
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComputer-aided design of triazolo-cages as anion receptors†
Minwei
Che
 ,
Sibali
Debnath‡
,
Amar H.
Flood
and
Krishnan
Raghavachari
*
,
Sibali
Debnath‡
,
Amar H.
Flood
and
Krishnan
Raghavachari
*
Department of Chemistry, Indiana University, Bloomington, IN 47405, USA. E-mail: mche@iu.edu; sdebnath@iu.edu; aflood@iu.edu; kraghava@indiana.edu
First published on 12th February 2025
Abstract
Molecular cages with three-dimensional cavities have garnered significant interest due to their enhanced encapsulation abilities. In this study, we computationally investigate the binding behavior of a triazolo-cage receptor composed of alternating triazole and phenyl building blocks. With six different anions, including atomic (F−, Cl−, Br−, and I−), linear (SCN−), and trigonal planar (NO3−) geometries, we analyze the binding selectivity of the parent cage with DFT calculations. The influence of solvation on binding strength is investigated by calculating binding free energies in both gas phase and six solvent environments of progressively increasing dielectric constants. Symmetry-Adapted Perturbation Theory (SAPT) analysis reveals that electrostatic interactions dominate the binding process. Additionally, we perform computer-aided design to generate a series of new cage receptors with diverse functionalities, and our findings highlight the tunable chloride affinity achieved by adjusting various cage properties. Overall, this study offers insights into the design of novel cage receptors with versatile functionalities and provides a strategic approach to the rational design of anion receptors.
Introduction
Supramolecular receptors that selectively bind anions are critical in applications such as anion sensing,1–4 anion extraction,5–8 and catalysis.9–12 These synthetic hosts feature molecular segments designed to selectively bind guest anions within well-defined intramolecular cavities, primarily through various non-covalent interactions.13–16 Hydrogen bonding, one of the most extensively studied supramolecular interactions, has traditionally been facilitated in anion receptors through the incorporation of NH groups, e.g., ureas,6,17,18 amides,19–21 and pyrroles.22,23 Early investigations into the CH hydrogen bonding motif were carried out in the 1960s by Sutor.24 However, the full significance of CH hydrogen bonds has only been recently recognized, as demonstrated through a series of preorganized macrocyclic and cage receptors that only bind anions using activated CH donors.25–31With the advancement in software and hardware in the past few years, computational methods have been applied to provide valuable insights into hydrogen-bonded chemical systems.32–34 Supramolecular methods35 calculate the interaction energy by determining the difference between the energy of the complex and the sum of the energies of its individual monomers.36 Commonly used methods in this category include dispersion-corrected density functional theory (DFT-D),37–39 Møller–Plesset perturbation theory (MP2),40–42 and coupled-cluster theory.43–45 In contrast, perturbative methods calculate the interaction energy by treating it as a perturbation to the Hamiltonian of the monomers.46 One of the most popular perturbative methods is symmetry-adapted perturbation theory (SAPT),47–49 which offers a physically meaningful decomposition of intermolecular interactions into electrostatic, induction, dispersion, and exchange-repulsion components.
Computer-aided design has revolutionized the way new materials are developed, providing tools that allow for the efficient screening50,51 and optimization of molecular structures.52–54 By leveraging computational techniques to predict the properties55 and behaviors56 of potential materials, researchers can significantly reduce the time and cost associated with experimental trial-and-error. In the context of anion receptors, computer-aided design enables the fine-tuning of structures51,57–59 after the use of computational methods help to understand supramolecular features like preorganization60–62 and interactions.63–65 This knowledge helps to provide a basis for proposing candidates with enhanced selectivity and binding affinities. By simulating interactions between receptors and anions, researchers can optimize receptor structures for improved performance in target applications.
Effective anion binding depends on the complementary fit between the receptor and the anion in terms of size, shape, and interactions.66 Therefore, when designing receptors, it is crucial to focus on functionalization, preorganization, and the interaction types. Molecular cages with three-dimensional (3D) cavities have received considerable attention in this regard since the creation of Katapinand66 and amine- or amide-based cryptands.67–69 These cages offer enhanced encapsulation for anions compared to two-dimensional (2D) hosts,70–73 leading to excellent selectivity.74,75 For example, the small hemicryptophane tris-urea cage synthesized by Delecluse et al.76 shows exclusive selectivity for F− over other halides. Xu et al.77 introduced a tricationic amide cage for anion recognition and catalysis in water. Jiang et al.78 synthesized a conformation-adaptive phenazine-based Pd2L4 cage receptor which exhibits very strong halide binding affinity. In the context of the structures investigated in this study, Liu et al.28 created a cryptand-like triazolo-cage that shows attomolar affinity for Cl−. A recent computational study by Li et al.79 explored the rigidity and binding cavity of cryptand-like cages, including this triazolo-cage, focusing on their interactions with atomic halides in the gas phase. However, their work did not examine these interactions in solution or with larger molecular anions of varying shapes.
In this work, we investigate the binding behavior of the parent triazolo-cage receptor (vide infra) in solution to six anions of various geometries, including atomic (F−, Cl−, Br−, I−), linear (SCN−), and trigonal planar (NO3−). We evaluate the influence of solvent on the interaction strength by calculating binding free energies in the gas phase and in six different solvent environments of progressively increasing dielectric constants. Additionally, we perform in silico design to generate a series of modified cage receptors with diverse functionalities, guided by various complementarity criteria. Furthermore, we predict the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 chloride binding free energies for these newly designed cage receptors. Overall, our study presents a strategic approach to designing new cage receptors with versatile functionalities.
:1 chloride binding free energies for these newly designed cage receptors. Overall, our study presents a strategic approach to designing new cage receptors with versatile functionalities.
