
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Vetting molecular candidates posited for the first diffuse interstellar bands (5780 and 5797 Å): a quantum chemical study†
Halis
Seuret
ab,
Ailish D.
Sullivan
b,
Cercis
Morera-Boado
*g,
Tina A.
Harriott
 *bc,
Daniel
Majaess
*bc,
Lou
Massa
d and
Chérif F.
Matta
*bef
*bc,
Daniel
Majaess
*bc,
Lou
Massa
d and
Chérif F.
Matta
*bef
aCentro de Investigaciones Químicas, IICBA, Universidad Autónoma del Estado de Morelos, Cuernavaca, 62209, Morelos, México
bDepartment of Chemistry and Physics, Mount Saint Vincent University, Halifax, Nova Scotia B3M2J6, Canada. E-mail: daniel.majaess@gmail.com; daniel.majaess@msvu.ca; cherif.matta@msvu.ca
cDepartment of Mathematics and Statistics, Mount Saint Vincent University, Halifax, Nova Scotia B3M2J6, Canada. E-mail: Tina.Harriott@msvu.ca
dHunter College & the PhD Program of the Graduate Center, City University of New York, New York, NY 10065, USA
eDepartment of Chemistry, Saint Mary's University, Halifax, Nova Scotia B3H3C3, Canada
fDépartement de Chimie, Université Laval, Québec, Québec G1V 0A6, Canada
gDepartamento de Físicoquímica Teórica, Secihti-Centro de Investigaciones Químicas, IICBA, Universidad Autónoma del Estado de Morelos, Cuernavaca, 62209, Morelos, México. E-mail: cercis.morerab@uaem.edu.mx
First published on 6th May 2025
Abstract
Diffuse interstellar bands (DIBs) comprise over 550 celestial absorption features whose molecular carriers remain largely unidentified or contested. In this study, we present a statistical analysis that identifies two previously overlooked families of strongly correlated lines associated with the original Heger features at 5780 and 5797 Å. Comprehensive UV-vis spectra were computed at several levels of theory (mainly TD-PBE0 and EOM-CCSD with an aug-cc-pVTZ basis set) for the following candidates posited as diffuse interstellar band carriers (in both their neutral and cationic forms): 2-cyclopenten-1-one, 3(2H)-thiophenone, 2(5H)-furanone, 3(2H)-selenophenone, 3-hydroxypropanamide, oxamic acid, lactamide, and glycolamide. Glycolamide is of particular interest since it has recently been detected in microwave (rotational) spectra of the comparatively dense molecular cloud G+0.693–0027. Importantly, the computations reveal that the anions exhibit marginal electron affinities despite producing improbable lines (i.e., with excitations to levels above the ionization threshold) overlapping DIBs, whereas the neutral molecules yield lines shortward of DIBs and possibly linked to the broad 220 nm interstellar feature, and their cations produced too few lines in the DIB domain inspected. Further vetting of candidates awaits the construction of an expansive optical-infrared molecular ion database, which will facilitate concurrent matching to DIBs in the optical (electronic) and their energy differences in the mid-infrared (vibrational), thereby narrowing the parameter space.
Introduction
Quantum chemistry plays an increasingly central role in astronomical research, particularly in the quest to identify the carriers of the diffuse interstellar bands (DIBs). As Fortenberry emphasized, theoretical calculations often provide the only viable means of predicting the spectra of elusive or unstable molecular species that are inaccessible to direct laboratory study. Without accurate computed spectra, attempts to match observational data to molecular carriers would remain largely speculative.1,2 The identification of DIB carriers—a problem persisting for over a century—thus hinges critically on high-level quantum chemical methods capable of reliably predicting electronic and vibrational transitions under astrophysical conditions. As Sousa-Silva et al. have noted, obtaining experimental spectra of candidate interstellar molecules presents significant challenges.3 Many relevant species are transient, reactive, or exist predominantly in ionic forms, complicating their isolation. Laboratory techniques such as matrix isolation spectroscopy, pioneered by Y. P. Lee and collaborators,4,5 have been employed to stabilize such molecules, but the interaction with the matrix environment can induce spectral shifts that obscure gas-phase signatures. Consequently, robust theoretical predictions remain indispensable. In light of these challenges, the present study seeks to vet molecular candidates for the 5780 Å and 5797 Å diffuse interstellar bands—the first DIBs discovered by Heger6,7—through a combination of statistical correlation analyses and quantum chemical computations.Diffuse interstellar bands are absorption features observed in spectra of diverse celestial targets. When observed in binary star spectra these lines do not undergo the expected periodic Doppler shifts due to the stars' motion, and thus these features arise in the intervening interstellar medium.6–13 These absorptions are molecular in origin, since they are generally broader than atomic lines. The broadening of molecular lines in DIBs spectra arises from several factors including (but not necessarily limited to) overlapping rotational transitions (especially for large molecules with dense rotational energy levels), thermal Doppler broadening due to random molecular motions, turbulence (non-thermal) Doppler broadening due to the collective currents of large masses within a given cloud in different directions, the short lifetimes of the excited states, the presence of molecules with different isotopic compositions (isotopologues), and many more effects. These factors contribute significantly in making the identification of DIB carriers difficult.9–13
A century ago, Mary Lea Heger identified the first DIBs at 5780 and 5797 Å.6,7,14 Since this seminal discovery more than 550 DIBs have been identified.15 There is yet to exist an undisputed molecular carrier for any DIB, though a consensus is continuing to coalesce around the connection between C60+ and two DIBs (9577 and 9632 Å).16–19
Identifying groups of correlated DIBs is a crucial first step in detecting a molecular carrier.20–25 The “correlation” referred-to here is that between the ratios of the equivalent widths (defined below) of pairs of DIBs within a group (also known as “family”) regardless of the observational sightline. In other words, while the individual equivalent widths of a set of “correlated lines” may vary from one sightline to the next, the pairwise ratios of these equivalent widths are constant – within a high statistical confidence – irrespective of the dust cloud being observed. Diverse groups of interrelated lines (e.g., via Pearson correlations), and the sheer number of DIBs, imply there are numerous molecules responsible for these lines.
The purpose of this effort is to identify families of correlated lines, and propose a procedure to identify possible candidate carriers. The following procedure was adopted, and could be pertinent for similar endeavors:
1. Identification of correlated DIBs based on equivalent widths. The equivalent width (EW) of a DIB is the area of the absorption feature in a normalized spectrum. It is related to the “extinction” or strength (loosely speaking, the “intensity”) of the absorption line.
