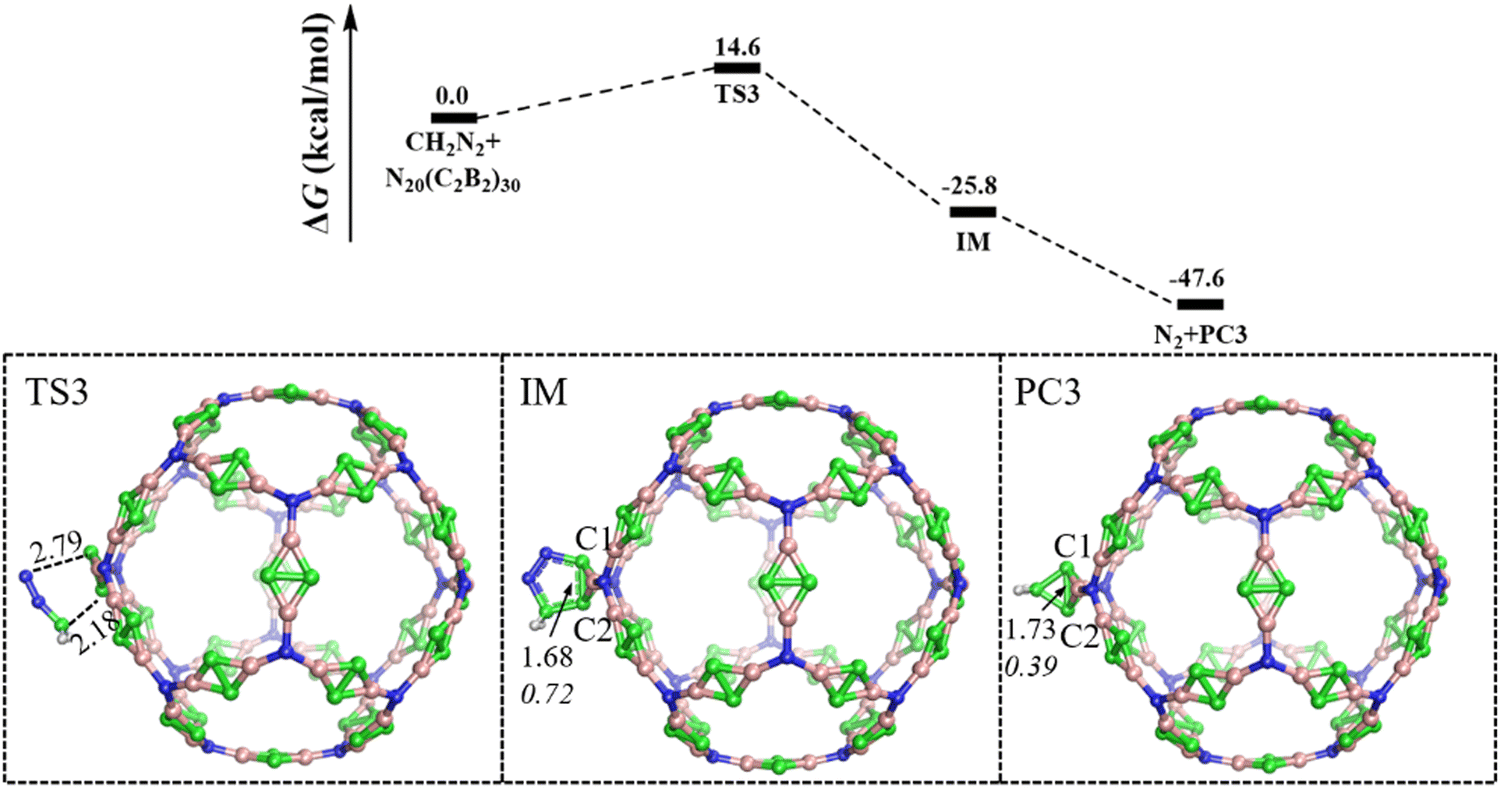 | ||
| Fig. 4 The predicted relative free energy profile (in kcal mol−1) for the [3+2] cycloaddition of N20(C2B2)30 with diazomethane is shown. Optimized structures, key bond lengths (Å, in plain), and WBIs (in italic). Color scheme: H atom (in white), B atom (in pink), C atom (in green), and N atom (in blue). | ||
The lengths (WBIs) of the C1–C2 bond in PC1, PC2, IM, and PC3 are 1.73 (0.66), 1.69 (0.74), 1.68 (0.72), and 1.73 Å (0.39), respectively. Thus, the C1–C2 bond is a weak single bond, as found in the products of the Diels–Alder reaction of BHCs.40
We have further investigated the reaction mechanism of the successive addition of three butadiene molecules to N20(C2B2)30. The results show that the free energy barriers at different stages of addition remain essentially unchanged, and the reaction exhibits sustained exergonicity, although the trend of exergonicity slows down. Therefore, it can be inferred that N20(C2B2)30 may undergo successive addition with butadiene molecules until saturation (see Fig. S5 in the ESI†). In addition, we have compared the addition reaction activities of the CC bonds in the exohedral and endohedral additions of the cluster with butadiene. The results indicate that both have comparable addition energy barriers. However, it should be noted that due to the spatial effects within the cluster, the stability of the endohedral addition product is slightly lower than that of the corresponding exohedral product (see Fig. S6 in the ESI†).
3.3. Binding interactions of N20(C2B2)30 and its ionic complexes with small molecules
Due to the CS bonding character of the inverted CC bond in N20(C2B2)30, the carbon atoms can serve as the active sites to bind small molecules. Accordingly, the binding behaviors of N20(C2B2)30 toward small gas molecules, such as H2, CO, NO, and NO2, are considered here. Optimized structures of complexes N20(C2B2)30(H2)2, N20(C2B2)30(CO)2, N20(C2B2)30(NO)2, and