
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLigand impacts on band edge energies and excited state splittings of silicane†
Guoying
Yao
 ab,
Ekadashi
Pradhan
b,
Zhenyu
Yang
*a and
Tao
Zeng
*b
ab,
Ekadashi
Pradhan
b,
Zhenyu
Yang
*a and
Tao
Zeng
*b
aMOE Laboratory of Bioinorganic and Synthetic Chemistry, Lehn Institute of Functional Materials, School of Chemistry, IGCME, Sun Yat-Sen University, Guangzhou, Guangdong, 510275, China. E-mail: yangzhy63@mail.sysu.edu.cn
bDepartment of Chemistry, York University, Toronto, Ontario M3J1P3, Canada. E-mail: tzeng@yorku.ca
First published on 6th February 2025
Abstract
Two-dimensional (2D) silicanes are promising semiconductors for applications in optoelectronics and photochemistry. Covalent functionalization presents a facile strategy to customize silicanes to attain desired properties. A comprehensive understanding of the impacts of ligands on silicanes is thus pivotal. In this study, we perform density functional theory (DFT) and time-dependent DFT (TDDFT) calculations to investigate the effects of three typical classes of ligands: (1) σ-withdrawing and π-donating, (2) σ-withdrawing and π-withdrawing, and (3) σ-donating and π-withdrawing, on the geometric structure, electronic structure, and band edge excited states of silicanes. Covalent functionalization of silicanes enables a wide range of band edge energies. The band gaps can be tuned between indirect and direct, and the underlying mechanism is explained for the first time. Additionally, TDDFT calculations confirm that the band edge optical absorptions can be adjusted by the ligands broadly from the near-infrared to the visible light region. These desirable properties enhance the functionalities of silicanes. Methodologically, we discuss the applicability of using the conventional one-particle orbital model to describe the excited states of silicanes, and nail down the ∼0.1 eV inaccuracy of the model in describing excited state splittings. We present empirical functions for quick estimations of band edge energies of covalently functionalized silicanes. Through careful comparisons, we justify the use of the carbon-adapted σ/π-donating/accepting indices of typical ligands in silicon chemistry and present a set of silicon-based indices for future prudent applications in inorganic computational studies of silicon-based materials.
I. Introduction
Over the past two decades, there has been a surge of research in two-dimensional (2D) materials, largely driven by the groundbreaking discovery of graphene in 20041 and the subsequent synthesis of silicene.2,3 Especially, silicene has found extensive applications in field-effect transitors4–6 and spintronics.7–9 Silicanes—a group of hexagonal sp3-silicon-based networks—can be viewed as functionalized silicenes and have been a focus of research due to their desirable optoelectronic properties, high biocompatibility, and synthetic readiness.10–12 These attributes suggest their wide applications in micro- and nano-electronics,13 optoelectronics,14,15 photocatalysis,16,17 and batteries.18–20Covalent surface functionalization (i.e., replacing surface Si–H by Si–R bonds21,22) is critical in silicon chemistry, particularly for enhancing solution processability and chemical stability.23,24 It is also a handle to modify and control properties such as conductivity, hydrophilicity, lubricity, and biocompatibility.25,26 The properties of silicon-based materials change significantly when reduced to the 2D nanoscale.27,28 However, compared to other 2D nanostructures such as MXenes and ionic semiconductor nanoplates,29,30 ligand functionalization for silicanes is less studied; there have been only a few reports on R being alkenyl,15 aryl,22,31 and amino32,33 groups. Consequently, the understanding of the ligand influences on the electronic and optical properties of silicanes is far behind the advance of the functionalization techniques.
Computational chemistry offers atomistic understanding of these influences. Previous computational studies have demonstrated that the band gap is tuned by various ligands from 1.2 to 2.5 eV, from the intrinsic indirect band gap to direct gap,21,34,35 and the gap is dependent on ligands coverage,35–37 strain,38 and external electric fields.39 The mechanisms underlying these gap modulations, however, remain elusive. Furthermore, the band gap is not the only key property; the band edge energies of silicanes are equally important as they influence the separation, recombination, and migration of photogenerated charge carriers.40 These important properties have not yet been studied. In terms of methodology, precedented computational studies of the excited states of silicanes have all relied on the conventional one-particle orbital description, i.e., energy gaps between hole and particle orbitals are viewed as excitation energies. While this description is usually applicable for extended systems without local formations, given the spatial confinement of their ∼4 Å thinness, the excitation energies of silicanes may not be solely determined by the orbital energy gaps. In this respect, silicanes resemble finite-sized molecules, and we may employ similar method and language developed for molecules with explicit electron correlation to investigate and interpret the excited states of silicanes. Surprisingly, no such study has been performed for such an important class of spatially confined materials.
Given these motivations, we performed density functional theory (DFT) and time-dependent DFT (TDDFT) calculations for pristine silicane (SiH) and substituted SiR species with three representative types of ligands: (1) σ-withdrawing and π-donating (sWpD: Cl, SH, OH, OCH3, and NH2), (2) σ-withdrawing and π-withdrawing (sWpW: CN), and (3) σ-donating and π-withdrawing (sDpW: BH2, Li). More sWpD ligands were considered since those are typical oxidative ligands for silicanes. “σ-withdrawing”, “π-donating”, etc. means that a ligand pulls or pushes electrons from or to its bonding partner through σ- or π-type covalent interaction. As shown below, the results fill the knowledge gaps mentioned above.
II. Methods
All ground state DFT calculations were performed using the Vienna ab initio Simulation Package (VASP 5.4.4)41 and the projector augmented-wave (PAW) pseudopotentials. The Perdew–Burke–Ernzerhof (PBE) functional42,43 was used for structural optimizations, while the HSE06 hybrid functional44 was used to calculate densities of states (DOS) and band structures. The energy cut-off for the plane wave basis set was set to 500 eV. The monolayer is on the ab-plane. A 40 Å high vacuum region was added in the c direction to isolate periodic images of the SiR monolayer along the c-axis. The first Brillouin zone (FBZ) was sampled using a 9 × 9 × 1 Monkhorst–Pack grid of k vectors. The structural optimization convergence criterion was 0.01 eV Å−1. All crystal structures and orbitals were visualized using VESTA.45 To compare the calculated orbital energy levels of different SiR species, an inert He atom was placed 20 Å above each SiR layer (Fig. S1, ESI†), and the He 1s orbital energies in the different calculations were aligned. This calibration is an alternative to the use of the vacuum energy level, which is also used here to calculate work functions of the investigated species. The effective masses of hole and excited electron (meeff and mheff) at band extrema were calculated using VASPKIT.46 Throughout this work, symbols such as VBMΓ, VBMM, CBMΓ, and CBMM are used to denote the valence band maximum (VBM) and the conduction band minimum (CBM) at the subscripted specific k points in FBZ. The subscripted VBM and CBM may not be genuine VBM and CBM in the whole FBZ, which are labeled as the unsubscripted “VBM” and “CBM”. CBMΓ + 1, VBMΓ − 1, etc., are used to label orbital levels that are higher or lower than the explicitly specified ones.To estimate the Si-adapted σ and π electron donating-accepting (sEDA and pEDA) capabilities of the ligands, natural population analyses were performed at the B3LYP/cc-pVTZ level.47,48 The sEDA and pEDA values of the ligands studied are listed in Table S2 (ESI†), defined as per the pioneering work of Oziminski and Dobrowolski (OD) for carbon chemistry, in which the same functional was employed.49 Additionally, Si13R13H9 models (Fig. S2, ESI†) were used to evaluate the natural atomic charge of Si atoms in SiR at the ωB97X-D/cc-pVDZ level.48,50 The central Si–R unit in the molecular model closely mimics each Si–R unit in the SiR monolayer. All these molecular calculations were performed using the GAMESS-US program.51,52
TDDFT calculations for the SiR species were carried out using the CP2K-8.2 package53 at the B3LYP47/GPW54 level with the Goedecker–Teter–Hutter (GTH) pseudopotential.55,56 The GPW hybrid basis set contains the TZVP-MOLOPT atomic basis57 and the plane waves up to 350 Ry. The use of the B3LYP functional is motivated by its success in TDDFT calculations of semiconductors and confined system such as Si nanoparticle.58,59 Our confidence on this functional also comes from the good agreement between the TDDFT-calculated lowest absorption energy of SiH and the experimental value (vide infra). The SCF convergence criterion was 1 × 10−6 atomic unit (a.u.). 4 × 4 × 1 supercells were employed to partially counteract the shrinkage of the FBZ to the Γ point in our TDDFT calculations. Two Fortran codes were developed to process the cube files of the orbitals from the supercell calculations: one returns the orbitals’ k vectors in the FBZ of the genuine SiR unit cell, and the other returns two-electron integrals of four read-in orbitals:  , through numerical integration. Setting ϕ1 = ϕ2 and ϕ3 = ϕ4 (ϕ1 = ϕ3 and ϕ2 = ϕ4), we can calculate Coulomb (exchange) integral between two orbitals. The two-electron integrals mentioned below were obtained using the code. The two codes are available upon request.
, through numerical integration. Setting ϕ1 = ϕ2 and ϕ3 = ϕ4 (ϕ1 = ϕ3 and ϕ2 = ϕ4), we can calculate Coulomb (exchange) integral between two orbitals. The two-electron integrals mentioned below were obtained using the code. The two codes are available upon request.