2. Bolstering the existence of these families by determining the correlations between the EWs of DIBs and interstellar reddening. Optical reddening arises from blue light being preferentially scattered when traversing dust clouds.
3. Using energy differences between correlated DIBs (a family) as a probe of the vibrational level spacings of the source ((non-)linear vibronic progression).
4. Using a molecular database to examine functional groups exhibiting infrared vibrations that agree with those implied by the wavelength differences between DIBs within a family.
5. Completing quantum chemistry computations of neutral molecules identified from comparisons to molecular databases, to assess if there are lines which directly overlap with observed DIBs. Evaluating their ion counterparts.
6. Benchmarking of the quantum chemical levels of theory, and assess their accuracy, precision, and systematic offsets.
Two families of DIBs, identified in steps (1) and (2), are of particular interest. These are the separate families tied to the first DIBs discovered by Mary Lea Heger at 5780 and 5797 Å. The identified 5780 Å family consists of 11 lines: 5236.27, 5487.64, 5609.82, 5705.12, 5779.59, 6195.99, 6203.58, 6269.89, 6284.05, 6993.12, and 7224.16 Å. DIBs associated with the 5797 Å family are 8 lines at: 5494.10, 5545.08, 5797.18, 5849.82, 6108.06, 6113.22, 6439.51, and 6449.27 Å.26 (Fig. 1).
 | ||
| Fig. 1 A display of the optical and near infrared (NIR) DIBs, showing all lines with equal emphasis (top); those DIBs belonging to the presumed 5780 Å family (middle) with lines of the family emphasized in red. The rest are in faint gray, and those belonging to the 5797 Å family (bottom) are similarly emphasized in red. (The number of lines associated with the 5780 Å family is 11 (middle), but the image appears to have only 9 and this is because the pairs [(6196, 6204) and (6270, 6284)] are overlapping at the scale of the figure. The same is true for the 5797 Å family (bottom) which should have 8 lines but appears to have only 6 due to the closeness of the pairs (6108, 6113), and (6440, 6449). | ||
Methodology
Families of correlated DIBs were identified using data from the Apache Point Observatory (APO) catalog of DIBs.15 The catalog samples DIBs in the wavelength domain between 4000 and 9000 Å, and provides data for 557 DIBs from 25 different sightlines. Smith et al.25 found the EW–EW Pearson correlation coefficients for 154![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 846 DIB pairs. An analysis of the results led to the identification of a number of families of strongly correlated DIBs. Each of these families of DIBs might arise from a single molecular carrier.25
846 DIB pairs. An analysis of the results led to the identification of a number of families of strongly correlated DIBs. Each of these families of DIBs might arise from a single molecular carrier.25
Common correlations within a family should emerge relative to the EWs and reddening.27–31 For example, Merrill and Wilson29 demonstrate that the EWs of the DIB 6283 Å showed an increasing linear dependence on reddening. Optical reddenings for the stars in the APO catalog were adopted,15 and a comparatively common correlation was found between optical reddening and the EWs of the lines in each proposed family.32 This increases confidence in those two families tied to the Heger lines (5780 and 5797 Å), prompting further analysis.
The energy difference between highly correlated DIBs have been used to probe vibrational transition quanta.21 That approach is adopted here to search for possible molecular sources behind the DIB families. Energy differences between DIBs within each family were compared with (approximate) IR molecular spectra. Accurate spectra are only known for a small percentage of molecules.11,13,33 To circumvent this limitation, approximate molecular spectra will be used in this work. The Rapid Approximate Spectral Calculations for ALL (RASCALL 1.0)3 database contains approximate IR spectra of more than 16000 neutral molecules. To create this database Sousa-Silva et al.3 analysed 120 functional groups associated with 19 different bond types, and found the properties for their spectrally-active rovibrational modes. The spectrum of each molecule was then estimated by considering the contributions of these functional groups to the overall molecular spectrum.
The energy offsets between DIBs within a given family were converted to the corresponding wavelengths (μm), denoted as λoffset. These wavelengths were then compared to those featured in RASCALL (λRASCALL). Molecules that featured lines matching at least three predicted vibrational energies to within a threshold of |λoffset − λRASCALL| = 0.013 μm were saved. The selected molecules suggest certain functional groups that the bona fide family may contain, rather than identifying the broader specific carrier for that family. The veracity of the results was then tested using Monte Carlo simulations.
The Monte Carlo simulations were performed using an in-house Python code whereby 10000 independent random families of DIBs within the range of 4000–9000 Å (where DIBs are primarily observed) were generated, each family containing one of the two Heger lines. This was done separately for each of the real families tied to the lines 5780 Å and 5797 Å. The random families contained the same number of lines as in the real family associated with each of the lines 5780 Å and 5797 Å. That is, the random families created for 5780 Å had 11 lines, and that for 5797 Å had 8 lines. These 10000 random families were analyzed using the same methods as the families associated with 5780 Å and 5797 Å and compared to molecules within the RASCALL 1.0 database using the same threshold |λoffset − λRASCALL| = 0.013 μm. The emergence of the candidate molecules associated with 5780 Å and 5797 Å from these random families were monitored. It is found that the candidate molecules are observed in less than 10% of the results for random families, suggesting that the results for the real families of DIBs are not coincidental.
The candidate neutral molecules were subsequently subjected to quantum chemical calculations to obtain their optimized molecular geometries, along with their UV-vis spectra at different levels of theory.
All calculations were conducted using the Gaussian16 suite of programs.34 The molecular geometries of all species were optimized in their lowest electronic state, followed by a harmonic frequency calculation to ensure that the optimized geometries are minima on the potential energy hypersurfaces (PES). The minima of the family consistent with the 5780 Å molecules have been previously determined (Ref. 59 and 60), while the family consistent with the 5797 Å molecules corresponds to rigid structures. Therefore, an extensive exploration of the PES was not considered necessary to locate the respective global minima.
None of the identified molecules in their neutral ground electronic states exhibit absorptions in the optical, only in the UV. Since ionization by electron loss or capture may shift their electronic spectra sufficiently to the Vis range, both cations and anions were considered. These ionized species were modelled by subtracting or adding one or two electron(s) (and not, for example by adding or removing protons). Thus, all singly-charged ionic species in this work are open-shell doublets, while the doubly-charged and the neutral species are singlet closed-shell systems.
Both vertical and adiabatic ionizations were considered. The vertical ionization is one in which the ion is frozen at the geometry optimized for the neutral species, and the adiabatic is one where the geometry of the ion is optimized before the final electronic structure is generated. The calculated adiabatic energy shifts include the zero-point vibrational energies (ZPEs). The ionization potential (IP), whether vertical or adiabatic, is defined as:
| IP = Ecation − Eneutral, | (1) |
| EA = Eanion − Eneutral. | (2) |
Since total energies are negative, when EA > 0, this implies that the energy of the neutral species is lower (i.e., more stable) than the anion.
