
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Simple carbenes as hydrogen bond acceptors: ab initio determination of nucleophilicities and reduced nucleophilicities†
Ibon
Alkorta
 *a and
Anthony
Legon
*b
*a and
Anthony
Legon
*b
aInstituto de Química Médica (IQM-CSIC), Juan de la Cierva, 3, E-28006 Madrid, Spain. E-mail: ibon@iqm.csic.es; Tel: +34 915622900
bSchool of Chemistry, University of Bristol, Cantock's Close, Bristol BS8 1TS, UK. E-mail: a.c.legon@bristol.ac.uk; Tel: +44 (0)117 331 7708
First published on 20th November 2024
Abstract
Nucleophilicities for a range of simple carbene molecules acting as hydrogen bond acceptors B in forming complexes B⋯HX are reported. The carbenes chosen to fulfil the roles of a Lewis base are B = R2M, cyclo-(CH)2M, H2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CM and two N-heterocyclic carbenes, where M is one of the group 14 tetrel atoms, C, Si, Ge or Sn and R = H, CH3, and F. All the carbenes but CH2 have a singlet electronic ground state. The Lewis acids, HX, involved are HF, HCl, HBr, HI and HCN, HCCH, and HCP. Nucleophilicities, NB, of the carbenes were determined graphically from equilibrium dissociation energies, De, for the process B⋯HX = B + HX by using the equation De = c·NB·EHX, where c = 1.0 kJ mol−1 and the EHX are known numerical electrophilicities of the Lewis acids HX. De values were calculated ab initio at the CCSD(T)-F12c/cc-pVDZ-F12 level of theory, which for CH2 refers to the singlet electronic excited state. It was established that NR2M values lie in the order of M = C ≫ Si ∼ Ge ∼ Sn for a given R and in the order R = CH3 > H > F for a given M. Reduced nucleophilicities, NB/σaxial, were determined by using the molecular electronic surface potential σaxial at atom M (which lies on the C2 axis) on the 0.001 e Bohr−3 iso-surface of each carbene molecule, as calculated at the MP2/aug-cc-pVTZ level. For R2M having R = CH3 and H and cyclo-(CH)2-M carbenes, the determined values of NB/σaxial are shown to be independent of R and M.
CM and two N-heterocyclic carbenes, where M is one of the group 14 tetrel atoms, C, Si, Ge or Sn and R = H, CH3, and F. All the carbenes but CH2 have a singlet electronic ground state. The Lewis acids, HX, involved are HF, HCl, HBr, HI and HCN, HCCH, and HCP. Nucleophilicities, NB, of the carbenes were determined graphically from equilibrium dissociation energies, De, for the process B⋯HX = B + HX by using the equation De = c·NB·EHX, where c = 1.0 kJ mol−1 and the EHX are known numerical electrophilicities of the Lewis acids HX. De values were calculated ab initio at the CCSD(T)-F12c/cc-pVDZ-F12 level of theory, which for CH2 refers to the singlet electronic excited state. It was established that NR2M values lie in the order of M = C ≫ Si ∼ Ge ∼ Sn for a given R and in the order R = CH3 > H > F for a given M. Reduced nucleophilicities, NB/σaxial, were determined by using the molecular electronic surface potential σaxial at atom M (which lies on the C2 axis) on the 0.001 e Bohr−3 iso-surface of each carbene molecule, as calculated at the MP2/aug-cc-pVTZ level. For R2M having R = CH3 and H and cyclo-(CH)2-M carbenes, the determined values of NB/σaxial are shown to be independent of R and M.
1. Introduction
Carbenes fulfil an important role in synthetic organic chemistry. There exists a voluminous literature of books and reviews, with just one example of each1,2 referred to here. Carbenes can act either as electrophiles or as nucleophiles and are known to be excellent hydrogen bond acceptors.3–10 In this article, we examine the nucleophilicities of some simple carbenes when forming hydrogen-bonded complexes. Two groups of carbenes investigated here are shown in Fig. 1, which illustrates the specific examples with Si as the tetrel atom. The first group (A, B, and C in Fig. 1) can be represented by the general formula R2M, where M is a group 14 (tetrel) atom, C, Si, Ge, or Sn, that is one from each of the first four rows of the Periodic Table. R is H, F or CH3. The second group consists of the related cyclic C2V molecules cyclo-(CH)2M (together with their isomers H2CCM), and two N-heterocyclic carbenes, cyclo-(CH)2(NH)2M (M = C and Si). Each carbene considered has C2V symmetry. We investigate hydrogen-bonded complexes formed with the HX molecules (X = F, Cl, Br, I, CN, CCH, CP). In each case, the HX molecule lies along the C2 axis of the carbene.
 | ||
| Fig. 1 Molecular diagrams of the carbenes discussed in this article. The molecules shown contain silicon as the ‘carbene’ atom but those based on other tetrel atoms C, Ge and Sn are also investigated. A = silylene, B = difluorosilylene, C = dimethylsilylene, D = silacyclopropenylidene, E = vinylidenesilanediyl (an isomer of D), and F = N-heterocyclic silylene. | ||
It is well-known that CH2 has a triplet electronic ground state, with energy separation from the singlet calculated by Schaefer et al.11 to be 46(8) kJ mol−1. The parent carbene CH2 can be included in the present discussion however because the ab initio calculation carried out yields the singlet state. Evidently, carbenes (CH3)2C and F2C have a singlet ground state, for which Hu12 calculates the singlet states to be lower in energy than the triplet states by 224 and 3 kJ mol−1, respectively. All other group 14 analogues of carbenes shown in Fig. 1 possess a singlet ground state. Consequently, the tetrel atom in each carbene considered here carries a non-bonding electron pair along the C2 axis.