III. Results and discussion
1. Geometrical structures
Fig. 1(a) shows a generic SiR structure with a hexagonal unit cell defined by the lattice vectors a and b. The SiR plane is buckled to the height h defined there. The first Brillouin zone (FBZ) is shown in Fig. 1(b), with the reciprocal lattice vectors ã and![[b with combining tilde]](https://www.rsc.org/images/entities/b_i_char_0062_0303.gif) , high-symmetry points and their connecting paths. The ligands are categorized in Fig. 1(c) according to their σ- and π-donating and withdrawing capabilities, quantified by the Oziminski–Dobrowolski (OD)49 sEDA and pEDA (s: σ, p: π, EDA: electron donator–acceptor descriptor) values. The sDpD quadrant in Fig. 1(c) is empty. Actually, we considered Ag atom as the sDpD ligand: SiAg was found to share similar structure with regular silicanes but exhibit no band gap. This hypothetical non-silicane metallic species is thus not discussed. SiLi, a salt with the Li+ cations located under (above) the buckled up (down) Si− anions (Fig. S1, ESI†), is also not of silicane type. However, this well-studied species is included in this work to highlight the differences between its anionic silicide framework and the regular silicane frameworks.
, high-symmetry points and their connecting paths. The ligands are categorized in Fig. 1(c) according to their σ- and π-donating and withdrawing capabilities, quantified by the Oziminski–Dobrowolski (OD)49 sEDA and pEDA (s: σ, p: π, EDA: electron donator–acceptor descriptor) values. The sDpD quadrant in Fig. 1(c) is empty. Actually, we considered Ag atom as the sDpD ligand: SiAg was found to share similar structure with regular silicanes but exhibit no band gap. This hypothetical non-silicane metallic species is thus not discussed. SiLi, a salt with the Li+ cations located under (above) the buckled up (down) Si− anions (Fig. S1, ESI†), is also not of silicane type. However, this well-studied species is included in this work to highlight the differences between its anionic silicide framework and the regular silicane frameworks.
 | ||
| Fig. 1 (a) Top and side views of the honeycomb structure of SiR, and (b) its first Brillouin zone with the special k points being labeled. (c) Classification of the investigated ligands in the four quadrants defined by σ/π-withdrawing/donating capabilities. | ||
The OD sEDA and pEDA were obtained for the C–R bonds, i.e., they are carbon-adapted. The use of the carbon-adapted OD EDA values for SiR may sound questionable in the beginning, but is fully justified in Section S2 (ESI†), where we calculated the Si-adapted sEDA and pEDA indices and compared them with the C-adapted ones proposed by OD. A good agreement between the two sets is seen. For comprehensiveness, the full list of ligands in the OD paper were considered,49 much more than the a few ligands studied in this work. Since the σ/π-withdrawing/donating are local covalent effects, in both finite-sized and extended systems, the EDA values obtained using molecules can be used to explain the ligands’ impacts to silicanes, in which they are covalently bonded to the Si framework.
The optimized structural parameters are summarized in Table 1. The parameters of SiH agree well with previous theoretical and experimental results.39,60–62 The hexagonal Si framework is maintained for SiCl, SiCN, and SiOCH3, with essentially a = b, and is slightly broken for SiSH (a = 3.909 Å, b = 3.979 Å), SiOH (3.882, 3.917 Å), SiNH2 (3.832, 3.903 Å), and SiBH2 (3.923, 3.884 Å). The Si–Si bond lengths in all SiR species are marginally elongated (<2.5%) from the SiH analogue, within the tolerable range of 17% elongation under equibiaxial strain.63 The bulky OCH3 leads to additional transversal stress and the largest a and b. The sWpD ligands slightly increase h from the SiH analogue, due to their π* antibonding (π-donating) interactions with the Si–Si bonds; their h values correlate well with their pEDA values, except for the bulky OCH3, whose extra transversal stress suppresses the buckling. The sWpW CN ligand slightly reduces h due to the lack of π-donating capability. The sDpW BH2 ligand, through its dominating σ-donating capability, makes the Si atoms less siliconium-like (see the +0.003 Si charge in Table 1) and hence significantly increases h. Naturally, the h value is even much larger in the purely silicide framework in SiLi.
| SiH | SiCl | SiSH | SiOH | SiOCH3 | SiNH2 | SiCN | SiBH2 | SiLi | |
|---|---|---|---|---|---|---|---|---|---|
| a The first (second) entry was for the effective mass at CBMΓ or VBMΓ along the Γ-to-M (Γ-to-K) direction. For SiH (SiBH2), the third and fourth entries was obtained at CBMM (CBMK) for the M-to-Γ (K-to-Γ) and M-to-K (K-to-M) directions. All the effective masses were calculated using HSE06 functional. m0 is the atomic unit of mass, i.e., the electronic mass. | |||||||||
| a/Å | 3.888 | 3.918 | 3.909 | 3.882 | 3.977 | 3.832 | 3.935 | 3.923 | 3.730 |
| b/Å | 3.888 | 3.921 | 3.979 | 3.917 | 3.975 | 3.903 | 3.933 | 3.884 | 3.730 |
| Space group |
P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m1 m1 |
Pm1 |
P1 | P1 | P1 | P1 | P1 | P1 |
Pm1 |
| Average Si–Si bond length/Å | 2.357 | 2.378 | 2.391 | 2.374 | 2.411 | 2.374 | 2.381 | 2.393 | 2.393 |
| Average Si–R bond length/Å | 1.501 | 2.075 | 2.169 | 1.694 | 1.683 | 1.780 | 1.831 | 1.992 | 2.628 |
| h/Å | 0.719 | 0.732 | 0.749 | 0.763 | 0.723 | 0.787 | 0.713 | 0.791 | 1.043 |
| Si charge/atomic unit | +0.071 | +0.305 | +0.195 | +0.520 | +0.623 | +0.430 | +0.319 | +0.003 | −0.769 |
| Work function Φ/eV | 4.98 | 5.57 | 4.58 | 3.93 | 3.47 | 3.42 | 9.08 | 5.06 | 2.36 |
| m eeff/m0a | 0.170 | 0.247 | 0.194 | 0.274 | 0.273 | 0.296 | 0.175 | 0.190 | 0.862 |
| 0.169 | 0.247 | 0.199 | 0.289 | 0.271 | 0.284 | 0.175 | 0.205 | 0.861 | |
| 2.996 | 1.155 | ||||||||
| 0.122 | 0.659 | ||||||||
| m eeff/m0a | 0.570 | 1.789 | 1.196 | 0.690 | 0.487 | 0.314 | 0.772 | 0.153 | 0.465 |
| 0.540 | 1.435 | 0.553 | 0.474 | 0.660 | 0.583 | 0.856 | 0.235 | 0.451 | |
The atomic charge certainly reflects the electronegativity difference between Si and R. Shown in Fig. S3 (ESI†) is the anticorrelation between the quantities, where the electronegativity of the atoms directly connected to Si is chosen to represent the electronegativity of R. The −0.769 atomic unit (a.u., i.e., the amount of electronic charge) Si charge in SiLi is close to the −1 formal oxidation state of Si in the LiSi salt and reflects the ionicity of the Si–Li interaction. This ionicity is consistent with the negligible pDOS of Li in the valence band (vide infra, Fig. 2(i)). On the other hand, the +0.305 a.u. Si charge in SiCl is far from the +1 formal oxidation state of Si in this compound, reflecting the polar yet covalent nature of the Si–Cl bond. Despite the overall anticorrelation, the Si charge depends on the detailed covalent interaction, and the electronegativity of the atom in contact with Si may not fully represent the electronegativity of R. For instance, given the same O atom connected to Si, the Si charge is higher in SiOCH3 than in SiOH. Overall, atomic charge is only a qualitative indicator.