N20(C2B2)30(NO2)2 are illustrated in Fig. 5, and the vibrational analysis reveals that they are all stable without any imaginary frequency. In addition, the evaluated binding energies of N20(C2B2)30 with H2, CO, NO, and NO2 are −3.96, −3.16, −1.72, and −2.57 eV, respectively, indicating that N20(C2B2)30 can effectively capture H2, CO, NO, and NO2. Moreover, since the binding energies are significantly different from each other, N20(C2B2)30 might be used to separate H2, CO, NO, and NO2.
 | ||
| Fig. 5 The complexes of N20(C2B2)30 with small gas molecules H2, CO, NO, and NO2 are shown. Selected bond lengths are given in Å. Color scheme: H atom (in white), B atom (in pink), C atom (in green), N atom (in blue), and O atom (in red). | ||
Every carbon atom of the C2B2 unit in N20(C2B2)30(H2)2 and N20(C2B2)30(CO)2 forms two C–H bonds with H2 and a CCO bond with CO, respectively, resulting in the cleavage of the inverted CC bond of N20(C2B2)30. Interestingly, CN (left) and C–N (right) in N20(C2B2)30(NO)2 are obviously different with the bond lengths of 1.20 and 1.42 Å, respectively. In order to better understand the bonding nature, the natural bond orbital analysis of N20(C2B2)30(NO)2 is performed. As seen from Fig. S7 (see the ESI†), there is a significant electron transfer from the N1 atom to the C2 atom, resulting in the C2 atom with a net negative charge of −0.64. The 2p orbitals of the left O, N, and C atoms form a linear structure with Π22 and Π34 bonds when the N atom loses an electron. In contrast, the 2p orbitals of the right O, N and C atoms form a V-like structure with the Π34 bond when the C atom accepts an electron. The lengths of two NO bonds in N20(C2B2)30(NO)2 are around 1.20 Å. Two carbon atoms in the C2B2 unit in N20(C2B2)30(NO2)2 form two C–N bonds and an inverted C–C bond, and the latter bears a weak π bond character with a distance of 1.71 Å. Thus, N20(C2B2)30 accommodates two NO2 through the covalent form of the CS bond of the inverted C⋯C.