All molecules were optimized using density functional theory (DFT)'s Perdew–Burke–Ernzerhof (PBE)35,36 GGA-XC functional. Additionally, optical spectra were computed using time-dependent density functional theory37–40 (TD-DFT),41,42 equation-of-motion coupled cluster with single and double excitations (EOM-CCSD)43–46 and state averaged complete-active-space-self-consistent-field (SA-CASSCF)47,48 with a (6,6) active space for neutral glycolamide (M0). Only for CASSCF calculations was the MOLPRO v2012.149,50 used. The hybrid PBE0 (Perdew–Burke–Ernzerhof exchange & 25% Hartree–Fock exchange plus full PBE energy correlation),51,52 was used for the TD-DFT calculations. All calculations were performed using Dunning's aug-cc-pVTZ correlation-consistent polarized valence triple-zeta basis set, augmented with diffuse function.53,54
Findings
A. Molecular candidates from a comparison of DIB offset energies and RASCALL vibrational energies
Molecules of interest were identified by examining the energy differences between highly correlated DIBs, which probe the vibrational level spacings of the carrier.21 For the 5780 Å family these are: glycolamide (IUPAC: 2-hydroxyacetamide), lactamide (IUPAC: 2-hydroxypropanamide), 3-hydroxypropanamide, and oxamic acid. For the family tied to 5797 Å these are: 2-cyclopenten-1-one, 2(5H)-furanone, 3(2H)-thiophenone, and 3(2H)-selenophenone. Fig. 2 shows the chemical structures of these compounds. | ||
| Fig. 2 The chemical structures of the molecules of interest for the 5780 Å (left column) and 5797 Å families (right column). | ||
The molecules for the 5780 Å family revealed some commonalities amongst the functional groups they may possibly contain. They all appear to possibly contain a primary amide and a hydroxyl group. The identification of certain types of chemical bonds and functional groups in interstellar medium is another piece in the jigsaw that can shed light on the nature of the carriers (see discussions in ref. 55). For the 5797 Å family, the identified molecules are similar in structure to 2-cyclopenten-1-one, each differing by one atom within the five-membered ring.
B. Calculated spectra of neutral ground electronic states
The spectra calculated with time-dependent DFT of the eight neutral molecules are displayed in Fig. 3a, 4 and Fig. S1 (ESI†) at the PBE0/aug-cc-pVTZ levels of theory. None of the neutral species showed absorption bands in the visible spectrum as expected from an inspection of their chemical structures.56,57 Several plausible explanations may account for the discrepancies between our calculated electronic spectra and the observed DIBs, for example, it is conceivable that these small organic molecules act as building blocks for more complex interstellar species with absorptions in the Vis. | ||
| Fig. 3 UV-vis spectra of glycolamide calculated at the TD-PBE0/aug-cc-pVTZ level of theory. (Top: Neutral molecule) The observed lines are solely in the far UV part of the spectrum. (Bottom: +1 Cation): Most lines fall in the far UV part of the spectrum, with one intense optical band. | ||
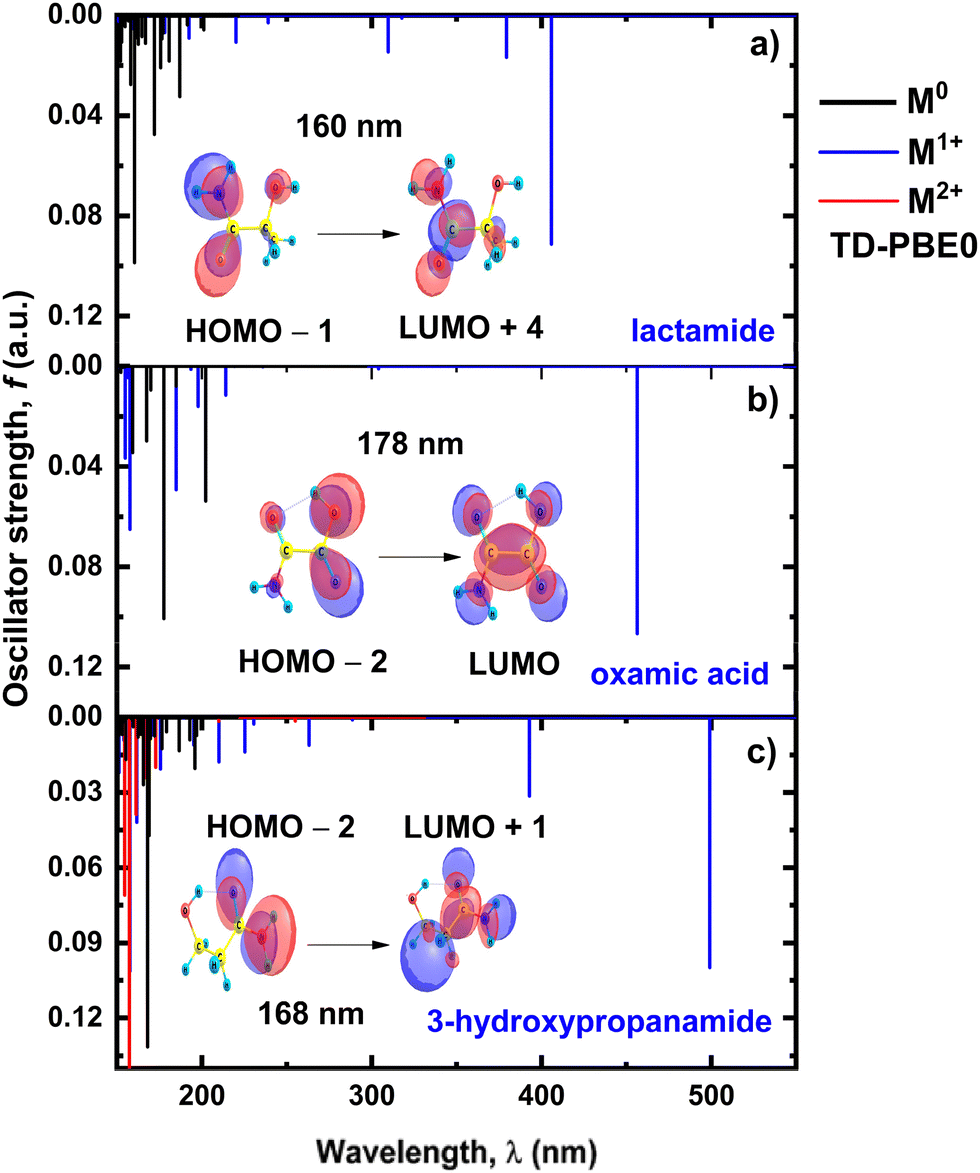 | ||
| Fig. 4 UV-vis spectra of candidate molecules associated with the 5780 Å DIB family: (a) lactamide, (b) oxamic acid, and (c) 3-hydroxypropanamide, in their neutral (black line) and their single (blue lines) and double (red lines) cationic forms, calculated at the TD-PBE0/aug-cc-pVTZ level of theory. | ||
The strongest absorption band of most neutral molecules is found between 187–217 nm (Fig. 3a, 4 and Fig. S1, ESI†). That hints that the prominent 220 nm interstellar absorption feature may arise in part from the superposition of the UV spectra of these molecules.58
Different minimum energy conformers are possible for the neutral molecules consistent with the 5780 Å family. Glycolamide, 3-hidroxypropanamide, and oxamic acid show the lowest energy as –syn conformations with an intramolecular OH⋯O hydrogen bond59 (Fig. 2). Conversely, lactamide’s minimum energy corresponds to a trans conformer, forming an NH⋯O intramolecular hydrogen bond.60 The neutral molecules consistent with the 5780 Å family show the most intense bands in the far and near UV spectrum. For glycolamide, several bands near 200 nm reflect n–π* transitions, typical of the amide moiety.61Fig. 3a depicts the n–π* HOMO–LUMO+5 transition of neutral glycolamide, and Fig. 4 shows other n–π* transitions for the rest of the 5780 Å family.