 | ||
| Fig. 2 The electronic band structures, densities of states, and atom-projected densities of states of (a) SiH, (b) SiCl, (c) SiSH, (d) SiOH, (e) SiOCH3, (f) SiNH2, (g) SiCN, (h) SiBH2, and (i) SiLi. The red and blue arrows indicate the indirect and direct band gaps, respectively. All band structures and densities of states have been shifted so that the Fermi energy is at 0 eV. | ||
2. Electronic structures
The ligand-induced changes in band structure and density of state (DOS) are far more significant (Fig. 2) than in geometric structure. The valence band maximum (VBM) and conduction band minimum (CBM) of SiH are located at Γ and M in FBZ (Fig. 2(a)), i.e., VBMΓ and CBMM. VBMΓ is doubly degenerate. The degeneracy arises from the e-type σSi–Si bonding orbitals (BOs), a result of the C3v site symmetry of each Si–H moiety (Fig. 3(a) and 5(a), (b)). CBMΓ is slightly higher than CBMM by 0.21 eV, which is the true CBM. Consequently, SiH features a 2.93 eV Γ-to-M indirect band gap and a 3.14 eV Γ-to-Γ direct gap, consistent with previous experimental and computational results.31,63–65 The atom-projected density of states (pDOS) of SiH in Fig. 2(a) shows almost no H contribution at the band edges. The CBMΓ orbital exhibits antibonding (AB) character between adjacent SiH units (see the red lines in Fig. 3(a)), while primarily nonbonding (NB) character for each Si–H bond. Our numerical orbital analysis indicates that the periodic modulation function of the CBMM orbital is a 45:55 mixture of the a1-type CBMΓ and the e-type degenerate CBMΓ + 1,2 orbitals (the second and third lowest CB orbitals at Γ, plotted in Fig. S4, ESI†). Since the CBMΓ + 1,2 orbitals are delocalized on the hollow centres of the 6-membered rings, CBMM is transversally polarized towards the ring centers. The effective mass of excited electron and hole for Γ-to-M (M-to-Γ) were calculated to be 0.170 (2.996) and 0.570 m0 (Table 1), respectively. These values imply higher mobility of the excited electron at Γ than at M, and higher electron mobility than hole mobility. | ||
| Fig. 3 The top and side views of the VBMΓ, CBMΓ, and CBMM orbitals of (a) SiH, (b) SiCl, (c) SiOH, and (d) SiOCH3. The characters of the orbitals with respect to certain pairs of atoms are specified: BO = bonding orbital, NB = nonbonding, and AB = antibonding. | ||
The five sWpD ligands reduce the Γ-to-Γ gaps more than the Γ-to-M gaps (the blue vs. red arrows in Fig. 2(a)–(f)), leading to direct band gaps for these functionalized SiR species. Their band edge orbitals resemble the SiH counterparts but exhibit π lone pair lobes on the π-donating ligands in the VBMΓ orbitals (Fig. 3(b)–(d)). The doubly degenerate VBMΓ level in SiH and SiCl is split to the VBMΓ and VBMΓ − 1 levels in all SiR species with non-atomic or non-linear ligands in the lack of the 3-fold site symmetry. The energy inversion between CBMM and CBMΓ results from their different spatial distributions. The CBMM orbital, distributed in the hollow space of the Si 6-membered rings, is less stabilized by the more positively charged Si framework induced by the oxidation (see Table 1 for the Si charges). On the contrary, the CBMΓ localized on the Si atoms is more stabilized. The pDOS of the five substituted SiR species in Fig. 2(a)–(f) show greater contributions of the ligands to the band edges than in SiH, due to the aforementioned π* interaction around VBM (Fig. 3 and Fig. S5, ESI†), which raises or prevents further decrease of VBM and contributes to the band gap reductions in these SiR species. The effective masses of excited electron at CBMΓ of the five SiR species only slightly change from the SiH analogues. This holds for all SiR species under consideration, except SiLi. The effective hole masses of the five are all larger than the corresponding effective excited electron masses. This holds for all species except SiBH2 and SiLi. The effective hole mass at VBMΓ becomes the most anisotropic for SiSH, with twice difference in the Γ-to-M direction vs. the Γ-to-K direction. This is consistent with the significant anisotropy of the π* interaction at VBMΓ (vide infra, Fig. 6(g) and (h)). The most significant anisotropy in effective mass of excited electron is seen at CBMM of SiH, with ∼30 times difference in the M-to-Γ vs. M-to-K directions. This is attributed to the especially flat M-to-Γ CBM section in Fig. 2(a), which is responsible for the 2.996 m0 large effective mass of excited electron. This flatness reflects the substantial interaction between the periodic modulation functions of CBMΓ and CBMΓ + 1,2 (vide supra) along the path between Γ and M in FBZ.
With the sWpW ligand, the VBM and CBM of SiCN are both at Γ, giving a 2.75 eV direct band gap (Fig. 2(g)). However, CBMM is only 0.01 eV higher, suggesting a competition between direct and indirect band gaps. As mentioned above, the periodic modulation functions of CBMM feature a mixture of CBMΓ and CBMΓ + 1,2. The CBMΓ + 1,2 component brings Si–C π character into CBMM, which is stabilized by the π-withdrawing CN (Fig. S6 and S7, ESI†) to be pseudo-degenerate with CBMΓ. The effective masses of excited electron in the Γ-to-M and Γ-to-K directions are similar to the SiH analogues, while the effective masses of hole are noticeably higher than the SiH analogues. SiCN can serve as an electron transport layer like SiH.
BH2 and Li were chosen as representative sDpW ligands. The SiLi VBM and CBM are located at Γ and are localized separately on Si− and Li+ (Fig. S6(c), ESI†), a feature of salts, with a direct gap of 0.73 eV. For SiBH2, the CBM is shifted to K, resulting in an indirect band gap of 1.58 eV (Fig. 2(h)). The VBMΓ and CBMΓ orbitals of SiBH2 resemble those of the other SiR species (Fig. S6(b), ESI†). CBMK is the genuine CBM of SiBH2, as the bonding interaction between the 2p vacant orbitals of adjacent BH2 ligands is oriented along the b direction and is maximized by this specific k vector. The effective masses of excited electron at CBMK in the K-to-Γ and K-to-M directions differ by about twice, which is consistent with the anisotropy of the bonding interaction mentioned above.
3. Excited states, the applicability of the one-particle description
Because of the Born-von Karman boundary condition imposed by running the TDDFT calculations only at the Γ point of the 4 × 4 × 1 supercell (vide supra, see also Section S3, ESI†), only the k vectors of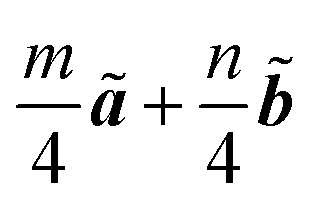 in FBZ of the true SiR unit cell were sampled. The TDDFT-DOS of the SiR species are shown in Fig. 4. For SiH, the lowest excited state (labeled ① in Fig. 4(a)) is a doubly degenerate optically bright state, with excitation from the doubly degenerate VBMΓ (Fig. 5(a) and (b)) to CBMΓ (Fig. 3(a)). The calculated absorption energy of 3.00 eV is in agreement with the experimental absorption onset at 3.0 eV,31 which reflects the accuracy of using the B3LYP functional in describing this class of materials. In Fig. 4(a), the doubly degenerate Γ state is immediately followed by a triply degenerate M state (the lower ② state in the panel) with excitations from the doubly degenerate VBMΓ to the triply degenerate CBMM. Another triply degenerate state of the same excitation scheme (the higher ② state) lies 0.10 eV higher. According to the one-particle orbital description, a 6-fold degeneracy should have arisen from the VBMΓ-to-CBMM (2 × 3 = 6) transitions, which is however split to the two 3-fold degenerate ② levels. The degenerate VBMΓ component orbitals can be linearly combined into one symmetric and one antisymmetric orbital with respect to the symmetry plane containing the long diagonal axis of the rhombic unit cell, which we call VBMΓ′ and VBMΓ′′ orbitals (Fig. 5(a) and (b)). The prime (double-prime) denotes evenness (oddness) with respect to the symmetry plane. The CBMM(1/2, 1/2) orbital shown in Fig. 5(c) has
in FBZ of the true SiR unit cell were sampled. The TDDFT-DOS of the SiR species are shown in Fig. 4. For SiH, the lowest excited state (labeled ① in Fig. 4(a)) is a doubly degenerate optically bright state, with excitation from the doubly degenerate VBMΓ (Fig. 5(a) and (b)) to CBMΓ (Fig. 3(a)). The calculated absorption energy of 3.00 eV is in agreement with the experimental absorption onset at 3.0 eV,31 which reflects the accuracy of using the B3LYP functional in describing this class of materials. In Fig. 4(a), the doubly degenerate Γ state is immediately followed by a triply degenerate M state (the lower ② state in the panel) with excitations from the doubly degenerate VBMΓ to the triply degenerate CBMM. Another triply degenerate state of the same excitation scheme (the higher ② state) lies 0.10 eV higher. According to the one-particle orbital description, a 6-fold degeneracy should have arisen from the VBMΓ-to-CBMM (2 × 3 = 6) transitions, which is however split to the two 3-fold degenerate ② levels. The degenerate VBMΓ component orbitals can be linearly combined into one symmetric and one antisymmetric orbital with respect to the symmetry plane containing the long diagonal axis of the rhombic unit cell, which we call VBMΓ′ and VBMΓ′′ orbitals (Fig. 5(a) and (b)). The prime (double-prime) denotes evenness (oddness) with respect to the symmetry plane. The CBMM(1/2, 1/2) orbital shown in Fig. 5(c) has  and is also a prime orbital. One state in the lower (higher) ② branch purely consists of the VBMΓ′′-to-CBMM(1/2, 1/2) (VBMΓ′-to-CBMM(1/2, 1/2)) excitation; it is an M′′ (M′) state. The other two states in the respective ② branch are related to the M′′ or M′ state by C6 and C3 rotations, and the relevant CBMM(1/2, 0) and CBMM(0, 1/2) orbitals are shown in (Fig. S9(e) and (f), ESI†). The splitting of the two ② branches is actually the M′′–M′ splitting.
and is also a prime orbital. One state in the lower (higher) ② branch purely consists of the VBMΓ′′-to-CBMM(1/2, 1/2) (VBMΓ′-to-CBMM(1/2, 1/2)) excitation; it is an M′′ (M′) state. The other two states in the respective ② branch are related to the M′′ or M′ state by C6 and C3 rotations, and the relevant CBMM(1/2, 0) and CBMM(0, 1/2) orbitals are shown in (Fig. S9(e) and (f), ESI†). The splitting of the two ② branches is actually the M′′–M′ splitting.