In particular, due to the empty 2pz orbital of the boron atom in N20(C2B2)30, boron atoms may bind CO through a dative bond interaction. The optimized structure of the binding complex N20(C2B2)30(CO) is depicted in Fig. 6, and it is a minimum on the PES predicted by the frequency analysis. The calculated adsorption energy of N20(C2B2)30 + CO → N20(C2B2)30CO is −0.02 eV, suggesting that the boron atom in N20(C2B2)30 is also an active site for CO adsorption.
 | ||
| Fig. 6 The adsorption of CO at the B site of N20(C2B2)30 is shown, where key bond lengths are given in Å. Color scheme: B atom (in pink), C atom (in green), N atom (in blue), and O atom (in red). | ||
Although it can bind with the aforementioned small molecules, N20(C2B2)30 cannot effectively bind with CO2 and N2O. Note that a zeolite recently reported can be used to separate CO2 and N2O through the Ag+ site.46 The carbon atoms of the inverted CC bond in N20(C2B2)30 can coordinate to the transition metal (TM) to form a complex (TM)N20(C2B2)30.39 Interestingly, it is found that the newly generated (TM)N20(C2B2)30+ (TM = Cu, Ag, and Au) could capture CO2 and N2O. The optimized structures of (TM)N20(C2B2)30+, (CO2)(TM)N20(C2B2)30+, and (N2O)(TM)N20(C2B2)30+, and the corresponding geometric parameters are illustrated in Fig. 7. Note that TM in (TM)N20(C2B2)30+ is coordinated with the terminal C atoms of three inverted CC units. Specifically, Cu+ in CuN20(C2B2)30+ binds with CO2 and N2O through the interaction of their O and N atoms with Cu, where the newly formed Cu–O or Cu–N bond replaces one Cu–C bond connected to the inverted CC unit. Differing from CuN20(C2B2)30+, the formation of the TM–O/N bond in (TM)N20(C2B2)30+ in (TM = Ag and Au) may weaken the Cu–C (inverted CC) bond to some extent. Calculated binding energies for (TM)N20(C2B2)30+ + CO2 → (CO2)(TM)N20(C2B2)30+ (TM = Cu, Ag, and Au) are −0.65, −0.38, and −0.09 eV, respectively. Similarly, the binding energies for (TM)N20(C2B2)30+ + N2O → (N2O)(TM)N20(C2B2)30+ (TM = Cu, Ag, and Au) are −0.68, −0.34, and −0.21 eV, respectively. Here the predicted binding energies of CO2 and N2O bound at the Ag+ site of AgN20(C2B2)30+ are well consistent with those of these small molecules bound at the Ag+ site of the reported zeolite.46 Accordingly, the introduction of transition metal ions can remarkably enhance the capability of N20(C2B2)30 toward the binding of inactive molecules, such as CO2 and N2O. Furthermore, the adsorption energy difference of 0.12 eV between AuN20(C2B2)30+ with CO2 and N2O indicates that these two gas molecules can be effectively separated. In order to better understand the interaction between TM (TM = Cu, Ag, and Au) and CO2, as well as TM and NO2, the electron density and Laplacian values of the critical points (CPs) of TM–O and TM–N bonds in (CO2)(TM)N20(C2B2)30+ and (N2O)(TM)N20(C2B2)30+ are calculated and listed in Table S1 (see the ESI†). The electron density and Laplacian values of the CPs of TM–O and TM–N bonds are in the range of 0.03–0.07 and 0.16–0.37, respectively, indicating that the TM–O and TM–N bonds are ionic bonds.43
 | ||
| Fig. 7 The optimized complexes of AgN20(C2B2)30+, CuN20(C2B2)30+, and AuN20(C2B2)30+ with CO2 and N2O, where key bond lengths are given in Å. Color scheme: B atom (in pink), C atom (in green), N atom (in blue), O atom (in red), Ag atom (in silver), Cu atom (in copper), and Au atom (in gold). | ||
4. Conclusions
A high-symmetry cage-like molecule N20(C2B2)30, resembling the fullerene C20 structurally, has been constructed by taking C2B2 and N motifs as linkers and nodes, respectively, and its stability and reactivity for cycloaddition and adsorption reactions are further demonstrated using density functional theory (DFT) calculations and AIMD simulations. The calculated results show that three cycloaddition reactions of N20(C2B2)30 with butadiene, cyclopentadiene, and diazomethane are all exothermic, and the corresponding free energy barriers are 20.9, 20.6, and 14.6 kcal mol−1, respectively, showing that these reactions are feasible, both thermodynamically and kinetically. For the adsorption reactions, the carbon atoms of the inverted CC bond in N20(C2B2)30 can effectively bind with H2, CO, NO, and NO2, whereas the boron atom site only accommodates CO. After the modification with transition metal (TM) ions (Cu+, Ag+, and Au+), CO2 and N2O can bind at the TM site of the obtained complexes (TM)N20(C2B2)30+. Among these transition metal complexes, only AuN20(C2B2)30+ shows obviously different adsorption energies for CO2 and N2O, and may be applied to effectively separate these two gas molecules. The present findings for novel structure and reactivity of N20(C2B2)30 may endow it with potential for applications in materials chemistry.