Computational methods
Density functional theory (DFT) calculations were performed using the Gaussian 16 suite of programs.80 The M06-2X functional81 was employed throughout the study. Solvent effects were accounted for using the conductor-like polarizable continuum model (CPCM).82 The Tight SCF convergence criteria and the UltraFine integration grid were used in all the DFT calculations. For geometry optimizations, the 6-311+G(d) basis set83,84 was used for the atoms of the cage receptors, whereas the larger 6-311+G(3df,p) basis set83,85 was applied for the atoms of the anion guests. A customized in-house basis set of comparable quality was used for iodine, with the coefficients provided in the ESI.† Vibrational frequencies and thermal corrections were evaluated using the same basis sets, and all optimized structures were verified as minima without imaginary frequencies. Natural population analysis was performed at the same level of theory using NBO 3.1.86–88 Single-point energies were obtained with larger basis sets to compute the binding affinities. In these calculations, the 6-311++G(3df,2p) basis set was used for the cage atoms, the aug-cc-pVTZ basis set for the anion atoms,89–91 except for bromine and iodine atoms, for which the aug-cc-pVTZ-PP basis set92,93 where pseudopotentials describing the inner core orbitals were used.Symmetry-adapted perturbation theory (SAPT) analysis was performed using the scaled SAPT0 method, sSAPT035 implemented in the Psi4 software.94 It incorporates scaling factors to improve the accuracy of exchange-type terms. The jun-cc-pVDZ basis set95 was used for all atoms except for bromine and iodine for which the jun-cc-pVDZ-PP basis set with pseudopotentials was applied.
Results and discussion
Anion complexes of the parent cage
The rigid triazolo-cage synthesized by Liu et al.28 shows an attomolar affinity for the chloride anion, and the crystal structure of is its chloride complex is illustrated in Fig. 1a and b. In this study, the triazolo-cage serves as the model structure. To reduce the computational cost, each acyl dicyclohexylamine group on the three phenyl groups is substituted by a hydrogen atom to form the simplified parent cage 1 (Fig. 1c and d). Cage 1 features a binding pocket of 3.6 Å in diameter, enveloped by a backbone of three arms. Each arm consists of two triazoles serving as polarized H-bond donors, along with one bridging phenylene that enhances its rigidity. With three equivalent arms, the cage has intrinsic three-fold symmetry (D3 point group). | ||
| Fig. 1 Crystal structure of the triazolo-cage: (a) side view and (b) top view. Optimized structure of cage 1 in the gas phase at the M06-2X/6-311+G(d) level of theory: (c) side view and (d) top view. | ||
In this study, we investigate the 1:1 binding of cage 1 with six anions to examine its binding preferences. The anionic guests range from 2.5 to 4.2 Å in size.96 The optimized structures of the complexes in the gas phase are shown in Fig. 2.
 | ||
| Fig. 2 Optimized structures of 1:1 1·X− in the gas-phase using. Side view of (a) 1·F−, (b) 1·Cl−, (c) 1·Br−, (d) 1·I−, (e) 1·NO3− and (f) 1·SCN−. Top view of (g) 1·F−, (h) 1·Cl−, (i) 1·Br−, (j) 1·I−, (k) 1·NO3− and (l) 1·SCN−. | ||
In the gas phase, complexes with F−, Cl−, and I− retain the D3-symmetry of the parent cage, where those halides are positioned at the center of the host cavity. Interestingly, the Br− complex shows a slight distortion that breaks the D3-symmetry. However, the energy lowering from the distortion is very small and is not physically meaningful. Thus, we suggest that it is an artifact resulting from the slight instabilities in the DFT numerical integration grids, not uncommon for such large molecules in high symmetries. Smaller anions like F−, Cl−, and Br− are stabilized by six H-bonds from triazoles and three H-bonds from phenylenes, whereas the large size of I− compels the triazole hydrogen bond donors to orient away from the center of the binding site, leading to ineffective hydrogen bonding. However, in solvents with increasing dielectric constants, the small fluoride anion (d = 2.5 Å) moves away from the cavity center towards one of the nitrogen atoms on the three-fold axis. As shown in Table 1, the distance between the F atom and the nearest N atom on the axis in the gas phase D3-symmetric structure is 4.38 Å. This distance decreases by 0.36 Å in CHCl3 (εr = 4.71) and an additional by 0.12 Å in DCM (εr = 8.93). Nevertheless, the trend levels off starting from acetone (εr = 20.49) to H2O (εr = 78.36), where the distance stabilizes at 3.87 Å. On the opposite side, the longer N⋯F distance on the other side is 4.38 Å in the gas phase, increasing to 4.79 Å in CHCl3 and 4.97 Å in DCM, before stabilizing at 5.0 Å from acetone onward. In contrast, larger spherical halides like Cl−, Br−, and I−, with diameters of 3.4 Å, 3.8 Å, and 4.2 Å, respectively, do not exhibit this drift along the axis due to their larger sizes, which prevent significant movement.
| Gas | CHCl3 | DCM | Acetone | MeCN | DMSO | H2O | |
|---|---|---|---|---|---|---|---|
| Dielectric constant (εr) | 1 | 4.71 | 8.93 | 20.49 | 35.69 | 46.83 | 78.36 |
| Distance between F and the nearest axis N atoms (Å) | 4.38 | 4.02 | 3.90 | 3.87 | 3.87 | 3.87 | 3.87 |
| Distance between F and the farther axis N atoms (Å) | 4.38 | 4.79 | 4.97 | 5.00 | 5.00 | 5.00 | 5.00 |
The D3-symmetry is also observed in the 1·NO3− complex, where the nitrogen atom of the nitrate is positioned at the center of the cavity. Each terminus oxygen atom forms two H-bonds (2.2 Å) with two opposing triazole units on different arms of the cage, while the hydrogen bond donors from the phenyl units are directed towards the nitrogen atom of the nitrate. Interestingly, despite NO3− being larger (d = 4.0 Å) than the spherical cavity, its trigonal planar structure is compatible with the three-arm structure of the cage receptor, though the oxygen atoms can be seen to be slightly outside the cavity.