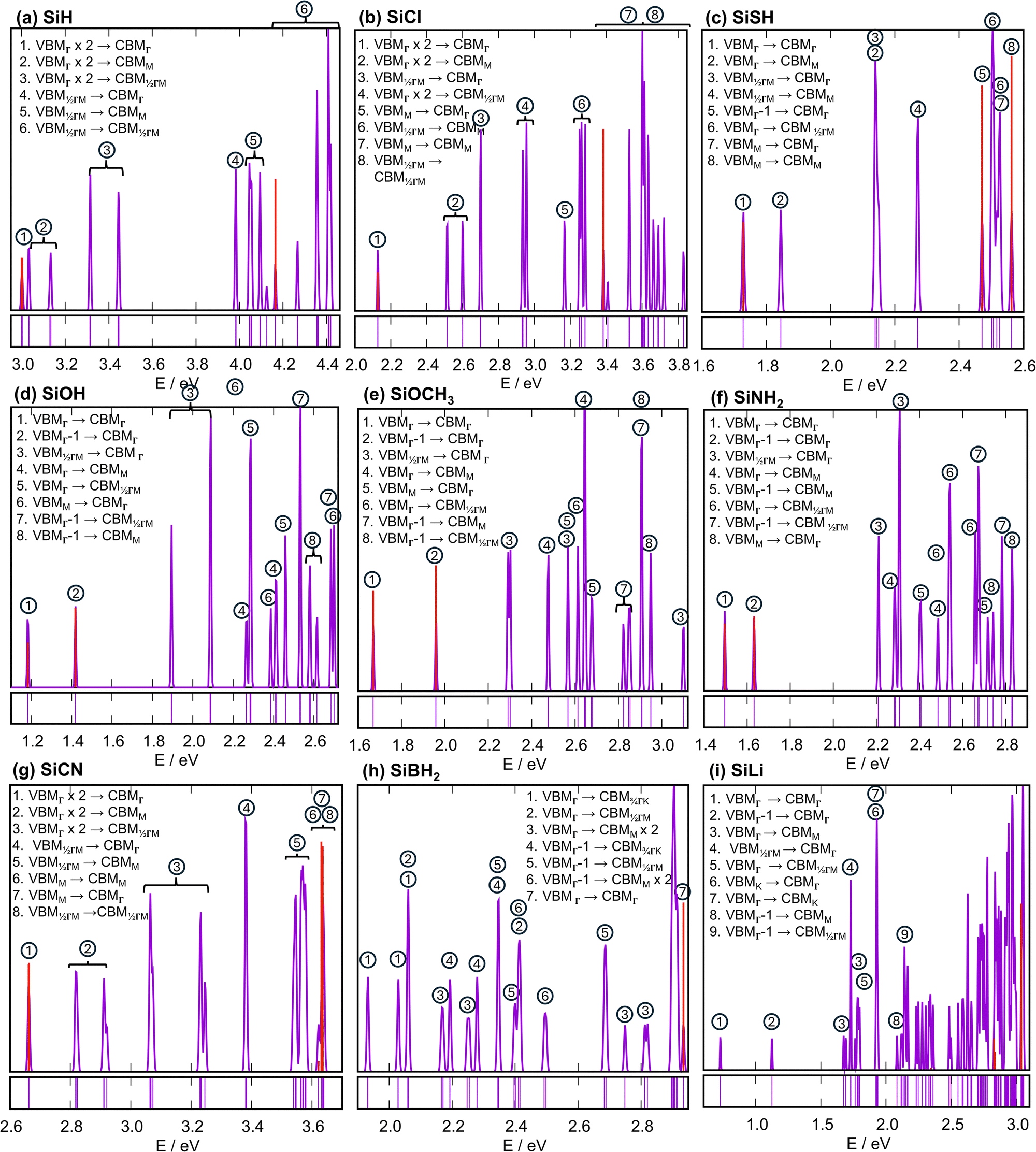 | ||
| Fig. 4 TDDFT-DOS of the six labeled silicane species obtained using their 4 × 4 × 1 supercells. They were obtained by Gaussian broadening with σ = 0.007 eV to each state's spike in the lower minor panels and summing over the Gaussians, and oscillator strengths are plotted as red spikes. States involving the same set of hole and particle orbitals are grouped and labeled by circled numbers. “VBMΓ × 2” in (a) and (b) indicates the doubly degenerate VBMΓ. Equal-height spikes in the lower minor panels indicate the existence of states at specific energies. | ||
 | ||
| Fig. 5 Frontier orbitals of the 4 × 4 × 1 SiH supercell. The orbitals are plotted with the same isosurface value of ±0.006 a.u. and are normalized in the supercell. The red dashed lines and the green dash line in (a) highlight a unit cell and the symmetry plane that cuts through the long diagonal axis of the super cell. | ||
According to the theory of configuration interaction with single excitations (CIS) for molecular systems, the energy (E(H → P)) of one singlet excitation scheme equals the energy gap between the hole (H) and particle (P) orbitals (εP − εH), subtract by the Coulomb integral between the two orbitals (JHP, which is the electrostatic attraction between the positively charged hole and the negatively charged electron), and plus twice their exchange integral (2KHP, which is the explicit correlation energy between the electron left in the hole orbital and the electron excited to the particle orbital),66i.e., E(H → P) = εP − εH − JHP + 2KHP. The M′ and M′′ states involve the same orbital energy gap. Numerical integration yields the same value of the VBMΓ′-CBMM(1/2, 1/2) and VBMΓ′′-CBMM(1/2, 1/2) Coulomb integrals. However, the exchange integral is 0.053 eV for the VBMΓ′-CBMM(1/2, 1/2) hole-particle pair, and 0.013 eV for the VBMΓ′′–CBMM(1/2, 1/2) pair. Twice the 0.04 eV difference qualitatively explains the 0.10 eV M′′–M′ splitting. Certainly, the TDDFT formalism is similar but not identical to the CIS formalism. The use of the CIS language to explain the TDDFT result is of qualitative nature, but insightful. This splitting of the extended SiH's excited state can be interpreted using the language of molecular systems. This is because the splitting here and the splittings of molecular electron configurations to term symbols are both driven by spatial confinement of orbitals.
The ③ states at the midpoint of the Γ-to-M path feature a similar magnitude of prime-double-prime splitting. Each ③ branch contains 6 degenerate states due to the 6-fold degeneracy of CBM1/2ΓM. The states grouped under ④, ⑤, and ⑥ in Fig. 4(a) can be viewed as the upshifted analogues of those under ①, ②, and ③, respectively, now with the hole on the lower-lying 6-fold degenerate VBM1/2ΓM. The second and third lowest state under ⑥ are two closely-lying optically bright Γ states. It is noteworthy that if we fully sample the FBZ by using even larger supercell, e.g., 16 × 16 × 1 and further broaden the already denser TDDFT-DOS peaks, the resultant continuous DOS will blur the splittings induced by the explicit electron. This justifies the use of the relatively small supercell and the discrete TDDFT-DOS as qualitative tools to deepen our understanding of the excited states. Now we have nailed down the ∼0.1 eV splitting of transitions involving degenerate hole and particle orbitals as the magnitude of explicit electron correlation effect. If the ∼0.1 eV energy difference does not matter for a specific study of SiR species, then the one-particle description is still applicable.
The TDDFT-DOS of SiCl in Fig. 4(b) shares features in Fig. 4(a): (1) a low-energy optically bright VBMΓ-to-CBMΓ transition; (2) the prime-double-prime splittings; (3) a high-energy optically bright VBM1/2ΓM-to-CBM1/2ΓM transition; (4) all low-energy excited states having hole and particle orbitals on the Γ-to-M path. Point (4) is shared by all SiR species considered in this work except SiBH2 and SiLi, since the Γ-to-M path contains all band edge orbitals in all species except the two. In Fig. 4(b), the VBMΓ-to-CBMM excitation lies substantially higher than the VBMΓ-to-CBMΓ excitation, as the CBMM does not compete for the true CBM. This is also seen in Fig. 4(c)–(g) for the same reason.
The rhombic primitive unit cell is slightly distorted to a parallelogram in SiOH, SiOCH3, SiSH, and SiNH2. Still, we use M (which is strictly applicable only to rhombic) to label the states at  ,
,  , and
, and  in their FBZs. Certainly, the 3-fold degeneracy of the M states is lifted, and the six Γ-to-M paths are no longer symmetrically related. Correspondingly, the TDDFT-DOS peaks grouped under the same numeric label are more broadly distributed in Fig. 4(c)–(g) than in Fig. 4(a), (b). Another feature in Fig. 4(d)–(f) is the splitting of the band edge optically bright state of 0.24 eV in SiOH, 0.29 eV in SiOCH3, and 0.14 eV in SiNH2 (Table S1, ESI†). They arise from the splitting of the originally doubly degenerate VBMΓ level due to the loss of the 3-fold site symmetry, which leads to the 0.25, 0.30, and 0.14 eV respective VBMΓ − 1-VBMΓ gaps (Fig. 2). The splittings in orbital energy levels match the splittings in excited states, i.e., the one-particle orbital description explains the splittings in those Γ excited states.
in their FBZs. Certainly, the 3-fold degeneracy of the M states is lifted, and the six Γ-to-M paths are no longer symmetrically related. Correspondingly, the TDDFT-DOS peaks grouped under the same numeric label are more broadly distributed in Fig. 4(c)–(g) than in Fig. 4(a), (b). Another feature in Fig. 4(d)–(f) is the splitting of the band edge optically bright state of 0.24 eV in SiOH, 0.29 eV in SiOCH3, and 0.14 eV in SiNH2 (Table S1, ESI†). They arise from the splitting of the originally doubly degenerate VBMΓ level due to the loss of the 3-fold site symmetry, which leads to the 0.25, 0.30, and 0.14 eV respective VBMΓ − 1-VBMΓ gaps (Fig. 2). The splittings in orbital energy levels match the splittings in excited states, i.e., the one-particle orbital description explains the splittings in those Γ excited states.