Author contributions
All authors contributed to the writing of the manuscript and approved the final version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Science Foundation of China (22033006 and 92372105).Notes and references
- H. C. Longuet-Higgins and M. D. Roberts, Proc. R. Soc. London, Ser. A, 1955, 230, 110–119 CAS.
- A. R. Pitochelli and F. M. Hawthorne, J. Am. Chem. Soc., 1960, 82, 3228–3229 CrossRef CAS.
- J. A. Wunderlich and W. N. Lipscomb, J. Am. Chem. Soc., 1960, 82, 4427–4428 CrossRef CAS.
- X. Zhao, Z. Yang, H. Chen, Z. Wang, X. Zhou and H. Zhang, Coord. Chem. Rev., 2021, 444, 214042 CrossRef CAS.
- H. W. Kroto, J. R. Heath, S. C. O’Brien, R. F. Curl and R. E. Smalley, Nature, 1985, 318, 162–163 CrossRef CAS.
- K. M. Kadish and R. Ruoff, Fullerenes: Chemistry, Physics, and Technology, Wiley-VCH, New York, 2000 Search PubMed.
- A. W. Hains, Z. Liang, M. A. Woodhouse and B. A. Gregg, Chem. Rev., 2010, 110, 6689–6735 CrossRef CAS PubMed.
- S. Goodarzi, T. Da Ros, J. Conde, F. Sefat and M. Mozafari, Mater. Today, 2017, 20, 460–480 CrossRef CAS.
- G. Speranza, Nanomaterials, 2021, 11, 967 CrossRef CAS PubMed.
- T. Suzuki, Q. Li, K. C. Khemani, F. Wudl and O. Almarsson, Science, 1991, 254, 1186 CrossRef CAS PubMed.
- T. Suzuki, Q. Li, K. C. Khemani and F. Wudl, J. Am. Chem. Soc., 1992, 114, 7301–7302 CrossRef CAS.
- F. Wudl, Acc. Chem. Res., 1992, 25, 157–161 CrossRef CAS.
- A. B. Smith, R. M. Strongin, L. Brard, G. T. Furst, W. J. Romanow, K. G. Owens and R. C. King, J. Am. Chem. Soc., 1993, 115, 5829–5830 CrossRef CAS.
- Q. Sun, Q. Wang, P. Jena and Y. Kawazoe, J. Am. Chem. Soc., 2005, 127, 14582–14583 CrossRef CAS PubMed.
- R. N. Goyal, V. K. Gupta, N. Bachheti and R. A. Sharma, Electroanalysis, 2008, 20, 757–764 CrossRef CAS.
- J. Zheng, L. Huang, C.-H. Cui, Z.-C. Chen, X.-F. Liu, X. Duan, X.-Y. Cao, T.-Z. Yang, H. Zhu, K. Shi, P. Du, S.-W. Ying, C.-F. Zhu, Y.-G. Yao, G.-C. Guo, Y. Yuan, S.-Y. Xie and L.-S. Zheng, Science, 2022, 376, 288–292 CrossRef CAS PubMed.