Table 1 shows the results obtained for the transitions to the first two excited states (S1: n–π* and S2: π–π*) with the three methods used in this work, namely, TD-PBE0, EOM-CCSD, and CASSCF, for neutral gycolamide.
| Neutral glycolamide (M0) | ||||
|---|---|---|---|---|
| State | CASSCF E (eV), f | EOM-CCSD E (eV), f | TD-PBE0 E (eV), f | Exp.62E (eV) |
| S1 | 6.51, 0.0005 (ΔE = 0.86) | 6.02, 0.0001 (ΔE = 0.37) | 5.93, 0.0002 (ΔE = 0.28) | 5.65 |
| S2 | 7.41, 0.0112 (ΔE = 0.09) | 6.90, 0.0116 (ΔE = 0.42) | 6.42, 0.0006 (ΔE = 0.90) | 7.32 |
| ΔES2–S1 | 0.90 | 0.88 | 0.49 | 1.67 |
As we could not find experimental gas-phase absorption spectra of glycolamide, we compared the first two excited states of neutral glycolamide with the experimental UV-vis data for the simplest amide, i.e., formamide, which should be similar given the similarity of the electronic environments in the two amides and that the transition being compared occur between corresponding energy levels.
Hirst et al.62 reported the experimental spectra of NH2CHO and identified the lowest energy n–π* transition at 5.65 eV and the most intense π–π* transition at 7.32 eV. The EOM-CCSD for glycolamide shows a better agreement with the corresponding experimental transitions in formamide than CASSCF (6,6) and TD-PBE0. The three methods agree with the experiment in that the S2 π–π* transition is the most intense. For glycolamide, agreement with experiment is in the following decreasing order: EOM-CCSD > SA-CASSCF (6,6) > TD-PBE0.
For the neutral molecules matching the 5797 Å family, the intense bands in the UV correspond to a transition from the HOMO or HOMO−1 orbitals to the LUMO, which corresponds to a n–π* transition (Fig. S1, ESI†). M. Christianson et al. obtained the experimental UV-vis spectra of 2-cyclopenten-1-one with the n–π* band at 203 nm, which is comparable with the TD-PBE0/aug-cc-pVTZ predicted value (208 nm, Fig. S1a, ESI†). In addition to n–π* type bands, this family also presents π–π* transitions.
C. Calculated spectra of molecular ions
Molecules in the interstellar medium can frequently exist in ionized form owing to bombardment from ionizing radiation. Stray electrons detached from one species can be captured by another, and even form metastable states.63–66 Since none of the identified molecules exhibit absorption in the visible part of the spectrum, their ionized forms were explored.
Fig. 3b displays the TD-DFT spectra of glycolamide in its +1 cation state. The calculated spectra of the remaining cation species (M = +1, +2) of the 5780 and 5797 Å families can be found in Fig. 4 and Fig. S1 (ESI†). Often, ionized molecules can be less stable than their neutral parents.67 For the 5780 Å family, all closed-shell singlet dicationic species dissociate into the alcohol radical (˙CH2OH for glycolamide), and the carbamoyl radical (˙CONH2). These two radicals have structural similarity to formaldehyde and formamide, respectively. Formaldehyde is present in the interstellar medium as Goldanskii demonstrated, especially relative to the tunneling of the formaldehyde monomer to produce polyoxymethylene (POM).68–71 Formamide is also believed to be present in the interstellar medium (ISM),72–77 and so are aminomethanol (NH2CH2OH) and the iminium ion (CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH2+), in addition to two open-shell (radical) molecules, the carbamoyl radical as a fragmentation product of di-ionization of glycolamide, in tandem with its isomer HCONH˙. Moreover, glycolamide is an isomer of glycine, which has also been found in the ISM. Glycine dications have been studied in gas-phase, and its fragmentation into two singly-charged cations is possible.67 Therefore, the possible fragmentation of the glycolamide dication in the ISM may likewise be feasible, or glycolamide-glycine isomerization could lead to new ISM species.
NH2+), in addition to two open-shell (radical) molecules, the carbamoyl radical as a fragmentation product of di-ionization of glycolamide, in tandem with its isomer HCONH˙. Moreover, glycolamide is an isomer of glycine, which has also been found in the ISM. Glycine dications have been studied in gas-phase, and its fragmentation into two singly-charged cations is possible.67 Therefore, the possible fragmentation of the glycolamide dication in the ISM may likewise be feasible, or glycolamide-glycine isomerization could lead to new ISM species.
For the 5780 Å family, Fig. 4 shows that although for the M+1 species the transitions shift toward the visible region, there is no match with the DIBs observed for this family. For the 5797 Å family, two ionized states could be obtained. Fig. S1 (ESI†) shows the TD-DFT spectra obtained with a PBE0/aug-cc-pvtz level of theory. The radical (doublet) singly-ionized and the (singlet) doubly-ionized cations of the four molecules exhibit bands with oscillator strength f > 0.0002 below 500 nm. The most intense bands of the cation are located in the UV (Fig. S1, ESI†).