A convenient aid when discussing the propensity of a simple carbene to form hydrogen-bonded complexes is the so-called molecular electrostatic surface potential, MESP.13 Diagrams of the 0.001 e Bohr−3 iso-electron density surface for each of F2Si, H2Si and (CH3)2Si are shown in Fig. 2. The corresponding MESP diagrams for M = C (including H2C), Ge and Sn are shown in Fig. S1 of the ESI.† The red regions in each case correspond to the negative (and therefore nucleophilic) regions of the iso-surface. The value of σaxial of the MESP on the C2 axis of each molecule is shown and coincides with the non-bonding electron pair of the R2M molecule. Diatomic or linear Lewis-acid molecules HX (X = F, Cl, Br, I, CN, CCH or CP) are therefore likely form hydrogen-bonded complexes R2M⋯HX in which the electrophilic H atom of HX interacts with the non-bonding electron pair. Hence, the equilibrium geometries of the R2M⋯HX complexes should (like the parent molecules) have a C2 axis of symmetry, with HX lying along that axis. It should be note that most of the F2M are electrophilic (positive MESP) along the C2 axis at M and are therefore expected to form, at best, weak complexes of the required symmetry with the HX molecules of interest.
 | ||
| Fig. 2 Molecular electrostatic surface potentials of F2Si, H2Si, and (CH3)2Si calculated at the MP2/aug-cc-pVTZ level of theory. The values of the MESP (the potential energy of a unit positive point charge on the 0.001 e Bohr−3 iso-surface) on the C2 axis near the Si atom and its non-bonding electron pair are shown. | ||
The nucleophilicity of a given carbene molecule acting as a Lewis base B is determined by the following method. It has been shown14 that the equilibrium energy De required for dissociation of a hydrogen-bonded complex B⋯HX of a Lewis base B and a Lewis acid HX into infinitely separated B and HX can be described by eqn(1), where NB is the nucleophilicity of B and EHX is the electrophilicity of HX:
| De = c·NB·EHX | (1) |
The constant c is chosen as 1 kJ mol−1 for convenience, so that, if De is given in kJ mol−1, NB and EHX are dimensionless. Many NB and EHX values have been generated15 by least-squares fitting of ab initio-calculated De values to eqn (1), including the set of EHX for HF, HCl, HBr, HI, HCN, HCCH and HCP. According to eqn (1), if, for a series of complexes B⋯HX in which B is fixed but HX is varied, De/c is plotted as the ordinate and EHX as the abscissa, and then the gradient of the straight line gives the nucleophilicity NB of B. By such means, new values of NB not included in the initially generated set can be obtained. Recent applications of this approach include the series of Lewis bases ethyne, ethene and cyclopropane16 and monohydrides of group 13 elements B, Al and Ga.17 Alternatively, the gradient of the De/c versus NB graph for a series of complexes B⋯A, in which A is a fixed Lewis acid and the B are a series of Lewis bases (such as N2, CO, C2H2, C2H4, HCN, H2O, and NH3) of known NB values, yielding the electrophilicity EA of A. Applications of this type have recently generated the electrophilicities of various interhalogens (via halogen-bonded complexes B⋯XY18), and of the group 14 elements Si, Ge and Sn in tetrel-bonded complexes B⋯H3M–X (M = Si, Ge, Sn).19
The nucleophilicities of the Lewis bases shown in Fig. 1 were calculated by using the approach set out in the preceding paragraph, namely by calculating the ab initio De values of the hydrogen-bonded complexes (e.g. R2M⋯HX, where X = F, Cl, Br, I, CN, CCH, and CP) and finding the gradients of De/c versus EHX plots. We also investigate here whether a reduced nucleophilicity can be defined by the gradient of a graph of De/σaxialversus EHX, as established for other series of Lewis bases.20 This quantity is of interest because it has been shown to be a property only of the atom of the Lewis base B that is directly involved in, for example, a hydrogen bond and is independent of the remainder of B. Moreover, together with its analogous quantity reduced electrophilicity of a Lewis acid, it provides a method21 of calculating the De values from the properties of the separate molecules B and HX.
2. Theoretical methods
The geometries of the systems have been optimized at the CCSD(T)-F12c computational level22,23 with the cc-pVDZ-F12 basis.24 The CCSD(T) level of calculation is known as the gold-standard for the calculation of medium sized systems. The CCSD(T)-F12c approach has been shown to improve the basis set convergence of correlation and reaction energies25 and thermochemistry26 and provide results similar to those at the CCSD(T)/cc-pV(T/Q)Z level.23 In addition, the cc-pVXZ-F12 basis set has shown broad applicability, including the estimation of interaction energies.27 The standard frozen-core approximation was used for all the complexes, except for those containing tin, where 10 electrons have been correlated instead of the default (4 electrons). This avoids the mixing of the core and valence electrons in complexes where a tin atom is present. The inherent basis set superposition error (BSSE) has been corrected with the Boys and Bernardi full counterpoise method.28 These calculations have been carried out with the scientific MOLPRO program.29 The molecular electrostatic potentials on the 0.001 au electron density iso-surface (MESP) of the isolated monomers were calculated with the Gaussian-16 program30 at the MP2/aug-cc-pVTZ level, analysed with the Multiwfn program31 and represented with the Jmol program.323. Results
3.1. Comparison of the nucleophilicities along the C2 axis direction of R2M molecules (M = Si, Ge, Sn; R = CH3, H and F)