The comparison of the VBMΓ and VBMΓ − 1 orbitals of SiOH in Fig. 6(a) and (b) attributes their splitting to the different O lone pairs involved in their π* interactions. In the VBMΓ − 1 (VBMΓ) orbital, the O σ(π) lone pairs that are in plane with (perpendicular to) the OH bonds provide the π* interactions on both sides of the Si framework. The σ lone pair is well known to be more stable and thus exerts weaker π* interaction to VBMΓ − 1, resulting in the VBMΓ − 1–VBMΓ splitting and also the splitting of the band edge bright state. This explanation also applies to the similar splitting in SiOCH3.
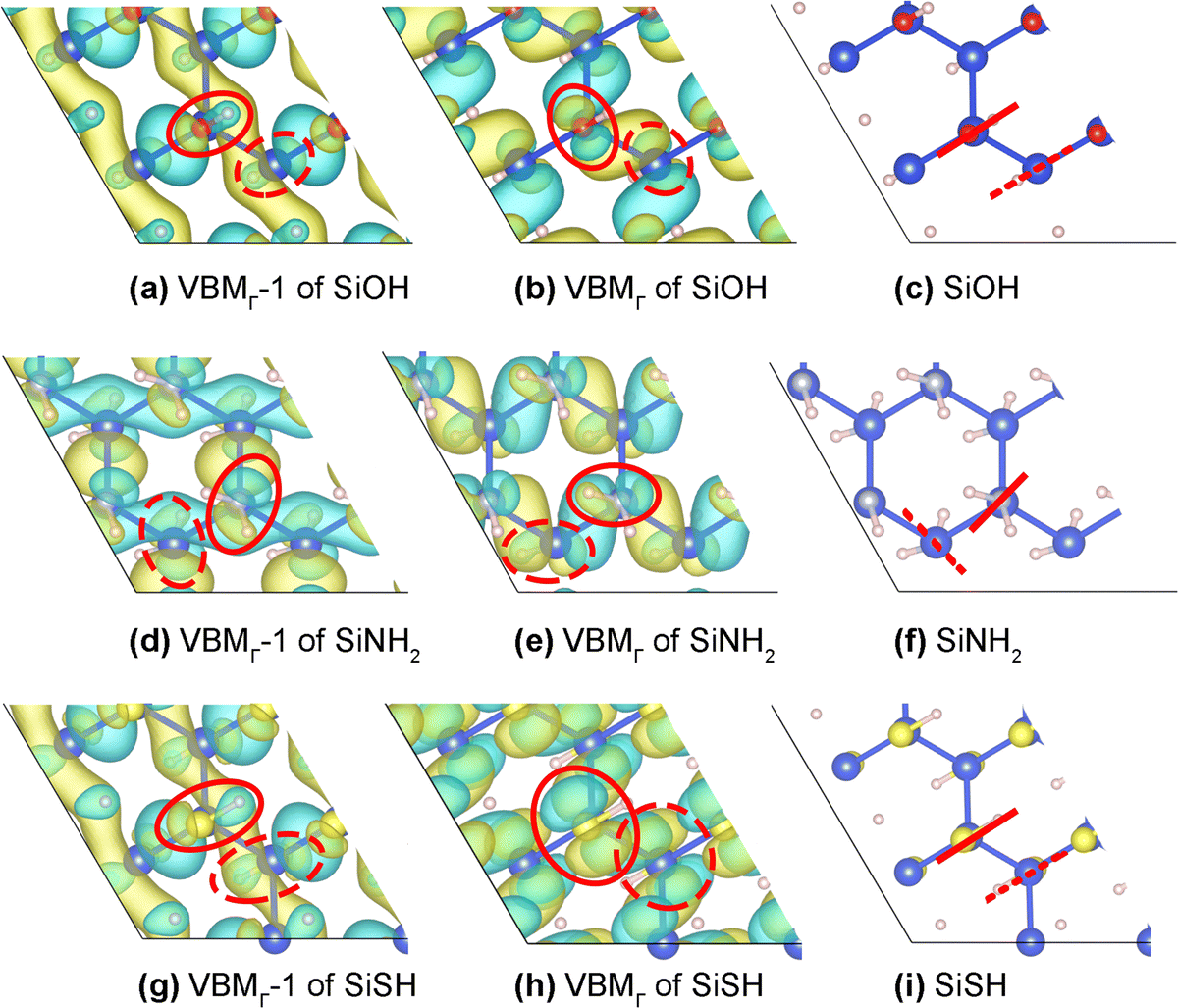 | ||
| Fig. 6 The VBMΓ − 1 and VBMΓ orbitals of SiOH ((a) and (b)), SiNH2 ((d) and (e)), and SiSH ((g) and (h)). The solid (dashed) ovals highlight the orientations of lone pair lobes of the ligands above (under) the Si framework. Red solid (dashed) lines in (c), (f), and (i) represent the planes that bisect the H–N–H angles or contain the O–H or S–H bonds above (below) the Si framework. | ||
The NH2 and SH ligands differ from OH and OCH3 by having only one active lone pair in exerting the π* interactions. The two themselves differ: the NH2 ligands above and under the Si framework are neither trans nor cis to each other, while the SH ligands on two sides are trans to each other (see the red lines in Fig. 6(f) and (i)). Thus, the antibonding interaction in SiNH2 cannot be concentrated in one VBMΓ orbital as in SiSH. Also, unlike in a free NH2 radical, the lone pair lobe of the NH2 ligand is not symmetric with respect to the plane that bisects the H–N–H angle. It can be polarized through mixing with one of the N–H bonding lobes to adjust its π interaction with the Si framework. This flexibility, absent in OH, OCH3, and SH ligands, allows the NH2 ligand to exert similar π* interactions in VBMΓ − 1 and VBMΓ, resulting in their smaller splitting, and the corresponding smaller splitting of band edge bright excitations.
The SiSH TDDFT-DOS in Fig. 4(c) shows a distinct difference from the other panels: the VBMΓ − 1-to-CBMΓ excitation (labeled ⑤ in Fig. 4(c)) is not a band edge excitation, as it lies 0.75 eV higher. This is due to the 0.70 eV substantial VBMΓ − 1–VBMΓ splitting in SiSH, which pushes the VBMΓ − 1 orbital deep into the VB (the band structures in Fig. 2(c)vs.Fig. 2(d), (f)). The large splitting is ascribed to: (1) each SH ligand mainly exerts π* interaction on VBMΓ through its π lone pair. The σ lone pair with S 3s and SH bonding character is inactive in pushing up VBMΓ − 1 (Fig. 6(g)vs.Fig. 6(h)); (2) the SH ligands above and under the Si framework are trans to each other, like the OH ligands in SiOH (Fig. 6(c) and (i)). The SH π lone pair lobes above and below are hence parallel and concertedly push up the VBMΓ energy.
Except for the red-shifts due to its smaller band gaps, the TDDFT-DOS of SiCN in Fig. 4(g) shares similar features as the SiH analogue in Fig. 4(a). The splittings of the ② states and ③ states in SiCN are for the same reason as the analogous splittings in SiH. In SiBH2, the BH2 2pπ vacant (σB–H) orbital lobes form bonding (antibonding) interaction with the σSi–Si bonding orbital lobes from both above and under the Si framework (see Fig. S10(a) and (b), ESI†) and stabilize (destabilize) the VBMΓ − 1 (VBMΓ) orbital, leading to their 0.25 eV orbital energy gap. Correspondingly, there is a 0.26 eV gap between the lowest excited states with the hole on the VBMΓ orbital and the VBMΓ − 1 orbital, i.e., the lowest ① and ④ states in Fig. 4(h). All these band edge states are optically dark states since the electron is excited to the CBM3/4ΓK orbital without conserving the crystal momentum. The true lowest unoccupied orbital is the CBMK orbital, which is not captured by our 4 × 4 × 1 supercell model. The CBM3/4ΓK (1/4, 1/2) orbital is plotted in Fig. S10(c) (ESI†) as an approximate of the CBMK orbital. The 1/4 index determines the largely nonbonding interaction along the a-axis, while the 1/2 index determines the maximum bonding interaction between the BH2 2pπ vacant lobes along the b-axis, as all the vacant lobes, above or under the Si framework, are pointing in the b-direction. The maximum bonding character along b and largely nonbonding character along a explain the shift of the true CBM from Γ or M in the other SiR species to K in SiBH2.
The lowest optically bright state (⑦ in Fig. 4(h)) lies 1 eV above the lowest band edge state and has the CBMΓ orbital as the particle orbital, which is similar to the analogue of SiH. Since both the CBMΓ and CBMK orbitals are mostly not within the Si framework, they have substantial spatial overlap, as illustrated by their product in Fig. S8 (ESI†). This spatial overlap should exist between CBMΓ and the other CBM orbitals on the Γ-to-K path. The substantial spatial overlaps suggest that after the VBMΓ-to-CBMΓ optical excitation, the electron can non-adiabatically relax to the CBMK orbital, resulting in a long-living electron on the BH2 group and a long-living hole in the Si framework. The long-living spatially separated holes and electrons will find extensive use in photovoltaics and photochemistry.