The M−1 and M−2 anions of the studied species are such that the added electron is either marginally bound or marginally unbound (Table 2 lists the IPs and EAs of the molecules of interest in their neutral closed-shell states). Hence, the absorption of an energetic photon necessary to induce an optical transition is most likely to eject that excess electron conferring the rest of the energy to the departing fragments' kinetic energy. The anions are, therefore, not pursued further in this paper.
Fig. 4 and Fig. S1 (ESI†) show that the single and double cationic forms of these two families do not exhibit bands above 500 nm, which again invalidates the cationic species as DIBs carriers.
| Moleculea | IP (eV) | EA (eV) |
|---|---|---|
| Adiab./ZPE/Vert. | Adiab./ZPE/Vert. | |
| a Adiabatic (Adiab.): energy difference between the ground state of the neutral system and the ground state of the ionized system, including structural relaxation. Vertical (Vert.): energy difference at the frozen geometry of the neutral system, excluding relaxation of the ion. ZPE: short for “adiabatic energy differences including zero-point vibrational corrections for both neutral and ionized states”. | ||
| Glycolamide | 9.01/9.01/9.68 | 0.06/0.03/0.07 |
| Lactamide | 8.68/8.66/9.20 | 0.11/0.06/0.15 |
| 3-Hydroxypropanamide | 8.73/8.73/9.08 | 0.03/0.01/0.06 |
| Oxamic acid | 9.62/9.58/9.88 | −0.45/−0.54/−0.16 |
| 2-Cyclopenten-1-one | 8.95/8.89/9.02 | −0.06/−0.13/0.07 |
| 2(5H)-Furanone | 9.90/9.84/10.07 | −0.06/−0.15/0.02 |
| 3(2H)-Thiophenone | 8.69/8.67/8.79 | −0.22/−0.32/0.01 |
| 3(2H)-Selenophenone | 8.49/8.47/8.56 | −0.31/−0.41/−0.10 |
It is pertinent here that we quote Herbst,78 who, in turn, quotes Bill Klemperer—who proposed Sn- (n = 2, 3) in oxide glasses as DIBs carriers—that “there is no better way to lose a scientific reputation than to speculate on the carrier of the diffuse bands”. While no definitive carriers emerged, the work, nevertheless, suggests a roadmap combining quantum chemistry and observational astronomical spectroscopy in the quest for this century-old open problem. This study also underscores the importance of compiling electronic and vibrational spectra of, not only neutral molecules, but equally importantly of their ionized states. The difficulty of obtaining extensive tabulations of such spectra especially for ions bring to the fore the role of quantum chemistry in this problem especially with the “astronomical” advances in computer power, algorithms, and machine learning. An analysis of rovibronic spectra of charged species could perhaps yield more definite confirmations of DIB correlations, especially in the light of additional information gathered from the rotational substructure of each absorption peak.
D. Ionization potentials and electron affinities
Table 2 lists the first ionization potentials and first electron affinities calculated at DFT with the PBE exchange–correlation functional. The experimental values of IP for: 2-cyclopenten-1-one (IP = 8.47 ± 0.05 eV),79 2(5H)-furanone (IP = 10.65 eV),80 and oxamic acid (IP = 10.51 eV)81 are all in closer agreement with the PBE result (ΔIP < 1 eV).Discussion and conclusions
The effort to identify carriers for DIBs has remained a challenge for a number of possible reasons. For example, degeneracies exist where more than one molecule can produce lines matching a small DIB family, particularly given the enormity of chemical space (>1060 possible chemical compounds). On the one hand, quantum chemistry calculations suffer from random and systematic errors (which have not been quantified yet at the time of writing, in the sense that the results of quantum chemistry do not have error bars). On the other hand, small perturbations, such as intense radiation or external fields, may alter an observational spectrum. This, in part, explains the barriers faced, and why a sole molecule has a consensus coalescing regarding its status as a carrier for only two of more than 550 DIBs. This effort aims, in its essence, to minimize the chemical and the parameter spaces to eventually close-in on potential carriers.We report two new families of highly correlated lines tied to the two Heger lines (5780 and 5797 Å). The correlated families are then analyzed, as described above, to posit a number of small organic molecules that may be related to these DIBs. Thus, electronic spectra were calculated for: glycolamide, 2-cyclopenten-1-one, 3(2H)-thiophenone, 2(5H)-furanone, 3(2H)-selenophenone, lactamide, 3-hydroxypropanamide, and oxamic acid. These particular molecules were selected on the basis of a comparison of the spacings between the DIBs within each family to a molecular database (RASCALL). The calculated spectra for the neutral and the cationic states of these molecules show that they are not DIB carriers. However, the same calculations suggest that these species absorb in the UV range within the broad 220 nm shoulder to which they may conceivably be among its carriers. Notably, glycolamide has been recently detected in the interstellar medium using high sensitivity Yebes 40 m and IRAM 30 m telescopes, which observed microwave radiation from the molecular cloud designated G+0.693–0027.82
Rivilla et al. report a column density for glycolamide of 7.4 ± 0.7 × 1012 cm−2, more than 11 orders of magnitudes less concentrated than molecular hydrogen (H2).82 Despite its comparative rarity with respect to the most abundant element in the universe (hydrogen), the presence of glycolamide within interstellar clouds is in tandem with the work conveyed here and suggests the that this molecule could be responsible—perhaps as a cation—for a subsample of DIB absorptions. Moreover, the detection of carbamoyl radicals in ISM,72 which are products of the Coulomb explosion of glycolamide after losing two electrons, bolsters the case for the glycolamide's presence in ISM and also for the propensity of this molecule to lose electrons in interstellar conditions.
Recently, an artificial intelligence reverse-engineering approach has been used in the context of drug-design, regressing from known properties to an actual compound with that set of properties.83 The amount of data required to obtain a high level of certainty for a one-to-one match of properties to a specific molecule requires a massive amount of properties data. A key objective moving forward is to reduce the expansive parameter space, thereby mitigating the number of candidate molecules. One pathway requires that a carrier emerge from simultaneously matching its ions in the electronic and vibrational (mid-IR) domains. Even stricter constraints could likewise be explored, whereby the selection is restricted to those molecules whose rotational signatures were observed in the microwave/radio (e.g., glycolamide).