Graphs of De/c versus the electrophilicities EHX of the Lewis acids HX (X = F, Cl, Br, I, CN, CCH, and CP) are shown for the four series of complexes H2M⋯HX (M = C, Si, Ge or Sn) in Fig. 3. Values of EHX are from ref. 15, and except for HI and HBr, refined versions of which are reported in ref. 33. For the reason already discussed in Section 1, M = C can be included in Fig. 3 since the calculation refers to the singlet state. The gradients of the straight lines give the axial nucleophilicities NH2M of the H2M molecules and therefore of the non-bonding electron pair carried on the C2 axis in each case. The determined values are 6.36(16), 2.71(7), 2.35(7) and 1.99(7) for M = C, Si, Ge and Sn, respectively. The value for M = C is higher than the rest, but the other three values are close to each other. It should be noted that the nucleophilicity decreases as the size of the atom M increases. | ||
| Fig. 3 Graphs of De/c versus EHX for the four series of hydrogen-bonded complexes H2M⋯HX, having M = C, Si, Ge or Sn and X = F, Cl, Br, I, CN, CCH, and CP. The gradients give the nucleophilicities of the non-bonding electron pair that lies along the C2 axis of H2M. | ||
The corresponding diagram for the series (CH3)2M⋯HX when M = C, Si, Ge or Sn is shown in Fig. 4. It should be noted that, in Fig. 4, the origin is no longer included as a point for (CH3)2C⋯HX because of a significant non-zero intercept (This behaviour seems to be a characteristic of Lewis bases that have methyl groups attached, see for example Fig. 6 of ref. 16.). The points for (CH3)2C⋯HX when X = Cl, Br, and I are excluded because of unduly large De values resulting from significant (CH3)2CH+⋯X− characters, as indicated by short distances r(C–Haxial) and lengthened distances r(H–X). The (CH3)2C⋯HX points having X = F, CN, CCH and CP seem to be better behaved and indicate that the axial nucleophilicity NB of M = C in this series is considerably greater than those having M = Si, Ge and Sn. It is noted that the three molecules associated with rows 2, 3 and 4 of the Periodic Table again have very similar axial nucleophilicities. In fact, the ratios of NB are 3.19(11)/1.36(8)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.18(8):1.00 for H2C/H2Si/H2Ge/H2Sn and 3.25(19):1.40(14):1.19(13):1.00 for (CH3)2C/(CH3)2Si/(CH3)2Ge/(CH3)2Sn; these ratios are therefore identical for H2M and (CH3)2M within the standard errors.
:1.18(8):1.00 for H2C/H2Si/H2Ge/H2Sn and 3.25(19):1.40(14):1.19(13):1.00 for (CH3)2C/(CH3)2Si/(CH3)2Ge/(CH3)2Sn; these ratios are therefore identical for H2M and (CH3)2M within the standard errors.
 | ||
| Fig. 4 Graphs of De/c versus EHX for the four series of hydrogen-bonded complexes (CH3)2M⋯HX, having M = C, Si, Ge or Sn and X = F, Cl, Br, I, CN, CCH, and CP. The gradients give the nucleophilicities of the non-bonding electron pair that lies along the C2 axis of (CH3)2M. | ||
Graphs of De/c versus EHX for the series F2C⋯HX, F2Si⋯HX and F2Ge⋯HX are shown in Fig. 5 (data are unavailable for F2Sn⋯HX because the region of the MESP on the C2 axis is positive and data are limited for F2Ge⋯HX because of evidence of ionic characters for some of the HX complexes). The values of the nucleophilicity decrease from 3.39 for M = C, through small positive 0.93 for M = Si and becomes negative −0.39 for Ge. This is reflected in the MESPs at the tetrel atom on the C2 axes of F2C, F2Si and F2Ge which have the values (see Fig. S1, ESI†) of −85.0, +1.5 and +49.8 kJ mol−1, respectively. Such MESP values indicate that the complexes with HX molecules are likely to be weak, except for F2C⋯HX. This expectation is borne out by the gradients of the lines in Fig. 4. Presumably, for F2Ge⋯HX, this is negative because of repulsion between the positive axial MESP at Ge on the C2 axis of F2Ge and the electrophilic atom H of the Lewis acids HX.
 | ||
| Fig. 5 Graphs of De/c versus EHX for the three series of hydrogen-bonded complexes F2M⋯HX having M = C, Si, or Ge and X = F, Cl, Br, I, CN, CCH, and CP. The gradients give the nucleophilicities at the non-bonding electron pair that lies along the C2 axis of M. See the text for discussion of the negative gradient for the F2Ge⋯HX series. | ||
3.2. The inductive effects of R = CH3 and R = F relative to R = H on the nucleophilicities of R2M (M = Si, Ge, Sn)
Fig. 6 shows the De/c versus EHX graphs for the three series (CH3)2C⋯HX, H2C⋯HX and F2C⋯HX. The gradients of the three straight lines are 9.41(22), 6.36(16) and 3.39(26), respectively. Thus, the substitution of the two H atoms by methyl groups increases the axial nucleophilicity near C by 3.1, while the substitution of two H atoms by F atoms decreases it by 3.0. | ||
| Fig. 6 Graphs of De/c versus EHX for the three series of hydrogen-bonded complexes R2C⋯HX having R = CH3, H or F and X = F, Cl, Br, I, CN, CCH, and CP. The gradients give the nucleophilicities associated with the non-bonding electron pair that lies along the C2 axis of C. They confirm that CH3 has a +I effect relative to H while F has a −I (electron-withdrawing) effect. | ||
The corresponding graphs for the R2Si⋯HX series are shown in Fig. 7, while that for R2Ge⋯HX is shown in Fig. S2 of the ESI.† The patterns in Fig. 7 and Fig. S2 (ESI†) are similar to those in Fig. 6, except that it decreases in nucleophilicity when CH3 is replaced by H and then F becomes smaller from M = C to M = Si to M = Ge. As discussed in connection with Fig. 5, the gradient of the graph associated with the F2Ge⋯HX line in Fig. S2 (ESI†) is negative. For the R2Sn series, R = F is not available (for reasons discussed earlier), but the graphs for the (CH3)2Sn⋯HX and H2Sn⋯HX are shown in Fig. S3 (ESI†). The axial nucleophilicity decreases by ∼0.8 in this case when CH3 is replaced by H.