Since SCN− is too large (d = 4.2 Å) for the spherical cavity of the parent cage, and it is only partially stabilized by the receptor. While its nitrogen terminus remains in the cavity, the sulfur atom lies outside of the binding site, resulting in a C2-symmetric form of 1·SCN−. The N terminus is stabilized by three H-bonds from phenylenes, as well as two H-bonds from two alternating triazole units on the same arm of cage 1. Nonetheless, the rest of the four triazole units do not participate in intermolecular hydrogen bonding. Since the number of effective H-bonds formed in 1·SCN− is considerably fewer than what was found in halide complexes, weaker binding with thiocyanate is expected.
Binding energies of the parent cage
The 1:1 binding free energies of cage 1 with the six anions were calculated in the gas phase and across six solvents of varying dielectric constants: chloroform (εr = 4.71), dichloromethane (εr = 8.93), acetone (εr = 20.49), acetonitrile (εr = 35.69), dimethylsulfoxide (εr = 46.83) and water (εr = 78.36). Complexation reactions are represented by eqn (1), where X− = F−, Cl−, Br−, I−, NO3−, and SCN−:| 1 + X− ⇄ 1·X− | (1) |
The calculated binding free energies are presented in Fig. 3a and Table S1 (ESI†). Among the anions tested, fluoride exhibits the strongest binding, followed by spherical chloride and bromide. By comparison, iodide demonstrates significantly weaker binding. The decline in binding free energies from Br− to the larger I− highlights the size selectivity of this cage. Additionally, the receptor's shape selectivity is evident from the notably lower binding energies for the trigonal planar nitrate and the linear thiocyanate. Overall, the following order of anion binding energy and selectivity for cage 1 is observed: F− > Cl− > Br− > NO3− > SCN− > I−.
 | ||
| Fig. 3 (a) Computed 1:1 anion binding free energies, ΔG (kcal mol−1), of cage 1 with F−, Cl−, Br−, I−, NO3−, and SCN− in gas, CHCl3, DCM, acetone, MeCN, DMSO, and H2O. (b) The dependence of the binding energy of 1·Cl− on the inverse of the solvent dielectric constant. | ||
Across the solvents, binding energies decrease, consistent with the trend previously in computational and experimental studies.30,61,64 An excellent linear correlation (R2 = 0.995) between the binding energy and the inverse of dielectric constant (1/εr) is illustrated in Fig. 3b for 1·Cl−. Similar correlations for the other ions are shown in Fig. S2 (ESI†).
The computed binding free energies of cage 1 with Cl−, Br−, I−, and NO3− have been compared to the experimental values obtained from titrations in DMSO as reported by Liu et al.2 As shown in Table S4 and Fig. S1 (ESI†), the computational method successfully reproduces the experimental trend, yielding a slope of 0.99. While it consistently underestimates binding free energies by 1–3 kcal mol−1, we believe a correction based on the linear regression can account for this systematic error. Experimental results were not reported for F−, though its strongest binding affinity is consistent with the high charge density, and in agreement with the gas phase calculations of Li et al.79 To better understand the basis for this trend, we performed a SAPT energy decomposition analysis, which provide a detailed breakdown of the contributing forces.
As shown in Fig. 4a, the SAPT results provide a breakdown of the different fundamental interactions that contribute to the stabilization of anion complexes formed with cage 1. Among the three attractive forces—electrostatics, induction, and dispersion—electrostatic contributions overwhelmingly dominate, as shown by the energy breakdown. This trend is further emphasized in Fig. 4b, where the ratio of electrostatics to the total interaction energy clearly surpasses that of the other components. It is interesting that the three non-electrostatic contributions (exchange-repulsion, induction, and dispersion) sum up to a small value, mostly due to the repulsive nature of the exchange term. Due to this near-cancellation, the electrostatic contributions closely track the total interaction strength. Similar observations have been made previously by Liu et al.28 in their investigation of the analogous 2-D receptor with anions.