Data availability
All data can be found in the body of the article and/or the published ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the reviewers for pertinent suggestions and insights. The authors are grateful to Clara Sousa-Silva (Bard College, New York) and Islam K. Matar (Saint Mary's University) for helpful comments, and to the Natural Sciences and Engineering Council of Canada (NSERC), the Canadian Foundation for Innovation (CFI), Mount Saint Vincent University, the Autonomous University of Morelos States, for funding. The Global Affairs Canada's International Scholarships Program is thanked for an Emerging Leaders in the Americas Program (ELAP) scholarship awarded to one of us (H. S.). A. D. S. is grateful to have received and NSERC-Undergraduate Student Research Assistantship to support her research. C. F. M. is grateful to the Institut des Hautes Études Scientifiques (IHES), Université Paris-Saclay, for a sabbatical stay in the fall of 2024 during which much of the work has been completed. The Digital Research Alliance of Canada is acknowledged for computational resources used in this work.References
- R. C. Fortenberry, Interstellar Anions: The Role of Quantum Chemistry, J. Phys. Chem. A, 2015, 119, 9941–9953 CrossRef CAS PubMed.
- R. C. Fortenberry, A Vision for the Future of Astrochemistry in the Interstellar Medium by 2050, ACS Phys. Chem. Au, 2023, 4, 31–39 CrossRef PubMed.
- C. Sousa-Silva, J. J. Petkowski and S. Seager, Molecular simulations for the spectroscopic detection of atmospheric gases, Phys. Chem. Chem. Phys., 2019, 21, 18970–18987 RSC.
- I. Weber, P. R. Joshi, D. T. Anderson and Y.-P. Lee, Unique Applications of para-Hydrogen Matrix Isolation to Spectroscopy and Astrochemistry, J. Phys. Chem. Lett., 2024, 15, 11361–11373 CrossRef CAS PubMed.
- I. Weber, M. Tsuge, P. Sundararajan, M. Baba, H. Sakurai and Y.-P. Lee, Infrared and Laser-Induced Fluorescence Spectra of Sumanene Isolated in Solid para-Hydrogen, J. Phys. Chem. A, 2022, 126, 5283–5293 CrossRef CAS PubMed.
- M. L. Heger, Further Study of the Sodium Lines in Class B Stars. The Spectra of Certain Class B Stars in the Regions 5630–6680 Å and 3280–3380 Å. Note on the Spectrum of [Gamma] Cassiopeiae between 5860 Å and 6600 Å, Lick Obs. Bull., 1922, 10, 146–147 Search PubMed.
- M. L. Heger, The Occurrence of Stationary D Lines of Sodium in the Spectroscopic Binaries, β Scorpii and d Orionis, Lick Obs. Bull., 1919, 10, 59–63 Search PubMed.
- G. H. Herbig, Diffuse Interstellar Bands. 4 Region 4400–6850 Å, Astophys. J., 1975, 196, 129–160 CrossRef CAS.
- G. H. Herbig, The Diffuse Interstellar Bands, Annu. Rev. Astron. Astrophys., 1995, 33, 19–73 CrossRef CAS.
- J. Fulara and J. Krelowski, Origin of Diffuse Interstellar Bands: Spectroscopic Studies of their Possible Carriers, New Astron. Rev., 2000, 44, 581–597 CrossRef CAS.
- S. Kwok, Physics and Chemistry of the Interstellar Medium, University Science Books, Sausalito, California, 2007 Search PubMed.
- T. P. Snow and J. D. Destree. The Diffuse Interstellar Bands in History and in the UV, in PAHs and the Universe: A Symposium to Celebrate the 25th Anniversary of the PAH Hypothesis, ed. C. Joblin and A. G. G. M. Tielens, EAS (European Astronomical Scociety) Publications Series, 2011, vol. 46, pp. 341–347 Search PubMed.
- J. Tennyson, Astronomical Spectroscopy: An Introduction to the Atomic and Molecular Physics of Astronomical Spectra, World Scientific Publishing, Co. Pte. Ltd, Singapore, 2nd edn, 2011 Search PubMed.
- B. J. McCall and R. E. Griffin, On the Discovery of the Diffuse Interstellar Bands, Proc. R. Soc. A, 2013, 469, 20120604 CrossRef.
- H. Fan, L. M. Hobbs, J. A. Dahlstrom, D. E. Welty, D. G. York, B. Rachford, T. P. Snow, P. Sonnentrucker, N. Baskes and G. Zhao, The Apache Point Observatory Catalog of Optical Diffuse Interstellar Bands, Astrophys. J., 2019, 878, 151 CrossRef CAS.
- G. A. Galazutdinov and J. Krelowski, Looking for the Weak Members of the C60+ Family in the Interstellar Medium, Acta Astron., 2017, 67, 159–169 Search PubMed.
- G. A. Galazutdinov, G. Valyavin, N. R. Ikhsanov and J. Krelowski, Diffuse Bands 9577 and 9633: Relations to Other Interstellar Features, Astron. J., 2021, 161, 127 CrossRef CAS.
- L. Schlarmann, B. Foing, J. Cami and H. Fan, C60+ Diffuse Interstellar Band Correlations and Environmental Variations, Astron. Astrophys., 2021, 656, L17 CrossRef CAS.
- D. Majaess, T. A. Harriott, H. Seuret, C. Morera-Boado, L. Massa and C. F. Matta, Strengthening the Link between Fullerenes and a Subset of Diffuse Interstellar Bands, Mon. Not. R. Astron. Soc., 2025, 538, 2392–2395 CrossRef.
- B. J. McCall, M. M. Drosback, J. A. Thorburn, D. G. York, S. D. Friedman, L. M. Hobbs, B. L. Rachford, T. P. Snow, P. Sonnentrucker and D. E. Welty, Studies of the Diffuse Interstellar Bands. IV. The Nearly Perfect Correlation between λλ 6196.0 and 6613.6, Astrophys. J., 2010, 708, 1628–1638 CrossRef CAS.
- A. Bondar, The Well Correlated Diffuse Interstellar Bands at λλ 6196, 6614 Å and their Possible Companions, Mon. Not. R. Astron. Soc., 2020, 496, 2231–2240 CrossRef.
- J. Krelowski and G. A. H. Walker, Three Families of Diffuse Interstellar Bands?, Astrophys. J., 1987, 312, 860–867 CrossRef CAS.
- J. Cami, P. Sonnentrucker, P. Ehrenfreund and B. H. Foing, Diffuse Interstellar Bands in Single Clouds: New Families and Constraints on the Carriers, Astron. Astrophys., 1997, 326, 822–830 Search PubMed.
- F. Xiang, Z. Liu and X. Yang, A Study on the Correlations between Diffuse Interstellar Bands, Publ. Astron. Soc. Jpn., 2012, 64, 31 CrossRef.