SiLi is a well-studied species for its extensive use in Li-ion battery (LIB).18,67 It is a purely ionic system and adopts a completely different structure compared to the other aforementioned species. In a range of 0.69 eV, the bottom of CB is all dominated by Li atomic orbitals (Fig. 2(i) and Fig. S6, ESI†), while the VBM is Si-dominated. Consequently, all low-lying excited states labeled in Fig. 4(i) are optically dark states with an electron being transferred from the Si− framework to the Li+ cationic layer, which will be easily returned to the Si framework due to the electropositivity of Li. Therefore, SiLi is immune to photodegradation and is a promising LIB material, as already well known.67–69
The DFT and TDDFT band gaps are compared in Fig. S11 (ESI†). The excellent agreement between the two sets of independently obtained gaps corroborates their respective accuracies.
4. Band edge variation
We used the 1s energy of an extra added He atom to calibrate orbital energy levels of the nine SiR species (Fig. S12, ESI†). The so calibrated band edge energies are compared in Fig. 7. Replacing H with Cl downshifts both VBM and CBM due to the ∼4-fold more positively charged Si framework (compare the Si atomic charges of SiH and SiCl in Table 1). The VBM does not downshift as much due to the Cl π* interaction (Fig. 3(b)). The σ-withdrawing capability of SH is almost half that of Cl (Table S2, ESI†), downshifting VBM and CBM less in SiSH. This less VBM downshift is even counteracted by the stronger π-donating capability of SH, which raises the VBM higher than in SiH. The π-donating capabilities of OH, OCH3, and NH2 are nearly twice as strong as that of Cl (Table S2, ESI†), and the π-donating capabilities dominate over the σ-withdrawing capabilities of the three ligands. Consequently, the VBMs of SiOH, SiOCH3, and SiNH2 are pushed up by 0.93, 1.63 and 1.53 eV higher than in SiH, despite their more positively charged Si atoms (Table 1). The higher VBM of SiOCH3vs. SiOH can be attributed to: (1) the higher pDEA of OCH3; (2) the elongation of the Si–Si bond induced by the transversal stress of the bulky OCH3 groups, whose σSi–Si bonding character is hence weakened. | ||
| Fig. 7 Comparison of band edge energies of the nine investigated species. | ||
The antibonding character in the CBMΓ orbitals in Fig. 3 and Fig. S5, S6 (ESI†) are represented by the three red lines (1 solid + 2 dashed). This π* interaction is stronger in SiOH, SiOCH3, and SiNH2 than in SiCl due to the shorter Si–O and Si–N bonds (Table 1), leading to the higher CBMs of the former three. Additionally, the CBM of SiOCH3 is further raised by the extra antibonding interaction between the CH3 moieties and the Si atoms (the green dotted lines in Fig. 3(d)), which originates from steric hindrance of the CH3 moieties.
In summary, the sWpD ligands pull down (push up) band edges through their σ-withdrawing (π-donating) capabilities, and the balance between the opposite effects determines the final VBM and CBM energies. On the contrary, the σ- and π-withdrawing capabilities of CN synergistically downshift the VBM and CBM of SiCN to exceptionally low. The BH2 ligand raises (lowers) the SiBH2 VBM through its σ-donating (π-withdrawing) capability. The two effects largely cancel and result in the VBM marginally changed from the SiH analogue. The CBM of SiBH2 is significantly lower than SiH due to the bonding interaction between the BH2 vacant lobes (Fig. S6(b) and S10(c), ESI†). The VBM of SiLi is much higher than the other SiR species because of its purely anionic Si framework.
We also calculated the work functions (Φ) for the SiR species as the difference between the vacuum and Fermi levels. Their data in Table 1 are in excellent agreement with the He-calibrated VBM energies shown in Fig. 7. The He calibration provides an alternative method for comparing energy levels of 2D materials obtained in individual calculations.
The qualitative understanding the influence of the σ/π-withdrawing/donating capabilities of ligands on the band edge energies motivated us to quantify this impact. We fitted the calibrated VBM and CBM energies of the silicane species (excluding SiLi) using 2D polynomial functions of the sEDA (x) and pEDA (y) values of the R ligands. The resultant functions (in eV) for silicanes with sWpD R reads:
| f(x,y) = VBM/CBM(SiH) + a(cosθx + sinθy) + b(−sinθx + cosθy) + c(cosθx + sinθy)2 + d(−sinθx + cosθy)2, |
IV. Conclusions
We systematically investigated the geometrical structures, electronic structures, and band edge excited states of silicanes with ligands that fall into three classes based on their electron-withdrawing/donating capabilities. The sWpD ligands reduce the band gap of the substituted silicanes and also tend to change the mixture of direct and indirect gap in SiH to purely direct gaps. This is attributed to the different spatial distributions of the CBMΓ and CBMM orbitals and consequently their different responses to the more positively charged Si framework induced by the oxidative ligands. The sWpW ligand reduces and maintains the mixture of direct and indirect gaps. The sDpW ligand leads to an indirect band gap. These ligands, through their rich combinations of sW/D and pW/D capabilities, modulate the band edge energies in an enormous range of 6 eV. Empirical functions are developed for the quick prediction of band edge energies of silicanes using the readily obtainable sW/D and pW/D indices of ligands. The explanation of the indirect-to-direct gap conversion, the demonstration of the large range of band edge energies, and their predictive functions are unprecedented contributions of this work. The wide range of band gaps of the substituted silicanes can regulate the band edge absorption from the near-infrared to the visible spectrum. The wide range of band edge energies allow for all three types of hetero-stackings between silicane species: straddling, staggered, and broken. Silicanes with sDpW ligands also feature far separated conducting holes and electrons in both real and reciprocal spaces, which are likely to have longer lifetime decays of the excited charge carriers. These findings will inspire extensive applications of silicane-based materials in optoelectronics and photocatalytic applications.We performed TDDFT calculations to assess the applicability of the one-particle orbital description for band edge excited states in silicanes. Excited states with degenerate hole and degenerate particle orbitals feature ∼0.1 eV splitting, which quantifies the inaccuracy of the one-particle description. This inaccuracy is understandable since the splitting is induced by explicit electron correlation, which is pronounced when hole and particle orbitals are spatially confined. On the other hand, the one-particle description is satisfactory in explaining the band edge bright excited state splittings induced by orbital level splittings. The different ligands lead to different splittings of the frontier orbital levels and the associated different splittings of the band edge bright states, and this is attributed to the different π interactions between the ligands and the Si framework. Detailed analyses of frontier orbitals in the extended systems are indispensable to unravel the mystery in those splittings that can or cannot be described by the one-particle picture. Another methodological contribution is the justification of the use of the C-adapted σ/π-donating/accepting indices proposed by Ozimiński and Dobrowolski in silicon chemistry. We calculated the Si-adapted indices, and they are in close agreement with the C-adapted indices. Despite the close agreement, the Si-adapted indices are presented for future prudent examinations of ligand effects in silicon chemistry.
Author contributions
Conceptualization, G. Y., T. Z., Z. Y.; model construction and calculation, G. Y.; results analysis G. Y., T. Z., Z. Y., and E. P.; writing: G. Y., T. Z., and Z. Y.; funding acquisition, Z. Y., T. Z., and G. Y.; all authors read and agreed to the published version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.† The codes for generate unit cell's k vectors for orbitals obtained from supercell calculation performed on Γ point only, and for numerical calculation of 2-electron integral for orbitals in cube format are available on request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
All calculations were carried out using resources provided by the Digital Research Alliance of Canada and the Tianhe-2 (TH-2) supercomputer clusters (Guangzhou, Guangdong, China). T. Z. is grateful to York University (481333) and the Natural Sciences and Engineering Research Council (NSERC) of Canada (RGPIN-2024-06286) for financial support. Z. Y. would like to acknowledge financial support from the National Natural Science Foundation of China (22175201), the Pearl River Recruitment Program of Talent (2019QN01C108), Guangzhou Science and Technology Planning Project (2024A04J6360), the Guangdong Basic Research Center of Excellence for Functional Molecular Engineering (2024G0003), and the Sun Yat-sen University. G. Y. thanks the China Scholarship Council program (202206380126) for the financial support.References
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Electric Field Effect in Atomically Thin Carbon Films, Science, 2004, 306(5696), 666–669 CrossRef CAS PubMed.
- B. Aufray, A. Kara, S. Vizzini, H. Oughaddou, C. Léandri, B. Ealet and G. Le Lay, Graphene-like silicon nanoribbons on Ag(110): A possible formation of silicene, Appl. Phys. Lett., 2010, 96(18), 183102 CrossRef.
- B. Lalmi, H. Oughaddou, H. Enriquez, A. Kara, S. Vizzini, B. Ealet and B. Aufray, Epitaxial growth of a silicene sheet, Appl. Phys. Lett., 2010, 97(22), 223109 CrossRef.
- L. Tao, E. Cinquanta, D. Chiappe, C. Grazianetti, M. Fanciulli, M. Dubey, A. Molle and D. Akinwande, Silicene field-effect transistors operating at room temperature, Nat. Nanotechnol., 2015, 10(3), 227–231 CrossRef CAS PubMed.
- M. W. Chuan, M. A. Riyadi, A. Hamzah, N. E. Alias, S. Mohamed Sultan, C. S. Lim and M. L. P. Tan, Device performances analysis of p-type doped silicene-based field effect transistor using SPICE-compatible model, PLoS One, 2022, 17(3), e0264483 CrossRef CAS PubMed.