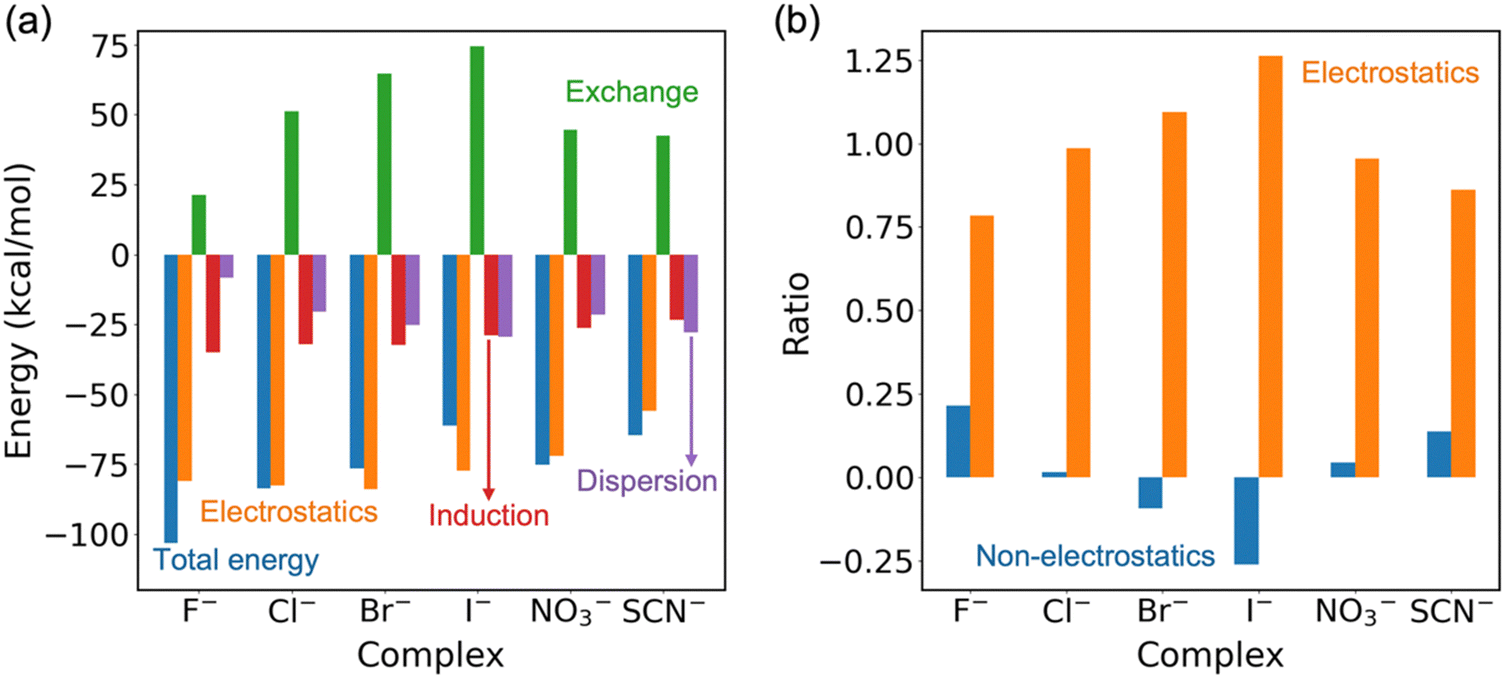 | ||
| Fig. 4 (a) SAPT calculations. (b) Ratios of non-electrostatics (exchange-repulsion, induction and dispersion) and electrostatics of the total sSAPT0 energies in the gas phase. | ||
A noticeable increase in exchange repulsion is observed as the size of the halide anion increase, from 1·F− to 1·Cl−, 1·Br−, and 1·I−. This trend reflects the growing wavefunction overlap between these anions and the cage. The largest iodide ion (I−) experiences the highest exchange repulsion, while the smallest fluoride (F−) exhibits considerably less. Although cage 1 was found to selectively bind chloride, with a cavity tailored in size and shape, the lower exchange repulsion in the 1·F− complex results in fluoride binding even stronger than chloride. Interestingly, 1·NO3− and 1·SCN− show reduced exchange repulsion due to the geometric mismatch between these non-spherical anions and the largely spherical binding cavity of cage 1.
To understand the secondary attraction contributions from induction interactions, we analyzed the partial charges of the anions in the complexes. The charges (Table 2), as determined using the natural population analysis, indicate that F− retains the highest partial negative charge, consistent with its strongest induction contribution. In comparison, the larger anions such as I− and NO3− show significantly lower charges in the complexes, which correlates with their reduced induction effects. Finally, dispersion forces, which arise from the dynamic correlation between electrons on the anion and the receptor, follow the expected trend based on anion size and polarizability. The least polarizable F− generates the weakest dispersion forces, while the larger, more polarizable I− and NO3− contribute significantly to the dispersion component.
| Anion | Atom | Natural charge |
|---|---|---|
| F− | F | −0.94249 |
| Cl− | Cl | −0.88624 |
| Br− | Br | −0.85536 |
| I− | I | −0.85201 |
| SCN− | S | −0.28759 |
| C | 0.08798 | |
| N | −0.72044 | |
| NO3− | N | 0.74705 |
| O | −0.54893 | |
| O | −0.54894 | |
| O | −0.54894 |
In summary, while electrostatic interactions dominate the binding in all six complexes, induction plays a secondary attractive role for highly polarizing anions. Meanwhile, dispersion becomes more important in stabilizing the larger and more polarizable anions. The trend in exchange mirrors the size and wavefunction overlap of the anions with the receptor.
Computer-aided design of cage receptors
While cage 1 exhibits remarkably strong binding with chloride, as described in the experimental studies by Liu et al.,28 its potential as a chloride extractant is limited due to its excessively high affinity, preventing the release of chloride after capture. Therefore, we performed computer-aided design and propose six new cage receptors by introducing various substituents and building blocks on the aryl group of 1 to achieve reduced chloride affinities. The design strategy is illustrated in Fig. 5. Substitutions are introduced to the phenylene subunit on one of the three arms based on five factors: electrostatics, sterics, flexibility, size, and electronics.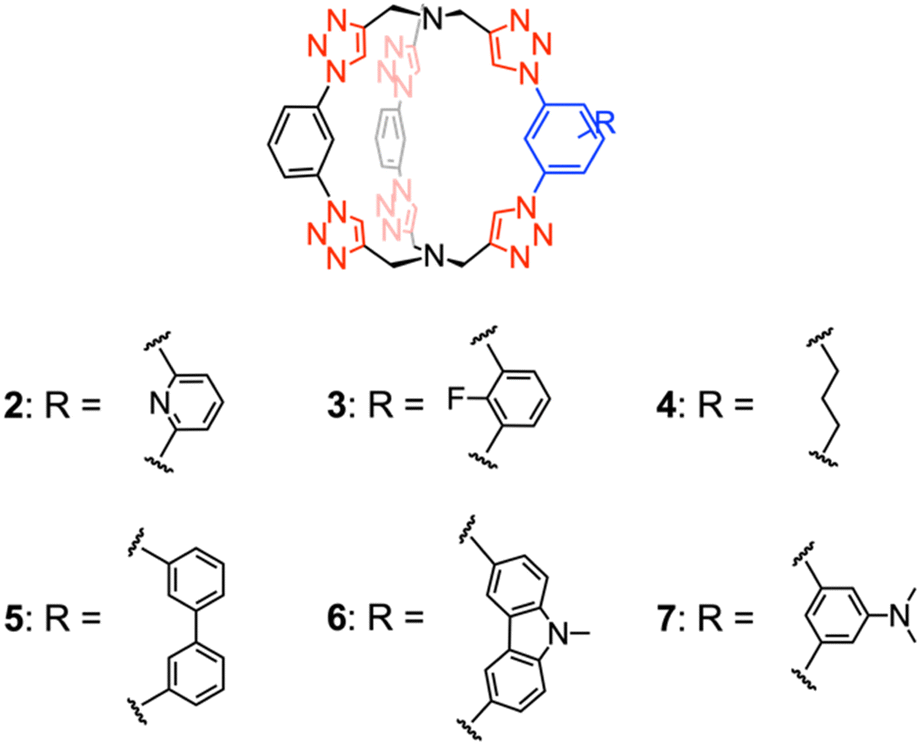 | ||
| Fig. 5 Design scheme for tuning chloride affinities for novel cage receptors. | ||
The optimized cage structures investigated in this study are shown in Fig. 6. Cages 2 and 3 are constructed by varying the electrostatic properties of the binding site. Specifically, 2 is designed by substituting the phenylene with a pyridine, and 3 is designed by substituting the phenylene C–H with a C–F unit. Based on the hypothesis that receptors with greater flexibility suffer from greater reorganization energy penalty, cage 4 is built with a flexible propyl chain substituting the rigid phenylene. Cages 5 and 6 feature larger binding cavities compared to 1, although 6 offers higher preorganization with a carbazole moiety as opposed to two phenylenes in 5. Finally, cage 7 is constructed by incorporating the electron donating NMe2 substituent. Fig. 7 shows the electrostatic potential (ESP) maps of cages 1–7, qualitatively demonstrating the positive electrostatic potentials at the center of the receptors.