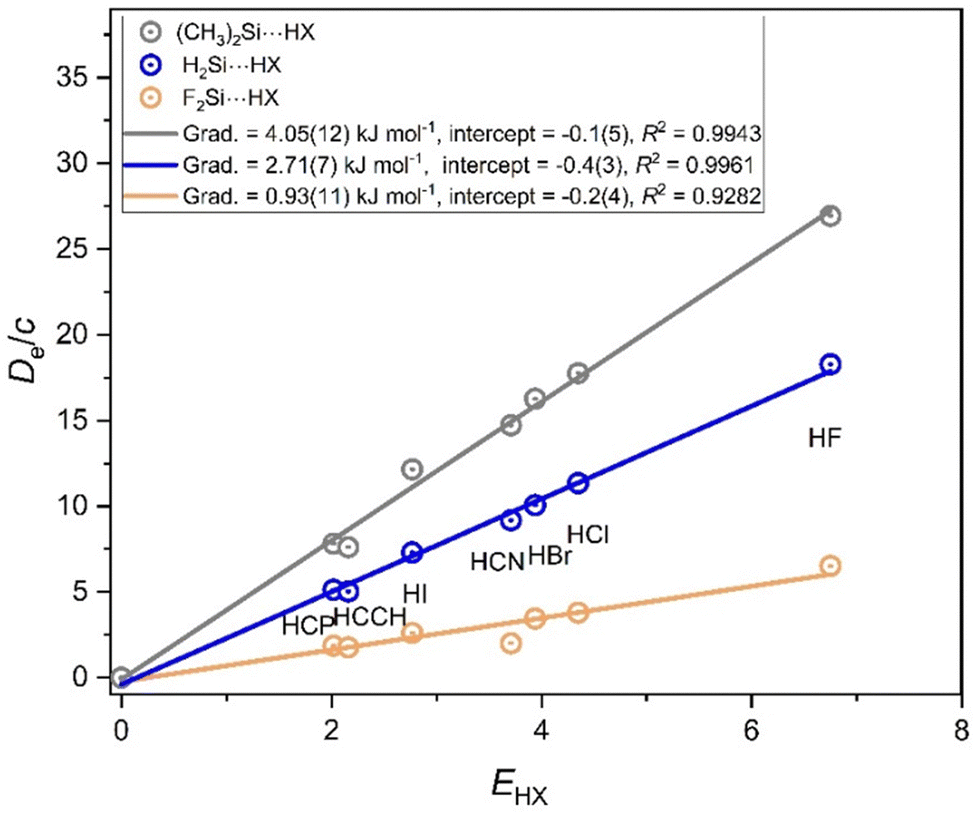 | ||
| Fig. 7 Graphs of De/c versus EHX for the three series of hydrogen-bonded complexes R2Si⋯HX having R = CH3, H or F and X = F, Cl, Br, I, CN, CCH, and CP. The gradients give the nucleophilicities associated with the non-bonding electron pair that lies along the C2 axis of Si. They again confirm that CH3 has a +I effect relative to H, while F has a −I (electron-withdrawing) effect. | ||
3.3. Axial nucleophilicities of the cyclic carbenes: cyclo-(CH)2M (M = C, Si, Ge)
Replacing the two methyl groups in the (CH3)2M molecule by the HCCH moiety leads to the cyclic carbene cyclo-(CH)2M. Cyclo-(CH)2C is the first member of cyclopropenylidenes,34 was initially characterised by its microwave spectrum,35 and is of significance in astrophysical spectroscopy. There is a singlet ground state for all M discussed here. To calculate ab initio the geometry of the cyclo-(CH)2M molecules and their hydrogen-bonded complexes, cyclo-(CH)2M⋯HX for M = C, Si and Ge is straight-forward, but for M = Sn there is evidence of ionic character and it is not included in what follows. Fig. 8 compares the plots of De/c versus EHX for the three series cyclo-(CH)2M⋯HX (M = C, Si, Ge). Fig. 8 illustrates clearly that, for the complexes cyclo-(CH)2M, the order of the nucleophilicities is M = C ≫ Si ∼ Ge. It should also be noted that nucleophilicities are in the order (CH3)2M > cyclo-(CH)2M for each of M = C, Si and Ge. Evidently, the inductive effect of two CH3 groups is greater than that arising from the HCCH moiety that replaces them.