- H. Emami-Nejad, A. mir, Z. Lorestaniweiss, A. Farmani and R. Talebzadeh, First designing of a silicene-based optical MOSFET with outstanding performance, Sci. Rep., 2023, 13(1), 6563 CrossRef CAS PubMed.
- L. Du, T. Hasan, A. Castellanos-Gomez, G.-B. Liu, Y. Yao, C. N. Lau and Z. Sun, Engineering symmetry breaking in 2D layered materials, Nat. Rev. Phys., 2021, 3(3), 193–206 CrossRef CAS.
- W.-F. Tsai, C.-Y. Huang, T.-R. Chang, H. Lin, H.-T. Jeng and A. Bansil, Gated silicene as a tunable source of nearly 100% spin-polarized electrons, Nat. Commun., 2013, 4(1), 1500 CrossRef PubMed.
- Y. Wei, T. Liu, C. Huang, Y. C. Tao and F. Qi, Controllable spin pairing states in silicene-based superconducting hybrid structures with noncollinear magnetizations, Phys. Rev. Res., 2021, 3(3), 033131 CrossRef CAS.
- S. Wang, X. Liu, M. Xu, L. Liu, D. Yang and P. Zhou, Two-dimensional devices and integration towards the silicon lines, Nat. Mater., 2022, 21(11), 1225–1239 CrossRef CAS PubMed.
- H. Okamoto, Y. Sugiyama and H. Nakano, Synthesis and Modification of Silicon Nanosheets and Other Silicon Nanomaterials, Chem. – Eur. J., 2011, 17(36), 9864–9887 CrossRef CAS PubMed.
- W. L. B. Huey and J. E. Goldberger, Covalent functionalization of two-dimensional group 14 graphane analogues, Chem. Soc. Rev., 2018, 47(16), 6201–6223 RSC.
- C. Wang, X. Xu, X. Pi, M. D. Butala, W. Huang, L. Yin, W. Peng, M. Ali, S. C. Bodepudi, X. Qiao, Y. Xu, W. Sun and D. Yang, Neuromorphic device based on silicon nanosheets, Nat. Commun., 2022, 13(1), 5216 CrossRef CAS PubMed.
- H. Nakano, K. Ito, A. Miura and Y. Majima, Solution-Processed Silicane Field-Effect Transistor: Operation Due to Stacking Defects on the Channel, Adv. Funct. Mater., 2020, 30(15), 1908746 CrossRef CAS.
- M. Stavrou, I. Papadakis, A. Stathis, M. J. Kloberg, J. Mock, T. Kratky, S. Günther, B. Rieger, M. Becherer, A. Lyuleeva-Husemann and S. Couris, Silicon Nanosheets versus Graphene Nanosheets: A Comparison of Their Nonlinear Optical Response, J. Phys. Chem. Lett., 2021, 12(2), 815–821 CrossRef CAS PubMed.
- C. Qian, W. Sun, D. L. H. Hung, C. Qiu, M. Makaremi, S. G. Hari Kumar, L. Wan, M. Ghoussoub, T. E. Wood, M. Xia, A. A. Tountas, Y. F. Li, L. Wang, Y. Dong, I. Gourevich, C. V. Singh and G. A. Ozin, Catalytic CO2 reduction by palladium-decorated silicon–hydride nanosheets, Nat. Catal., 2019, 2(1), 46–54 CrossRef CAS.
- S. Wang, C. Wang, W. Pan, W. Sun and D. Yang, Two-Dimensional Silicon for (Photo)Catalysis, Sol. RRL, 2021, 5(2), 2000392 CrossRef CAS.
- X. Deng, X. Zheng, T. Yuan, W. Sui, Y. Xie, O. Voznyy, Y. Wang and Z. Yang, Ligand Impact of Silicanes as Anode Materials for Lithium-Ion Batteries, Chem. Mater., 2021, 33(23), 9357–9365 CrossRef CAS.
- Y. An, Y. Tian, C. Wei, Y. Zhang, S. Xiong, J. Feng and Y. Qian, Recent advances and perspectives of 2D silicon: Synthesis and application for energy storage and conversion, Energy Storage Mater., 2020, 32, 115–150 CrossRef.
- C. Jia, F. Zhang, N. Zhang, Q. Li, X. He, J. Sun, R. Jiang, Z. Lei and Z.-H. Liu, Bifunctional Photoassisted Li–O2 Battery with Ultrahigh Rate-Cycling Performance Based on Siloxene Size Regulation, ACS Nano, 2023, 17(2), 1713–1722 CrossRef CAS PubMed.
- H. Nakano, H. Tetsuka, M. J. S. Spencer and T. Morishita, Chemical modification of group IV graphene analogs, Sci. Technol. Adv. Mater., 2018, 19(1), 76–100 CrossRef CAS PubMed.
- M. Ohashi, S. Shirai and H. Nakano, Direct Chemical Synthesis of Benzyl-Modified Silicane from Calcium Disilicide, Chem. Mater., 2019, 31(13), 4720–4725 CrossRef CAS.
- A. B. Sieval, A. L. Demirel, J. W. M. Nissink, M. R. Linford, J. H. van der Maas, W. H. de Jeu, H. Zuilhof and E. J. R. Sudhölter, Highly Stable Si–C Linked Functionalized Monolayers on the Silicon (100) Surface, Langmuir, 1998, 14(7), 1759–1768 CrossRef CAS.
- B. Jaeckel, R. Hunger, L. J. Webb, W. Jaegermann and N. S. Lewis, High-Resolution Synchrotron Photoemission Studies of the Electronic Structure and Thermal Stability of CH3- and C2H5-Functionalized Si(111) Surfaces, J. Phys. Chem. C, 2007, 111(49), 18204–18213 CrossRef CAS.
- L. J. Webb and N. S. Lewis, Comparison of the Electrical Properties and Chemical Stability of Crystalline Silicon(111) Surfaces Alkylated Using Grignard Reagents or Olefins with Lewis Acid Catalysts, J. Phys. Chem. B, 2003, 107(23), 5404–5412 CrossRef CAS.
- H. H. Arefi, M. Nolan and G. Fagas, Role of the Head and/or Tail Groups of Adsorbed –[Xhead
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) group]–Alkyl–[Xtailgroup] [X = O(H), S(H), NH(2)] Chains in Controlling the Work Function of the Functionalized H:Si(111) Surface, J. Phys. Chem. C, 2015, 119(21), 11588–11597 CrossRef CAS.
group]–Alkyl–[Xtailgroup] [X = O(H), S(H), NH(2)] Chains in Controlling the Work Function of the Functionalized H:Si(111) Surface, J. Phys. Chem. C, 2015, 119(21), 11588–11597 CrossRef CAS. - B. K. Teo and X. H. Sun, Silicon-Based Low-Dimensional Nanomaterials and Nanodevices, Chem. Rev., 2007, 107(5), 1454–1532 CrossRef CAS PubMed.
- G. Yao, E. Pradhan, Z. Yang and T. Zeng, Photo-Induced Long-Distance Charge Transfer in Silicanes: the Stacking Matters, Nano Lett., 2025, 25(4), 1697–1705 CrossRef CAS PubMed.
- T. Li, G. Liu, G. Yao, X. Ma, Z. He and Y. Cao, Ligand-Triggered MXene Quantum Dots with Tunable Work Function as Anode and Cathode Interlayers for Organic Solar Cells, Sol. RRL, 2024, 8(13), 2400270 CrossRef CAS.
- J. E. Kelm and J. L. Dempsey, Metal-Dictated Reactivity of Z-Type Ligands to Passivate Surface Defects on CdSe Nanocrystals, J. Am. Chem. Soc., 2024, 146(8), 5252–5262 CrossRef CAS PubMed.
- Y. Sugiyama, H. Okamoto, T. Mitsuoka, T. Morikawa, K. Nakanishi, T. Ohta and H. Nakano, Synthesis and Optical Properties of Monolayer Organosilicon Nanosheets, J. Am. Chem. Soc., 2010, 132(17), 5946–5947 CrossRef CAS PubMed.
- H. Okamoto, Y. Kumai, Y. Sugiyama, T. Mitsuoka, K. Nakanishi, T. Ohta, H. Nozaki, S. Yamaguchi, S. Shirai and H. Nakano, Silicon Nanosheets and Their Self-Assembled Regular Stacking Structure, J. Am. Chem. Soc., 2010, 132(8), 2710–2718 CrossRef CAS PubMed.
- J. Ohshita, K. Yamamoto, D. Tanaka, M. Nakashima, Y. Kunugi, M. Ohashi and H. Nakano, Preparation and Photocurrent Generation of Silicon Nanosheets with Aromatic Substituents on the Surface, J. Phys. Chem. C, 2016, 120(20), 10991–10996 CrossRef CAS.
- P. A. Denis, Stacked functionalized silicene: a powerful system to adjust the electronic structure of silicene, Phys. Chem. Chem. Phys., 2015, 17(7), 5393–5402 RSC.
- R. Wang, X. Pi, Z. Ni, Y. Liu and D. Yang, Density functional theory study on organically surface-modified silicene, RSC Adv., 2015, 5(43), 33831–33837 RSC.
- M. Sun, Q. Ren, S. Wang, J. Yu and W. Tang, Electronic properties of Janus silicene: new direct band gap semiconductors, J. Phys. D: Appl. Phys., 2016, 49(44), 445305 CrossRef.