 | ||
| Fig. 6 Optimized geometries at M06-2X/6-311+G(d) in the gas phase for new cages: Side views (top row) and top views (bottom row) of cages 2, 3, 4, 5, 6 and 7. | ||
 | ||
| Fig. 7 The electrostatic potential (ESP) maps at M06-2X/6-311++G(3df,2p) for cages (a) 1, (b) 2, (c) 3, (d) 4, (e) 5, (f) 6 and (g) 7. | ||
Binding of cages 2–7
We compare the number of hydrogen bonds (HBs) formed in 1:1 chloride complexes with cages 1–7 (Table 3), as well as the predicted binding free energies (Table 4). Both cages 1 and 7 offer nine hydrogen bonds in their respective 1:1 chloride complexes. However, eight hydrogen bonds are formed with 2, 3, and 4. With significantly larger cavities than the chloride guest, cages 5 and 6 are only able to provide six hydrogen bonds to stabilize the anion.
:1 chloride complexes
| 1·Cl− | 2·Cl− | 3·Cl− | 4·Cl− | 5·Cl− | 6·Cl− | 7·Cl− | |
|---|---|---|---|---|---|---|---|
| a Based on hydrogen atoms pointing toward the anion with H⋯Cl− distances <3.0 Å. b Average distances between Cl− and triazole H-bond donors. c Average Cl− and phenylene H-bond donors. | |||||||
| Number of H-bondsa | 9 | 8 | 8 | 8 | 6 | 6 | 9 |
| Average Tz H⋯Cl− H-bond distancesb (Å) | 2.50 | 2.47 | 2.50 | 2.50 | 2.65 | 2.68 | 2.50 |
| Average Ph H⋯Cl− H-bond distancesc (Å) | 2.63 | 2.62 | 2.59 | 2.63 | 2.90 | 2.91 | 2.63 |
:1 cage·Cl− binding free energies (kcal mol−1)
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | |
|---|---|---|---|---|---|---|---|
| Gas phase | −66.16 | −58.36 | −57.55 | −62.71 | −58.09 | −59.51 | −61.91 |
| DCM | −14.35 | −10.85 | −9.10 | −13.53 | −10.10 | −11.23 | −12.49 |
With the presence of the lone pair from the nitrogen atom in the binding cavity of cage 2, we observe a decrease of 7.8 kcal mol−1 in chloride binding free energy in the gas phase as compared to cage 1. Similarly, due to the highly electronegative fluorine atom, cage 3 is predicted to have the lowest Cl− binding energy in the series, with an 8.6 kcal mol−1 decrease. Cage 4, with one fewer phenylene CH and enhanced flexibility, exhibits a 3.5 kcal mol−1 decrease in binding energy. While cage 5 bears an additional phenyl unit compared to 1, the contribution of the CH proton from the phenyl group to the overall hydrogen bonding is minimal.97 Additionally, the increased cavity size renders 5 less favorable for Cl− binding, with a predicted binding energy 8.1 kcal mol−1 lower than that of 1. The presence of the carbazole unit in cage 6 increases the cavity size, resulting in a 6.7 kcal mol−1 decrease in the Cl− affinity. Lastly, although cage 7 offers a similar cavity size to 1, the electron donating NMe2 lowers the binding energy by 4.3 kcal mol−1. With this knowledge, it is possible for us to establish a systematic scheme to develop novel cage receptors with customizable anion affinities.
The SAPT energy decomposition analysis (Fig. 8) of the new cages reveals the dominance of electrostatic contribution to the gas phase binding. The strongest binding is observed for 1·Cl−, 4·Cl−, and 7·Cl−. Whereas 2·Cl−, 3·Cl−, 5·Cl−, and 6·Cl− have slightly less favorable overall interaction energies. The electrostatic interaction is strongest for 1·Cl−, 4·Cl−, and 7·Cl−, indicating the dominance of this term in these complexes. The exchange repulsion is highest for 3·Cl−, suggesting significant electron cloud overlap repulsion in this complex, which is within our expectation since a fluorine atom substitutes one of the phenylene hydrogen atoms. The induction interaction is relatively consistent across 1·Cl−, 2·Cl−, 3·Cl−, 4·Cl−, and 7·Cl−, indicating similar polarization effects. Dispersion interactions are strongest for 1·Cl−, 2·Cl−, 3·Cl−, 4·Cl−, and 7·Cl−, similar to the observations for induction. Overall, 5·Cl−, and 6·Cl−exhibit weaker exchange repulsion, induction, and dispersion interactions. Due to the dominance of electrostatics across all species, it is expected that the chloride binding for all seven cages will be weakened in DCM (εr = 8.93), where the electrostatic interactions will be partially screened in interactions. With the highest ratio of non-electrostatic contributions observed in 5·Cl−, and 6·Cl−, we expect the dielectric screening to be less prominent for them in DCM.