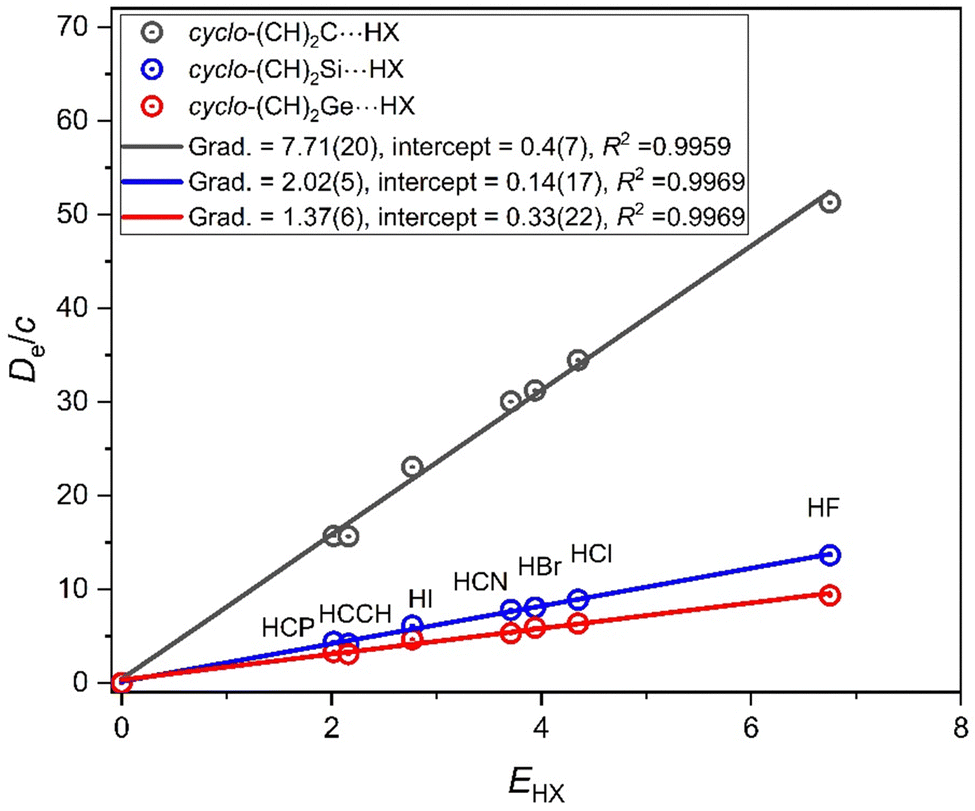 | ||
| Fig. 8 Graphs of De/c versus EHX for the three series of hydrogen-bonded complexes cyclo-(CH)2M⋯HX, where M = C, Si or Ge and X = F, Cl, Br, I, CN, CCH, CP. Each Lewis base is a carbene carrying the group 14 atom from the first three rows of the Periodic Table. The gradients give the nucleophilicities of the non-bonding electron pair on M that lies along the C2 axis and cyclo-(CH)2M can be viewed as generated from (CH3)2M by removing four H atoms and linking the two CH groups that remain. | ||
3.4. Reduced nucleophilicities of carbene molecules acting as Lewis bases
In an earlier article,20 we showed that the gradient of a graph of −De/σmin plotted against EHX for a series of hydrogen-bonded complexes B⋯HX in which X varies but B is the same Lewis base defines a reduced nucleophilicity ИB = NB/σmin of B. The term ‘reduced’ is taken to mean that ИB is a property only of the atom of B that is directly involved in the B⋯HX hydrogen bonds, independent of the remainder of B. In this section, we examine whether the reduced nucleophilicity is a term that can also be applied to the carbenes discussed earlier. The appropriate graphs here are those in which −De/σaxial is plotted against EHX, where σaxial is the value of the MESP along the C2 axis of each carbene, given that the HX molecules (X = F, Cl, Br, I, CN, CCH, CP) lie along the C2 axes when B = R2M (M = Si, Ge, Sn; R = H, CH3, F). The values of σaxial calculated at the MP2/aug-cc-pVTZ level of theory are set out in Table 1. Graphs of −De/σaxialversus EHX are shown in Fig. 9 for the complexes B⋯HX (X = F, Cl, Br, I, CN, CCH, CP) in which B = H2M, (CH3)2M or cyclo-(CH)2M for each of M = Si, Ge and Sn. None of the F2M complexes is included in Fig. 9 because, as already discussed, σaxial is positive or near-zero for these, resulting in very small (sometimes even negative) De values because of repulsion between the adjacent regions of the positive MESPs of B and HX.| σ axial/(kJ mol−1) | ||||
|---|---|---|---|---|
| Carbene | M = C | M = Si | M = Ge | M = Sn |
| H2M | −151.0 | −65.9 | −60.1 | −41.4 |
| F2M | −85.0 | +1.5 | +49.8 | +86.1 |
| (CH3)2M | −178.7 | −101.0 | −91.1 | −65.9 |
| cyclo-(CH)2M | −186.9 | −46.2 | −32.8 | −1.8 |
| H2CCM |
−168.0 | −20.2 | −17.8 | +6 |
| cyclo-NHM | −204.0 | −87.1 | — | — |
 | ||
| Fig. 9 Graphs of −De/σaxialversus EHX for the ten series of hydrogen-bonded complexes H2M⋯HX, (CH3)2M⋯HX, cyclo-(CH)2M⋯HX when M = C, Si, Ge or Sn, and X = F, Cl, Br, I, CN, CCH, CP. Each Lewis base molecule is a group 14 carbene. The gradients give the reduced nucleophilicities of the non-bonding electron pair on M, which lies on the C2 axis. | ||
It is clear from Fig. 9 that the ten straight lines of differing gradients previously generated in De/c versus EHX graphs have become parallel and effectively conflated when De is divided by σaxial. The lines are of almost equal gradient (except perhaps for that of H2Sn⋯HX). Otherwise, all gradient values fall within two standard errors of the mean value 0.0417 (standard deviation = 16).
This provides strong evidence that ИB = NB/σaxial can indeed be described as a reduced nucleophilicity that is common to the Lewis bases involved, namely, H2M, (CH3)2M and cyclo-(CH)2M, where M = C, Si, Ge or Sn. It should be noted that not only are the quantities ИB = NB/σaxial from Fig. 9 independent of the groups attached to a given tetrel atom, but they also appear to be independent of the group 14 atom.
Some series do not appear in Fig. 9. The cyclic carbene cyclo-(CH)2Sn has the very low value of σaxial of −1.8 kJ mol−1. Clearly, the quantity ИB = NB/σaxial increases rapidly as the σaxial value approaches zero and becomes infinite when the σaxial value reaches zero. The ИB value is therefore very sensitive to small errors in σaxial, is unreliable close to the singularity and becomes negative after the singularity. For these reasons, members of the series F2M⋯HX, in addition to cyclo-(CH)2Sn⋯HX, have been excluded from Fig. 9. For (CH3)2C⋯HX (as indicated earlier), there is evidence of ionic character in several points, with only four available, and consequently they are not included in Fig. 9.