- F. Smith, D. Majaess, T. A. Harriott, L. Massa and C. F. Matta, Establishing New Diffuse Interstellar Band Correlations to Identify Common Carriers, Mon. Not. R. Astron. Soc., 2021, 507, 5236–5245 CrossRef.
- A. D. Sullivan, E. R. Smith, F. M. Smith, T. M. Fields, T. A. Harriott, D. Majaess, L. Massa and C. F. Matta, Separate Molecular Carriers for the Mary Lea Heger DIBs at 5780 and 5797 Å, Bull. Am. Astron. Soc., 2023, 55, No. 2, e-id: 2023n2i213p05, 213-05 Search PubMed.
- J. Hartmann, Investigations on the Spectrum and Orbit of Delta Orionis, Astrophys. J., 1904, 19, 268–286 CrossRef.
- P. Merrill, Unidentified Interstellar Lines, Publ. Astron. Soc. Pac., 1934, 46, 206–207 CrossRef.
- P. W. Merrill and O. C. Wilson, Unidentified Interstellar Lines in the Yellow and Red, Astrophys. J., 1938, 87, 9–23 CrossRef CAS.
- H. Fan, D. E. Welty, D. G. York, P. Sonnentrucker, J. A. Dahlstrom, N. Baskes, S. D. Friedman, L. M. Hobbs, Z. Jiang, B. Rachford and T. P. Snow, The Behavior of Selected Diffuse Interstellar Bands with Molecular Fraction in Diffuse Atomic and Molecular Clouds, Astrophys. J., 2017, 850, 194 CrossRef.
- G. Galazutdinov, A. Bondar, B.-C. Lee, R. Hakalla, W. Szajna and J. Krelowski, Survey of Very Broad Diffuse Interstellar Bands, Astrophys. J., 2020, 159, 113 CAS.
- E. R. Smith, F. M. Smith, T. A. Harriott, D. Majaess, L. Massa and C. F. Matta, Novel Correlations between Diffuse Interstellar Bands and Optical Reddening, Res. Not. Am. Astron. Soc., 2022, 6(4), 82 Search PubMed.
- J. E. Dyson and D. A. Williams, The Physics of the Interstellar Medium, CRC Press, Taylor & Francis Group, Boca Raton, 3rd edn, 2021 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian Inc, Wallingford CT, 2019 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Errata: Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
- P. Hohenberg and W. Kohn, Inhomogeneous Electron Gas, Phys. Rev. B: Condens. Matter Mater. Phys., 1964, 136, 864–871 CrossRef.
- R. G. Parr and W. Yang, Density-Functional Theory of Atoms and Molecules, Oxford University Press, Oxford, 1989 Search PubMed.
- D. S. Scholl and J. A. Steckel, Density Functional Theory: A Practical Introduction, Wiley, Hoboken, New Jersey, 2009 Search PubMed.
- W. Koch and M. C. Holthausen, A Chemist's Guide to Density Functional Theory, Wiley-VCH, New York, 2nd edn, 2001 Search PubMed.
- E. Runge and E. K. U. Gross, Density-Functional Theory for Time-Dependent Systems, Phys. Rev. Lett., 1984, 52, 997–1000 CrossRef CAS.
- M. Petersilka, U. J. Gossmann and E. K. U. Gross, Excitation Energies from Time-Dependent Density-Functional Theory, Phys. Rev. Lett., 1996, 76, 1212–1215 CrossRef CAS PubMed.
- J. F. Stanton and R. J. Bartlett, Equation of Motion Coupled-Cluster Method: A Systematic Biorthogonal Approach to Molecular Excitation Energies, Transition Probabilities, and Excited State Properties, J. Chem. Phys., 1993, 98, 7029–7039 CrossRef CAS.
- D. C. Comeau and R. J. Bartlett, The Equation-of-Motion Coupled-Cluster Method. Applications to Open- and Closed-Shell Reference States, Chem. Phys. Lett., 1993, 207, 414–423 CrossRef CAS.
- A. Szabo and N. S. Ostlund, Modern Quantum Chemistry: Introduction to Advanced Electronic Structure Theory, Dover Publications, Inc., New York, 1989 Search PubMed.
- I. N. Levine, Quantum Chemistry, Pearson Education, Inc, Boston, 7th edn, 2014 Search PubMed.
- P. J. Knowles and H.-J. Werner, An Efficient Second-Order MC SCF Method for Long Configuration Expansions, Chem. Phys. Lett., 1985, 115, 259–267 CrossRef CAS.
- H.-J. Werner and P. J. Knowles, A Second Order Multiconfiguration SCF Procedure with Optimum Convergence, J. Chem. Phys., 1985, 82, 5053–5063 CrossRef CAS.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby and M. Schütz, MolPro: A General-Purpose Quantum Chemistry Program Package, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 242–253 CAS.
- H.-J. Werner, et al., MOLPRO, 2012.1. A Package of Ab Initio Programs, 2012, https://www.molpro.net Search PubMed.
- J. P. Perdew, M. Ernzerhof and K. Burke, Rationale for Mixing Exact Exchange with Density Functional Approximations, J. Chem. Phys., 1996, 105, 9982–9985 CrossRef CAS.
- C. Adamo and V. Barone, Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0 Model, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- R. A. Kendall, T. H. Dunning Jr. and R. J. Harrison, Electron Affinities of the First-Row Atoms Revisited. Systematic Basis Sets and Wave Functions, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS.
- D. E. Woon and T. H. Dunning Jr., Gaussian-Basis Sets for Use in Correlated Molecular Calculations. 3. The Atoms Aluminum through Argon, J. Chem. Phys., 1993, 98, 1358–1371 CrossRef CAS.
- D. Majaess, H. Seuret, T. A. Harriott, C. Morera-Boado, A. D. Sullivan, L. Massa and C. F. Matta, Potential Vibrational Modes Tied to Diffuse Interstellar Bands, Mon. Not. R. Astron. Soc., 2025, 539, in press Search PubMed.
- Y. Y. Paperno, V. P. Pozdnyakov, A. A. Smirnova and L. M. Elagin, Physico-chemical Laboratory Techniques in Organic and Biological Chemistry (English Translation by O. Glebov), Mir Publishers, Moscow, 1979 Search PubMed.
- N. J. Turro, Modern Molecular Photochemistry, University Science Books, Sausalito, CA, 1991 Search PubMed.
- F. Y. Xiang, A. Li and J. X. Zhong, Diffuse Interstellar Bands and the Ultraviolet Extinction Curves: The Missing Link Revisited, Astrophys. J., 2017, 835, 107 CrossRef.