 | ||
| Fig. 8 Energy decomposition of chloride complexes formed with cages 1–7. (a) Total sSAPT0, electrostatics, exchange repulsion, induction, and dispersion energies at sSAPT0/jun-cc-pVDZ. (b) Ratios of non-electrostatics and electrostatics in total sSAPT0 energies. | ||
The design of cages 2–7 demonstrates how structural modifications systematically alter chloride binding affinities and interaction profiles. Across all cages, electrostatics remain the dominant force for binding. A reduction in the electrostatic interactions results in lower binding affinities compared to cage 1. Additionally, steric hindrance from larger substituents can interfere with optimal hydrogen bonding, weakening the binding. Increased flexibility reduces binding energy due to a higher reorganization energy penalty (Table S3, ESI†). Larger cavity sizes lead to fewer hydrogen bonds with chloride, diminishing binding strength. Finally, altering the electronic properties of the cage slightly decreases chloride affinity by weakening electrostatic interactions. These insights emphasize how strategic manipulation of these factors can effectively fine-tune chloride binding properties, offering a framework for designing cage receptors with customizable anion affinities.
Conclusions
This study highlights the potential of molecular cages in the form of triazolo-based receptors for selective anion binding. Through a comprehensive computational investigation, we have demonstrated that the parent triazolo-cage receptor exhibits a strong preference for binding small, spherical anions such as fluoride and chloride, driven primarily by electrostatic interactions. Our findings indicate that the receptor's binding cavity is capable of some structural adaptation to accommodate smaller anions, while its cavity size enhances selectivity toward chloride and bromide. The use of computer-aided design offers a pathway to achieve tunable anion affinities, particularly for chloride, by fine-tuning structural elements and electronic properties. These results showcase the effectiveness of computational techniques in guiding receptor design and reducing experimental trial and error. Overall, this work provides a strategic framework for the rational design of molecular cages with tailored anion-binding capabilities.Data availability
The ESI,† provided with the manuscript includes the coordinates of all the structures investigated in the manuscript. This will allow the results to be reproduced using standard quantum chemical computer programs such as Gaussian 16, Q-Chem or ORCA. In addition, the dependence of the calculated receptor binding affinities of the different anions on the dielectric constants of the solvents is also shown.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported entirely by the Chemical Sciences, Geosciences, and Biosciences Division of the Basic Energy Sciences Program of the US Department of Energy Office of Science (DE-SC0002728).References
- P. Sokkalingam, J. Yoo, H. Hwang, P. H. Lee, Y. M. Jung and C. H. Lee, Eur. J. Org. Chem., 2011, 2911–2915 CrossRef
.
- H. C. Gee, C. H. Lee, Y. H. Jeong and W. D. Jang, Chem. Commun., 2011, 47, 11963–11965 RSC
- A. Kalaiselvan, A. Naniyil, R. M. Ipe, S. V. K. Isukapalli, S. R. Vennapusa, A. P. Andrews and S. Gokulnath, J. Org. Chem., 2023, 88, 14377–14387 CrossRef CAS PubMed
- C. Rando, J. Vázquez, J. Sokolov, Z. Kokan, M. Necas and V. Sindelár, Angew. Chem., Int. Ed., 2022, 61, e202210184 CrossRef CAS
- R. Andrews, S. Begum, C. J. Clemett, R. A. Faulkner, M. L. Ginger, J. Harmer, M. Molinari, G. M. B. Parkes, Z. M. H. Qureshi, C. R. Rice, M. D. Ward, H. M. Williams and P. B. Wilson, Angew. Chem., Int. Ed., 2020, 59, 20480–20484 CrossRef CAS PubMed
- R. Custelcean, P. V. Bonnesen, N. C. Duncan, X. H. Zhang, L. A. Watson, G. Van Berkel, W. B. Parson and B. P. Hay, J. Am. Chem. Soc., 2012, 134, 8525–8534 CrossRef CAS
- X. F. Ji, R. T. Wu, L. L. Long, C. X. Guo, N. M. Khashab, F. H. Huang and J. L. Sessler, J. Am. Chem. Soc., 2018, 140, 2777–2780 CrossRef CAS
- S. K. Kim, J. Lee, N. J. Williams, V. M. Lynch, B. P. Hay, B. A. Moyer and J. L. Sessler, J. Am. Chem. Soc., 2014, 136, 15079–15085 CrossRef CAS PubMed
- J. A. Birrell, J. N. Desrosiers and E. N. Jacobsen, J. Am. Chem. Soc., 2011, 133, 13872–13875 CrossRef PubMed
- S. Lin and E. N. Jacobsen, Nat. Chem., 2012, 4, 817–824 CrossRef PubMed
- G. Pupo and V. Gouverneur, J. Am. Chem. Soc., 2022, 144, 5200–5213 CrossRef
- P. Steinforth, M. Gómez-Martínez, L. M. Entgelmeier, O. G. Mancheño and M. Schönhoff, J. Phys. Chem. B, 2022, 126, 10156–10163 CrossRef PubMed