3.5. H2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) CM (M = C, Si, Ge) as carbene Lewis bases
CM (M = C, Si, Ge) as carbene Lewis bases
The molecules H2CCC, H2CCSi and H2CCGe are isomers of cyclo-(CH)2–M (M = C, Si, Ge). They too are singlet carbenes and have also been characterised via their microwave spectra [see ref. 36 and 37, respectively]. In this section, the nucleophilicities of H2CCM when forming hydrogen bonds with the HX series are determined and their reduced nucleophilicities examined.
Questions that arise for the carbenes H2CCM are (a) whether a reduced nucleophilicity can be assigned to these molecules, and, if so, (b) whether it is identical to those determined viaFig. 9? To answer these questions, plots of De/c versus EHX for the hydrogen-bonded complexes H2CCM⋯HX (M = C, Si or Ge and X = F, Cl, Br, I, CN, CCH, and CP) are displayed in Fig. 10. The series H2CCSn⋯HX is excluded because the Lewis base has a small positive σaxial (See Table 1 and earlier discussion). Fig. 10 indicates clearly that the order of the nucleophilicities of the three carbenes is NH2CCC ≫ NH2CCSi ∼ NH2CCGe.
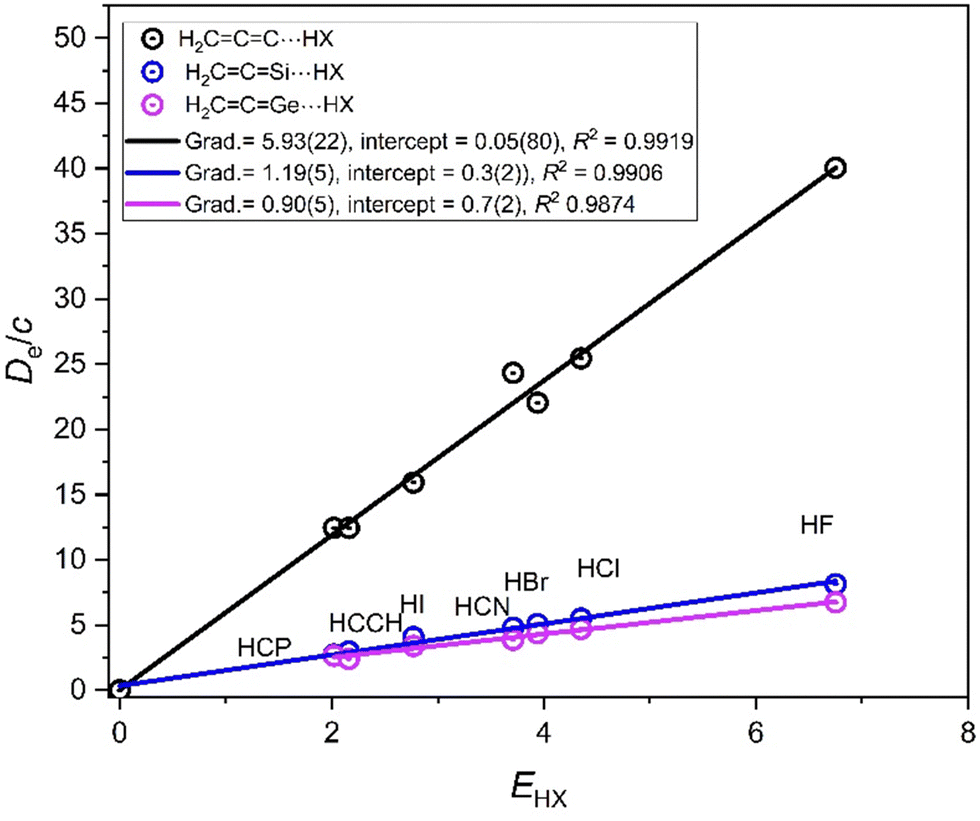 | ||
| Fig. 10 Graphs of De/c versus EHX for the series of hydrogen-bonded complexes H2CCM⋯HX (M = C, Si or Ge; X = F, Cl, Br, I, CN, CCH, and CP). Each Lewis base molecule is a carbene. The gradients give the nucleophilicities of these carbenes when forming a hydrogen bond to the series of HX molecules. | ||
It is immediately obvious from Fig. 11 that the division of the points in Fig. 10 by the appropriate σaxial values from Table 1 conflates only two of the straight lines (those associated with M = Si and Ge) to give a single gradient. Accordingly, the gradients are now the reduced nucleophilicities ИB = NB/σaxial of the two Lewis bases B = H2CCSi and H2CCGe and this quantity is a property that is common to the atoms Si and Ge. Their values ИB = 0.0586(26) and 0.0562(29) are identical within their standard errors and yield a mean of 0.0574(28). The value for H2CCC is certainly not identical to those for M = Si and Ge as shown in Fig. 9. Perhaps, this result is to be expected, given that the environment of the all the group 14 atoms C, Si and Ge in the Lewis bases in Fig. 9 consists of two single bonds at an angle of ∼90–110 degrees and, when the HX molecules are included, is in a trigonal environment. On the other hand, in the H2CCM molecules, the tetrel atom is in a diagonal environment, created by a double bond on one side and the HX molecule on the other side. The non-bonding pair on M is also in close contact with the π electrons in these molecules. It is not clear why the gradient for the H2CCC⋯HX graph is different from those with M = Si or Ge, but perhaps there is a stronger interaction between the axial non-bonding electron pair of C and the π-electron system than occurs with Si or Ge. It is interesting to note, however, that the value ИH2CCC = 0.0353(13) is identical (within standard errors) with those values of 0.0349(17) and 0.0337(18) obtained38 by similar means for hydrogen bonds to the C atoms in OC, SC, and SeC and HNC, CH3NC, and FNC, respectively.