- N. Gonzalez Szwacki, A. Sadrzadeh and B. I. Yakobson, Phys. Rev. Lett., 2007, 98, 166804 CrossRef PubMed.
- E. Scuseria, Chem. Phys. Lett., 1991, 176, 423–427 CrossRef.
- A. D. Zdetsis, Phys. Rev. B, 2007, 76, 075402 CrossRef.
- K. Geng, T. He, R. Liu, S. Dalapati, K. T. Tan, Z. Li, S. Tao, Y. Gong, Q. Jiang and D. Jiang, Chem. Rev., 2020, 120, 8814–8933 CrossRef CAS PubMed.
- J. Jian, W. Li, X. Wu and M. Zhou, Chem. Sci., 2017, 8, 4443–4449 RSC.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- D. Andrae, U. Häußermann, M. Dolg, H. Stoll and H. Preuß, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS.
- Y. Duan, C. Wu, S. Chowdhury, M. C. Lee, G. Xiong, W. Zhang, R. Yang, P. Cieplak, R. Luo, T. Lee, J. Caldwell, J. Wang and P. Kollman, J. Comput. Chem., 2003, 24, 1999–2012 CrossRef CAS PubMed.
- S. F. Boys and F. Bernardi, Mol. Phys., 2002, 100, 65–73 CrossRef.
- E. D. Glendening, C. R. Landis and F. Weinhold, J. Comput. Chem., 2019, 40, 2234–2241 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- P. V. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. Van Eikema Hommes, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg Williams, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. MontgomeryJr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian Inc., Wallingford, CT, 2016 Search PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B, 1993, 47, 558–561 CrossRef CAS PubMed.
- M. Torrent, N. A. W. Holzwarth, F. Jollet, D. Harris, N. Lepley and X. Xu, Comput. Phys. Commun., 2010, 181, 1862–1867 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Nosé, J. Chem. Phys., 1984, 81, 511–519 CrossRef.
- B. J. Levandowski and K. N. Houk, J. Am. Chem. Soc., 2016, 138, 16731–16736 CrossRef CAS PubMed.
- C. Zhang, W. Sun and Z. Cao, J. Chem. Phys., 2007, 126, 144306 CrossRef PubMed.
- B. N. Cyvin, E. Brendsdal, J. Brunvoll and S. J. Cyvin, J. Mol. Struct., 1995, 352, 481–488 CrossRef.
- G. Duškesas and S. Larsson, Theor. Chem. Acc., 1997, 97, 110–118 Search PubMed.
- C. Zhang and F. Li, J. Phys. Chem. A, 2012, 116, 9123–9130 CrossRef CAS PubMed.
- C. Zhang, Z. Wang, J. Song, C. Li and Y. Mo, Theor. Chem. Acc., 2019, 138, 106 Search PubMed.
- Z. Tian, C. Zhang, Z. Pei, J. Liang and Y. Mo, Mol. Syst. Des. Eng., 2023, 8, 85–91 RSC.
- C. Cao, C. Zhang, J. Gu and Y. Mo, Phys. Chem. Chem. Phys., 2024, 26, 28082–28090 RSC.
- S. Shaik, D. Danovich, W. Wu and P. C. Hiberty, Nat. Chem., 2009, 1, 443–449 CrossRef CAS PubMed.
- S. Shaik, D. Danovich, J. M. Galbraith, B. Braïda, W. Wu and P. C. Hiberty, Angew. Chem., Int. Ed., 2020, 59, 984–1001 CrossRef CAS PubMed.
- H. Ueno, H. Kawakami, K. Nakagawa, H. Okada, N. Ikuma, S. Aoyagi, K. Kokubo, Y. Matsuo and T. Oshima, J. Am. Chem. Soc., 2014, 136, 11162–11167 CrossRef CAS PubMed.
- L. Wang, C. Lin, I. Boldog, J. Yang, C. Janiak and J. Li, Angew. Chem., Int. Ed., 2024, 63, e202317435 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp04653f |
| This journal is © the Owner Societies 2025 |