- A. Maris, On the Conformational Equilibrium of Glycolamide: A Free Jet Millimetre-Wave Spectroscopy and Computational Study, Phys. Chem. Chem. Phys., 2004, 6, 2611–2616 RSC.
- A. Pikulska, K. H. Hopmann, J. Bloino and M. Pecul, Circular Dichroism and Optical Rotation of Lactamide and 2-Aminopropanol in Aqueous Solution, J. Phys. Chem. B, 2013, 117, 5136–5147 CrossRef CAS PubMed.
- N. De Silva, S. Y. Willow and M. S. Gordon, Solvent Induced Shifts in the UV Spectrum of Amides, J. Phys. Chem. A, 2013, 117, 11847–11855 CrossRef CAS PubMed.
- J. D. Hirst, D. M. Hirst and C. L. Brooks, Ab Initio Calculations of the Excited States of Formamide, J. Phys. Chem., 1996, 100, 13487–13491 CrossRef CAS.
- J. Simons, Roles Played by Metastable States in Chemistry, in Resonances in Electron-Molecule Scattering, van der Waals Complexes, and Reactive Chemical Dynamics, ed. D. G. Truhlar, American Chemical Society, Washington DC, ch. 1 ACS Symposium Series, No. 263, 1984. pp. 3–16 Search PubMed.
- J. D. Steill, Molecular Anion Spectroscopy and Stability, PhD Thesis, University of Tennessee, Knoxville, TN, 2007 Search PubMed.
- K. D. Jordan, V. K. Voora and J. Simons, Negative Electron Affinities from Conventional Electronic Structure Methods, Theor. Chem. Acc., 2014, 133, 1445 Search PubMed.
- T. J. Millar, C. Walsh and T. A. Field, Negative Ions in Space, Chem. Rev., 2017, 117, 1765–1795 CrossRef CAS PubMed.
- S. Maclot, D. G. Piekarski, A. Domaracka, A. Mery, V. Vizcaino, L. Adoui, F. Martķn, M. Alcami, B. A. Huber, P. Rousseau and S. Diaz-Tendero, Dynamics of Glycine Dications in the Gas Phase: Ultrafast Intramolecular Hydrogen Migration Versus Coulomb Repulsion, J. Phys. Chem. Lett., 2013, 4, 3903–3909 CrossRef CAS.
- V. I. Goldanskii, Quantum Chemical Reactions in the Deep Cold, Sci. Am., 1986, 254(2), 46–53 CrossRef CAS.
- V. I. Goldanskii, Molecular Tunnelling and Quantum Chemical Kinetics, in Problems in Chemical Kinetics, ed. N. M. Emanuel, English Translation, Mir Publishers, Moscow, 1981, ch. 17, vol. 268, pp. 204–215.
- V. I. Goldanskii, Facts and Hypotheses of Molecular Chemical Tunnelling, Nature, 1979, 279, 109–115 CrossRef CAS.
- V. I. Goldanskii, Mechanism for Formaldehyde Polymer Formation in Interstellar Space, Nature, 1977, 268, 612–613 CrossRef CAS.
- S. Thripati and R. O. Ramabhadran, Pathways for the Formation of Formamide, a Prebiotic Biomonomer: Metal-Ions in Interstellar Gas-Phase Chemistry, J. Phys. Chem. A, 2021, 125, 3457–3472 CrossRef CAS PubMed.
- A. Lopez-Sepulcre, A. A. Jaber, E. L. B. Mendoza, C. Ceccarelli, C. Vastel, R. Bachiller, J. Cernicharo, C. Codella, C. Kahane and M. Kama, Shedding Light on the Formation of the Pre-Biotic Molecule Formamide with ASAI, Mon. Not. R. Astron. Soc., 2015, 449, 2438–2458 CrossRef CAS.
- G. R. Adande, N. J. Woolf and L. M. Ziurys, Observations of Interstellar Formamide: Availability of a Prebiotic Precursor in the Galactic Habitable Zone, Astrobiol., 2013, 13, 439–453 CrossRef CAS PubMed.
- C. Kahane, C. Ceccarelli, A. Faure and E. Caux, Detection of Formamide, the Simplest but Crucial Amide, in a Solar-Type Protostar, Astrophys. J., Lett., 2013, 763, L38–L42 CrossRef.
- C. A. Gottlieb, P. Palmer, L. J. Rickard and B. Zuckerman, Studies of Interstellar Formamide, Astrophys. J., 1973, 182, 699–710 CrossRef CAS.
- R. H. Rubin, G. W. J. J. Swenson, R. C. Benson, H. L. Tigelaar and W. H. Flygare, Microwave detection of interstellar formamide, Astrophys. J., 1971, 169, L39–L44 CrossRef CAS.
- E. Herbst, Chemistry in the Interstellar Medium, Annu. Rev. Phys. Chem., 1995, 46, 27–53 CrossRef CAS.
- P. Linda, G. Marino and S. Pignataro, A Comparison of Sensitivities to Substituent Effects of Five-Membered Heteroaromatic Rings in Gas Phase Ionization, J. Chem. Soc. B, 1971, 1585–1587 RSC.
- D. Wang, D. Wang, S. Li and Y. Li, He I Photoelectron Spectroscopic Studies of the Electronic Structure of 2(3H) Furanone and 2(5H) Furanone, J. Electron Spectrosc. Relat. Phenom., 1994, 70, 167–172 CrossRef CAS.
- J. P. Huke and I. H. Hillier, The Electronic Structure of Chromones, Studied by Low-Energy Photoelectron Spectroscopy and Ab Initio Molecular Orbital Calculations, J. Chem. Soc., Perkin Trans. 2, 1985, 1191–1194 RSC.
- V. M. Rivilla, M. Sanz-Novo, I. Jiménez-Serra, J. Martín-Pintado, L. Colzi, S. Zeng, A. Megķas, Á. López-Gallifa, A. Martínez-Henares, S. Massalkhi, B. Tercero, P. de Vicente, S. Martín, D. San Andrés, M. A. Requena-Torres and J. L. Alonso, First Glycine Isomer Detected in the Interstellar Medium: Glycolamide (NH2C(O)CH2OH), Astrophys. J., Lett., 2023, 953(2), L20 CrossRef CAS.
- T. Le, R. Winter, F. Noe and D.-A. Clevert, Neuraldecipher – Reverse-Engineering Extended-Connectivity Fingerprints (ECFPs) to their Molecular Structures, Chem. Sci., 2020, 11, 10378–10389 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp04023f |
| This journal is © the Owner Societies 2025 |