 | ||
| Fig. 11 Graphs of −De/σaxialversus EHX for the three series of hydrogen-bonded complexes H2CCM⋯HX. (M = C, Si and Ge; X = F, Cl, Br, I, CN, CCH, and CP). Each Lewis base molecule carries a group 14 atom and is a singlet carbene. The gradients yield the reduced nucleophilicity associated with the tetrel atom M, which lies on the C2 axis of each H2CCM molecule. | ||
3.6. Cyclo-(CH)2(NH)2M (M = C or Si) acting as Lewis bases in forming hydrogen bonds
N-heterocyclic carbenes and N-heterocyclic silylenes, having formulae cyclo-(CH)2(NH)2M (M = C or Si) (hereafter referred to as NHC and NHSi, respectively), have recently fulfilled an important role in organic chemistry.39,40 The De values of hydrogen-bonded complexes NHC⋯HX and NHSi⋯HX formed by these two Lewis bases were therefore calculated at the same CCSD(T)(F12c)/cc-pVDZ-F12 level of theory as other systems discussed here. The resulting graphs of De/c and −De/σaxialversus EHX are shown in Fig. 12 and 13, respectively. Each of NHC and NHSi has a C2 symmetry axis, as do all other Lewis bases discussed in this article, and a high value of the MESP on the C2 axis, favouring the geometry of NHM⋯HX complexes of the type illustrated in Fig. 12 and 13.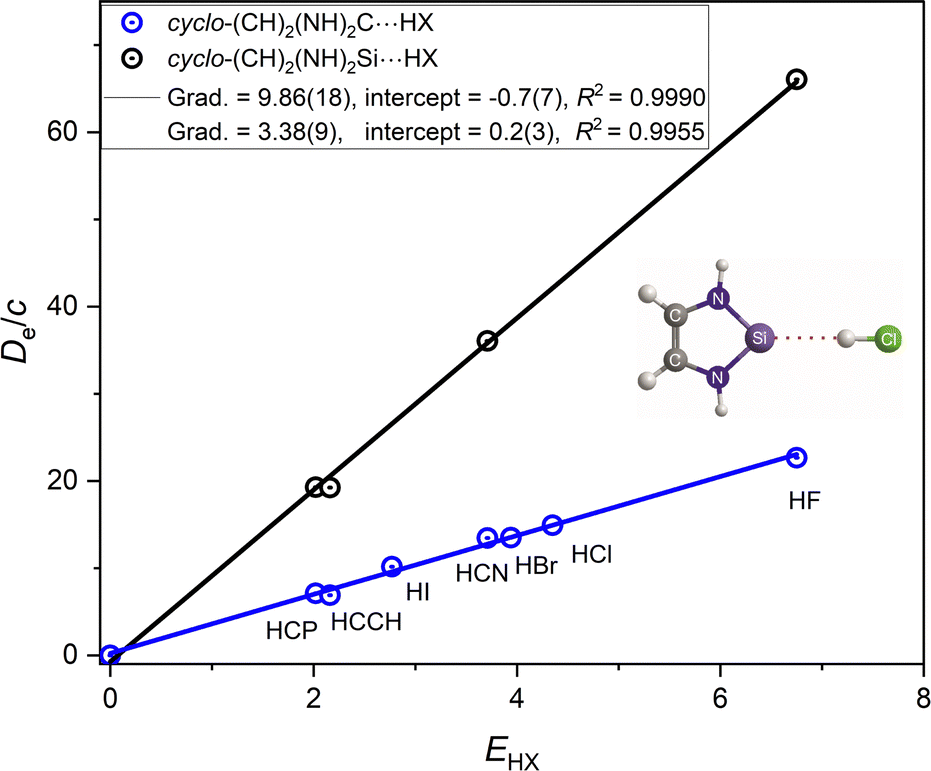 | ||
| Fig. 12 D e/c versus EHX for the complexes cyclo-(CH)2(NH)2M⋯HX (M = C and Si; X = F, Cl, Br, I, CN, CCH, and CP). | ||
 | ||
| Fig. 13 −De/σaxialversus EHX for the complexes cyclo-(CH)2(NH)2M⋯HX (M = C and Si; X = F, Cl, Br, I, CN, CCH, and CP). | ||
Fig. 12 reveals that the nucleophilicity of the NHC molecule is three times that of its Si analogue, a ratio that is higher than that observed for other R2M⋯HX series. Fig. 13 leads to the reduced nucleophilicities of 0.0388(11) for NHSi⋯HX and the higher value of 0.0483(9) for NHC⋯HX, indicating that the conflation of values is not as good as those reported in Fig. 9. The first value of the gradient ИNHSi falls within two standard errors of the mean of those in Fig. 9, but that for NHC⋯HX is somewhat higher. Whether it is significantly higher is not immediately clear and caution is necessary. First, there are only four different HX molecules involved, instead of the normal seven and moreover two of these points are essentially identical. Second, the small data set probably means that the standard error for ИNHC is too low. It is noted that there is a range of ∼0.01 in the ИB values as shown in Fig. 9.
4. Discussion and conclusions
The nucleophilicities NB of several simple carbenes acting as Lewis bases B, namely H2M, F2M, (CH3)2M, cyclo-(CH)2M, H2CCM (M is one of the tetrel atoms C, Si, Ge, or Sn) and cyclo-(CH)2(NH)2M (M = C or Si only), have been reported in this article. According to eqn (1), graphs of De/c versus EHX (the numerical electrophilicity of HX) for the series of hydrogen-bonded complexes B⋯HX (X = F, Cl, Br, I, CN, CCH, CP) are straight lines through the origin, having gradients NB. All but one (the prototype H2C) of the carbenes investigated possess a singlet electronic ground state, but H2C was calculated ab initio as a singlet and was therefore not the electronic ground state molecule. Its properties were consistent with those of the others examined. The equilibrium dissociation energies De of the hydrogen-bonded complexes (each having a C2 axis of symmetry) were calculated ab initio at the CCSD(T)(F12c)/cc-pVDZ-F12 level.