- J. M. Lehn, Acc. Chem. Res., 1978, 11, 49–57 CrossRef
- E. Mulugeta, Q. He, D. Sareen, S. J. Hong, J. H. Oh, V. M. Lynch, J. L. Sessler, S. K. Kim and C. H. Lee, Chem, 2017, 3, 1008–1020 Search PubMed
- P. Molina, F. Zapata and A. Caballero, Chem. Rev., 2017, 117, 9907–9972 CrossRef
- M. Chvojka, D. Madea, H. Valkenier and V. Sindelár, Angew. Chem., Int. Ed., 2024, 63, e202318261 CrossRef PubMed
- B. P. Hay, T. K. Firman and B. A. Moyer, J. Am. Chem. Soc., 2005, 127, 1810–1819 CrossRef
- S. J. Brooks, P. A. Gale and M. E. Light, Chem. Commun., 2005, 4696–4698 RSC
- S. Valiyaveettil, J. F. Engbersen, W. Verboom and D. N. Reinhoudt, Angew. Chem., Int. Ed. Engl., 1993, 32, 900–901 CrossRef
- M. P. Hughes and B. D. Smith, J. Org. Chem., 1997, 62, 4492–4499 CrossRef
- J.-H. Liao, C.-T. Chen and J.-M. Fang, Org. Lett., 2002, 4, 561–564 CrossRef PubMed
- J. L. Sessler, S. Camiolo and P. A. Gale, Coord. Chem. Rev., 2003, 240, 17–55 CrossRef
- K. A. Nielsen, W.-S. Cho, J. Lyskawa, E. Levillain, V. M. Lynch, J. L. Sessler and J. O. Jeppesen, J. Am. Chem. Soc., 2006, 128, 2444–2451 CrossRef PubMed
- D. J. Sutor, J. Chem. Soc., 1963, 1105–1110 RSC
- Y. Li and A. H. Flood, Angew. Chem., Int. Ed., 2008, 47, 2649–2652 CrossRef
- S. Lee, C.-H. Chen and A. H. Flood, Nat. Chem., 2013, 5, 704–710 CrossRef PubMed
- S. Lee, B. E. Hirsch, Y. Liu, J. R. Dobscha, D. W. Burke, S. L. Tait and A. H. Flood, Chem. – Eur. J., 2016, 22, 560–569 CrossRef PubMed
- Y. Liu, W. Zhao, C. H. Chen and A. H. Flood, Science, 2019, 365, 159–161 CrossRef PubMed
- S. Mirzaei, V. E. M. Castro and R. H. Sanchez, Chem. Sci., 2022, 13, 2026–2032 RSC
- N. Bhattacharjee, X. F. Gao, A. Nathani, J. R. Dobscha, M. Pink, T. Ito and A. H. Flood, Chem. – Eur. J., 2023, 29, e202302339 CrossRef PubMed
- S. Debnath, A. H. Flood and K. Raghavachari, J. Phys. Chem. B, 2024, 128, 1586–1594 CrossRef PubMed
- J. M. Herbert and K. Carter-Fenk, J. Phys. Chem. A, 2021, 125, 1243–1256 CrossRef PubMed
- J. Gorges, B. Bädorf, S. Grimme and A. Hansen, Synlett, 2023, 1135–1146 CrossRef
- B. H. Tang, M. Pauls, C. Bannwarth and S. Hecht, J. Am. Chem. Soc., 2023, 146, 45–50 CrossRef PubMed
- T. M. Parker, L. A. Burns, R. M. Parrish, A. G. Ryno and C. D. Sherrill, J. Chem. Phys., 2014, 140, 094106 CrossRef PubMed
- T. H. Dunning, J. Phys. Chem. A, 2000, 104, 9062–9080 CrossRef CAS
- S. Grimme, Chem. – Eur. J., 2012, 18, 9955–9964 CrossRef CAS
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef
- O. Marchetti and H. J. Werner, J. Phys. Chem. A, 2009, 113, 11580–11585 CrossRef CAS PubMed
- J. Rezác, C. Greenwell and G. J. O. Beran, J. Chem. Theory Comput., 2018, 14, 4711–4721 CrossRef
- P. Matczak and S. Wojtulewski, J. Mol. Model., 2015, 21, 41 CrossRef
- K. Raghavachari, G. W. Trucks, J. A. Pople and M. Headgordon, Chem. Phys. Lett., 1989, 157, 479–483 CrossRef CAS
- Q. L. Ma and H. J. Werner, J. Chem. Theory Comput., 2019, 15, 1044–1052 CrossRef CAS
- G. Bistoni, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2020, 10, e1442 CAS
- B. Jeziorski, R. Moszynski and K. Szalewicz, Chem. Rev., 1994, 94, 1887–1930 CrossRef CAS
- A. L. Ringer, M. O. Sinnokrot, R. P. Lively and C. D. Sherrill, Chem. – Eur. J., 2006, 12, 3821–3828 CrossRef CAS PubMed
- Y. Geng, T. Takatani, E. G. Hohenstein and C. D. Sherrill, J. Phys. Chem. A, 2010, 114, 3576–3582 CrossRef CAS PubMed
- C. D. Sherrill, Acc. Chem. Res., 2013, 46, 1020–1028 CrossRef CAS
- A. Tarzia and K. E. Jelfs, Chem. Commun., 2022, 58, 3717–3730 RSC
- B. W. McCann, N. De Silva, T. L. Windus, M. S. Gordon, B. A. Moyer, V. S. Bryantsev and B. P. Hay, Inorg. Chem., 2016, 55, 5787–5803 CrossRef CAS PubMed
- M. Foscato, V. Venkatraman, G. Occhipinti, B. K. Alsberg and V. R. Jensen, J. Chem. Inf. Model., 2014, 54, 1919–1931 CrossRef CAS PubMed
- M. Foscato, B. J. Houghton, G. Occhipinti, R. J. Deeth and V. R. Jensen, J. Chem. Inf. Model., 2015, 55, 1844–1856 CrossRef CAS PubMed
- E. R. Abdurakhmanova, D. Mondal, H. J. Drzejewska, P. Cmoch, O. Danylyuk, M. J. Chmielewski and A. Szumna, Chem, 2024, 10, 1910–1924 CAS
- I. Sandler, S. Sharma, B. Chan and J. M. Ho, J. Phys. Chem. A, 2021, 125, 9838–9851 CrossRef CAS PubMed
- N. J. Young and B. P. Hay, Chem. Commun., 2013, 49, 1354–1379 RSC
- R. O. Ramabhadran, Y. Liu, Y. R. Hua, M. Ciardi, A. H. Flood and K. Raghavachari, J. Am. Chem. Soc., 2014, 136, 5078–5089 CrossRef CAS
- X. X. Fan, D. Zhang, S. Y. Jiang, H. Wang, L. T. Lin, B. Zheng, W. H. Xu, Y. X. Zhao, B. P. Hay, Y. T. Chan, X. J. Yang, X. P. Li and B. Wu, Chem. Sci., 2019, 10, 6278–6284 RSC