- F. Bechstedt, P. Gori and O. Pulci, Beyond graphene: Clean, hydrogenated and halogenated silicene, germanene, stanene, and plumbene, Prog. Surf. Sci., 2021, 96(3), 100615 CrossRef CAS.
- W. Tang, M. Sun, Q. Ren, Y. Zhang, S. Wang and J. Yu, First principles study of silicene symmetrically and asymmetrically functionalized with halogen atoms, RSC Adv., 2016, 6(98), 95846–95854 RSC.
- S. Wang and J. Yu, Tuning electronic properties of silicane layers by tensile strain and external electric field: A first-principles study, Thin Solid Films, 2018, 654, 107–115 CrossRef CAS.
- Y. Wang, L. Wang, X. Zhang, X. Liang, Y. Feng and W. Feng, Two-dimensional nanomaterials with engineered bandgap: Synthesis, properties, applications, Nano Today, 2021, 37, 101059 CrossRef CAS.
- G. Kresse and J. Furthmüller, Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci., 1996, 6(1), 15–50 CrossRef CAS.
- P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50(24), 17953–17979 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77(18), 3865–3868 CrossRef CAS PubMed.
- A. V. Krukau, O. A. Vydrov, A. F. Izmaylov and G. E. Scuseria, Influence of the exchange screening parameter on the performance of screened hybrid functionals, J. Chem. Phys., 2006, 125(22), 224106 CrossRef PubMed.
- K. Momma and F. Izumi, VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data, J. Appl. Crystallogr., 2011, 44(6), 1272–1276 CrossRef CAS.
- V. Wang, N. Xu, J.-C. Liu, G. Tang and W.-T. Geng, VASPKIT: A user-friendly interface facilitating high-throughput computing and analysis using VASP code, Comput. Phys. Commun., 2021, 267, 108033 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields, J. Phys. Chem. C, 1994, 98(45), 11623–11627 CrossRef CAS.
- T. H. Dunning, Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen, J. Chem. Phys., 1989, 90(2), 1007–1023 CrossRef CAS.
- W. P. Ozimiński and J. C. Dobrowolski, σ- and π-electron contributions to the substituent effect: natural population analysis, J. Phys. Org. Chem., 2009, 22(8), 769–778 CrossRef.
- J.-D. Chai and M. Head-Gordon, Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections, Phys. Chem. Chem. Phys., 2008, 10(44), 6615–6620 RSC.
- A. E. Reed, R. B. Weinstock and F. Weinhold, Natural population analysis, J. Chem. Phys., 1985, 83(2), 735–746 CrossRef CAS.
- G. M. J. Barca, C. Bertoni, L. Carrington, D. Datta, N. De Silva, J. E. Deustua, D. G. Fedorov, J. R. Gour, A. O. Gunina, E. Guidez, T. Harville, S. Irle, J. Ivanic, K. Kowalski, S. S. Leang, H. Li, W. Li, J. J. Lutz, I. Magoulas, J. Mato, V. Mironov, H. Nakata, B. Q. Pham, P. Piecuch, D. Poole, S. R. Pruitt, A. P. Rendell, L. B. Roskop, K. Ruedenberg, T. Sattasathuchana, M. W. Schmidt, J. Shen, L. Slipchenko, M. Sosonkina, V. Sundriyal, A. Tiwari, J. L. Galvez Vallejo, B. Westheimer, M. Włoch, P. Xu, F. Zahariev and M. S. Gordon, Recent developments in the general atomic and molecular electronic structure system, J. Chem. Phys., 2020, 152(15), 154102 CrossRef CAS PubMed.
- T. D. Kühne, M. Iannuzzi, M. Del Ben, V. V. Rybkin, P. Seewald, F. Stein, T. Laino, R. Z. Khaliullin, O. Schütt, F. Schiffmann, D. Golze, J. Wilhelm, S. Chulkov, M. H. Bani-Hashemian, V. Weber, U. Borštnik, M. Taillefumier, A. S. Jakobovits, A. Lazzaro, H. Pabst, T. Müller, R. Schade, M. Guidon, S. Andermatt, N. Holmberg, G. K. Schenter, A. Hehn, A. Bussy, F. Belleflamme, G. Tabacchi, A. Glöβ, M. Lass, I. Bethune, C. J. Mundy, C. Plessl, M. Watkins, J. VandeVondele, M. Krack and J. Hutter, CP2K: An electronic structure and molecular dynamics software package – Quickstep: Efficient and accurate electronic structure calculations, J. Chem. Phys., 2020, 152(19), 194103 CrossRef PubMed.
- G. Lippert, J. Hutter and M. Parrinello, The Gaussian and augmented-plane-wave density functional method for ab initio molecular dynamics simulations, Theor. Chem. Acc., 1999, 103(2), 124–140 Search PubMed.
- S. Goedecker, M. Teter and J. Hutter, Separable dual-space Gaussian pseudopotentials, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54(3), 1703–1710 CrossRef CAS PubMed.
- C. Hartwigsen, S. Goedecker and J. Hutter, Relativistic separable dual-space Gaussian pseudopotentials from H to Rn, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58(7), 3641–3662 CrossRef CAS.
- J. VandeVondele and J. Hutter, Gaussian basis sets for accurate calculations on molecular systems in gas and condensed phases, J. Chem. Phys., 2007, 127(11), 114105 CrossRef PubMed.
- L. Bernasconi, S. Tomić, M. Ferrero, M. Rérat, R. Orlando, R. Dovesi and N. M. Harrison, First-principles optical response of semiconductors and oxide materials, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83(19), 195325 CrossRef.
- R. Derian, K. Tokár, B. Somogyi, Á. Gali and I. Štich, Optical Gaps in Pristine and Heavily Doped Silicon Nanocrystals: DFT versus Quantum Monte Carlo Benchmarks, J. Chem. Theory Comput., 2017, 13(12), 6061–6067 CrossRef CAS PubMed.
- R. B. Pontes, R. R. Mançano, R. da Silva, L. F. Cótica, R. H. Miwa and J. E. Padilha, Electronic and optical properties of hydrogenated group-IV multilayer materials, Phys. Chem. Chem. Phys., 2018, 20(12), 8112–8118 RSC.
- A. Weiss, G. Beil and H. Meyer, The Topochemical Reaction of CaSi2 to a Two-Dimensional Subsiliceous Acid Si6H3(OH)3 (= Kautskys’ Siloxene), Z. Naturforsch., B: J. Chem. Sci., 1980, 35(1), 25–30 CrossRef.
- J. R. Dahn, B. M. Way, E. Fuller and J. S. Tse, Structure of siloxene and layered polysilane (Si6H6), Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48(24), 17872–17877 CrossRef CAS PubMed.
- O. D. Restrepo, R. Mishra, J. E. Goldberger and W. Windl, Tunable gaps and enhanced mobilities in strain-engineered silicane, J. Appl. Phys., 2014, 115(3), 033711 CrossRef.
- J. S. Tse, J. R. Dahn and F. Buda, Electronic Structures of Layered Polysilanes, J. Phys. Chem. C, 1995, 99(7), 1896–1899 CrossRef CAS.
- S. Z. Butler, S. M. Hollen, L. Cao, Y. Cui, J. A. Gupta, H. R. Gutiérrez, T. F. Heinz, S. S. Hong, J. Huang, A. F. Ismach, E. Johnston-Halperin, M. Kuno, V. V. Plashnitsa, R. D. Robinson, R. S. Ruoff, S. Salahuddin, J. Shan, L. Shi, M. G. Spencer, M. Terrones, W. Windl and J. E. Goldberger, Progress, Challenges, and Opportunities in Two-Dimensional Materials Beyond Graphene, ACS Nano, 2013, 7(4), 2898–2926 CrossRef CAS PubMed.
- P. Kimber and F. Plasser, Toward an understanding of electronic excitation energies beyond the molecular orbital picture, Phys. Chem. Chem. Phys., 2020, 22(11), 6058–6080 RSC.
- S. Wei, T. Hartman, S. Mourdikoudis, X. Liu, G. Wang, E. Kovalska, B. Wu, J. Azadmanjiri, R. Yu, L. Chacko, L. Dekanovsky, F. M. Oliveira, M. Li, J. Luxa, S. Jamali Ashtiani, J. Su and Z. Sofer, Reaction Mechanism and Performance of Innovative 2D Germanane-Silicane Alloys: SiGe1-H Electrodes in Lithium-Ion Batteries, Adv. Sci., 2024, 11(24), 2308955 CrossRef CAS PubMed.
- S. Zhang, Y. Wang, G. Yang and Y. Ma, Silicon Framework-Based Lithium Silicides at High Pressures, ACS Appl. Mater. Interfaces, 2016, 8(26), 16761–16767 CrossRef CAS PubMed.
- W. He, W. Xu, Z. Li, Z. Hu, J. Yang, G. Qin, W. Teng, T. Zhang, W. Zhang, Z. Sun and X. Yu, Structural Design and Challenges of Micron-Scale Silicon-Based Lithium-ion Batteries, Adv. Sci., 2025, 2407540 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Figures and tables for discussion, discussions of the electron donating and accepting descriptors, comparison of TDDFT results using different supercells, and fitting functions for band edge energies. See DOI: https://doi.org/10.1039/d4cp04458d |
| This journal is © the Owner Societies 2025 |