- C. D. Jia, B. P. Hay and R. Custelcean, Inorg. Chem., 2014, 53, 3893–3898 Search PubMed
- A. Lutolli, M. W. Che, F. C. Parks, K. Raghavachari and A. H. Flood, J. Org. Chem., 2023, 88, 6791–6804 CrossRef CAS
- F. C. Parks, E. G. Sheetz, S. R. Stutsman, A. Lutolli, S. Debnath, K. Raghavachari and A. H. Flood, J. Am. Chem. Soc., 2022, 144, 1274–1287 Search PubMed
- F. C. Parks, Y. Liu, S. Debnath, S. R. Stutsman, K. Raghavachari and A. H. Flood, J. Am. Chem. Soc., 2018, 140, 17711–17723 CrossRef CAS
- A. Sengupta, Y. Liu, A. H. Flood and K. Raghavachari, Chem. – Eur. J., 2018, 24, 14409–14417 CrossRef CAS PubMed
- Y. Liu, A. Sengupta, K. Raghavachari and A. H. Flood, Chem, 2017, 3, 411–427 CAS
- J. H. Yang, J. Kim, B. P. Hay, K. Lee and S. K. Kim, Eur. J. Org. Chem., 2022, e202200808 CrossRef CAS
- C. Park and H. Simmons, J. Am. Chem. Soc., 1968, 90, 2431–2432 Search PubMed
- B. Dietrich, J. Lehn and J. Sauvage, Tetrahedron Lett., 1969, 10, 2889–2892 CrossRef
- S. O. Kang, J. M. Llinares, V. W. Day and K. Bowman-James, Chem. Soc. Rev., 2010, 39, 3980–4003 Search PubMed
- S. O. Kang, J. M. Llinares, D. Powell, D. VanderVelde and K. Bowman-James, J. Am. Chem. Soc., 2003, 125, 10152–10153 CrossRef
- N. A. Fakhre and B. M. Ibrahim, J. Hazard. Mater., 2018, 343, 324–331 CrossRef
- Y. J. Li and A. H. Flood, Angew. Chem., Int. Ed., 2008, 47, 2649–2652 CrossRef PubMed
- Y. L. Li and A. H. Flood, J. Am. Chem. Soc., 2008, 130, 12111–12122 CrossRef
- K. P. McDonald, R. O. Ramabhadran, S. Lee, K. Raghavachari and A. H. Flood, Org. Lett., 2011, 13, 6260–6263 CrossRef
- J. Lehn and J. Sauvage, J. Am. Chem. Soc., 1975, 97, 6700–6707 CrossRef
- S. Durot, J. Taesch and V. Heitz, Chem. Rev., 2014, 114, 8542–8578 CrossRef PubMed
- M. Delecluse, C. Colomban, B. Chatelet, S. Chevallier-Michaud, D. Moraleda, J.-P. Dutasta and A. Martinez, J. Org. Chem., 2020, 85, 4706–4711 CrossRef
- C. K. Xu, Q. G. Tran, D. X. Liu, C. J. Zhai, L. Wojtas and W. Q. Liu, Chem. Sci., 2024, 15, 16040 RSC
- W. L. Jiang, B. Huang, X. L. Zhao, X. L. Shi and H. B. Yang, Chem, 2023, 9, 2655–2668 Search PubMed
- J. Y. Li, C. W. Wang and Y. R. Mo, Chem. – Eur. J., 2023, 29 Search PubMed
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, 2016.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef PubMed
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef
- T. Clark, J. Chandrasekhar, G. W. Spitznagel and P. V. Schleyer, J. Comput. Chem., 1983, 4, 294–301 CrossRef
- A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef
- J. P. Foster and F. Weinhold, J. Am. Chem. Soc., 1980, 102, 7211–7218 CrossRef
- A. E. Reed and F. Weinhold, J. Chem. Phys., 1983, 78, 4066–4073 CrossRef
- A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735–746 CrossRef
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef
- R. A. Kendall, T. H. Dunning and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef
- D. E. Woon and T. H. Dunning, J. Chem. Phys., 1993, 98, 1358–1371 CrossRef
- K. A. Peterson, D. Figgen, E. Goll, H. Stoll and M. Dolg, J. Chem. Phys., 2003, 119, 11113–11123 CrossRef
- K. A. Peterson, B. C. Shepler, D. Figgen and H. Stoll, J. Phys. Chem. A, 2006, 110, 13877–13883 CrossRef
- J. M. Turney, A. C. Simmonett, R. M. Parrish, E. G. Hohenstein, F. A. Evangelista, J. T. Fermann, B. J. Mintz, L. A. Burns, J. J. Wilke, M. L. Abrams, N. J. Russ, M. L. Leininger, C. L. Janssen, E. T. Seidl, W. D. Allen, H. F. Schaefer, R. A. King, E. F. Valeev, C. D. Sherrill and T. D. Crawford, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 556–565 Search PubMed
- E. Papajak, J. J. Zheng, X. F. Xu, H. R. Leverentz and D. G. Truhlar, J. Chem. Theory Comput., 2011, 7, 3027–3034 CrossRef CAS PubMed
- H. K. Roobottom, H. D. B. Jenkins, J. Passmore and L. Glasser, J. Chem. Educ., 1999, 76, 1570–1573 CrossRef
- V. S. Bryantsev and B. P. Hay, J. Am. Chem. Soc., 2005, 127, 8282–8283 CrossRef
Footnotes |
| † Electronic supplementary information (ESI) available: Exponents and coefficients of the custom basis set used for Iodine. Calculated binding free energies of cage 1. SAPT energy decomposition of cage complexes. Benchmarking of calculations – Comparison with experiment. The 1/εr dependence of binding free energies of cage 1. The xyz atomic coordinates of the cages and anion-complexes investigated in this study are included in a zip file. See DOI: https://doi.org/10.1039/d4cp04589k |
| ‡ Current address: Department of Chemistry, Columbia University, New York 10027. |
| This journal is © the Owner Societies 2025 |