The results for the nucleophilicities are summarized in Table 2. These measure the propensity of the tetrel atoms involved to engage in hydrogen bonding to HX molecules. It is noted that for a given tetrel atom M, the order of the nucleophilicities for the R2M carbenes is as follows: (CH3)2M > H2M > F2M, a result consistent with the accepted order of the inductive effects of the groups CH3, H and F. The order across the (CH3)2M row in Table 2 is M = C ≫ Si ∼ Ge ∼ Sn; there are indications in the other R2M rows that this is also true, even though the remaining rows are incomplete. The entries in Table 2 for cyclo-(CH)2M and H2CCM, respectively, reveal a similar pattern, while, for a given M, the nucleophilicity of the carbene for cyclo-(CH)2M is greater than that of H2CCM. Evidently, in passing from row 1 to row 2 of the Periodic Table, there is a large drop in the nucleophilicity of the tetrel atom in these carbenes, but the changes from rows 2 to 3 to 4 are much smaller. (CH3)2M has the largest nucleophilicity for a given M, followed by cyclo-(CH)2M and H2CCM. It has also been demonstrated that the carbenes investigated here exhibit the phenomenon of reduced nucleophilicity when each point on the straight line from the Deversus EHX plot is divided by the value of the MESP on the 0.001 e Bohr−3 iso-surface on the C2 symmetry axis at the tetrel atom. In the present example, it appears that the reduced nucleophilicity is independent not only of the tetrel atom that participates in the hydrogen bond, but also of the remainder of the carbene molecule, at least for the R2M and cyclo-(CH)2M, which have a mean value ИB of 0.0417(16). This seems not to be the case for the H2CCM carbenes which have the somewhat larger value of 0.0530(26) (based on only the Si and Ge values, however). H2CCC is different. See Fig. 11 and associated discussion.
| Carbenes | M = C | M = Si | M = Ge | M = Sna |
|---|---|---|---|---|
|
a As indicated in Table 1, the axial MESPs of the Sn-based carbenes discussed are low or even positive and as a result precluded the analysis being applied to complexes involving F2Sn, cyclo-(CH)2Sn and H2CCSn.
b All carbenes in this table have a singlet electronic ground state, except H2C which has a triplet ground state. The excited singlet state of H2C was that calculated ab initio and employed here.
c Missing values in this column arise because of a repulsive interaction of a positive MESP at Sn on the C2 axis and the electrophilic H of HX. This leads to a distortion of results.
|
||||
| H2M | 6.36(12)b | 2.71(7) | 2.35(7) | 1.99(7) |
| F2M | 3.39(26) | 0.93(11) | −0.39(2)c | — |
| (CH3)2M | 9.41(22) | 4.05(18) | 3.44(9) | 2.89(16) |
| cyclo-(CH)2M | 7.71(20) | 2.02(5) | 1.37(6) | — |
| H2CCM |
5.93(22) | 1.19(5) | 0.90(5) | — |
| cyclo-NHM | 9.86(18) | 3.38(9) | — | — |
A further property of interest in connection with the reduced nucleophilicity ИB of a Lewis base B, such as the H2M discussed here, and its counterpart the reduced electrophilicity ΞHX of a Lewis acid is that they provide a route to the dissociation energy De from the properties of the separate component molecules. It was shown in ref. 21 that
| De = {σmax(HX)·σmin(B)}·ИB·ΞHX | (2) |
To illustrate the use of eqn (2) in this context, Fig. 14 shows a graph of De values (as calculated by eqn (2)) plotted against the ab initio calculated values of De for 49 complexes H2M⋯HX, (CH3)2M⋯HX, where M = C, Si, Ge and Sn. The F2M⋯HX were excluded for reasons already discussed, as were the (CH3)2C⋯HX because only 4 points are available in Fig. 6 and there is a large intercept on the De/c axis. The mean value of ИB = 0.0417 for the 10 carbenes involved in Fig. 9 was used, while the ΞHX values are those listed in Table 2 of ref. 21.
 | ||
| Fig. 14 Graph of De values calculated by eqn (2) for the complexes R2M⋯HX (R = H, CH3; M = C, Si, Ge, Sn; X = F, Cl, Br, I, CN, CCH, CP) versus De values calculated ab initio at the CCSD(T)F12C/cc-pVDZ-F12 level. | ||
Finally, the series of complexes H2C⋯HX have been a part of the main investigation, even though the electronic ground state of H2C is a triplet. However, the ab initio calculation for H2C referred to the closed shell version, that is the electronic singlet state, which is higher in energy than the triplet. The value of σaxial for singlet H2C is in Table 1. Fig. 9 establishes unambiguously that for singlet H2C the value of the reduced nucleophilicity is ИB = 0.0421(11), which is identical with the mean 0.0417(16) of the 10 values available from Fig. 9. This seems reasonable, as all the carbene molecules in Fig. 9 have singlet electronic ground states, even though all have different electronic energies.
Data availability
Some of the material associated with this article is made available in the ESI.†Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
IA thanks the Ministerio de Ciencia, Innovación y Universidades (PID2021-125207NB-C32) for a grant of financial support and ACL is pleased to acknowledge the University of Bristol for the award of a University Senior Research Fellowship and for the award of an account on the Blue Pebble HPC.References
- R. A. Moss and M. P. Dyle, Contemporary Carbene Chemistry, (Volume 7 of the Wiley Series of Reactive Intermediates in Chemistry and Biology), 2013 Search PubMed.
- Y. Mizuhata, T. Sasamori and N. Takitoh, Chem. Rev., 2009, 109, 3479–3511 CrossRef CAS.
- J. A. Pople, Chem. Phys. Lett., 1986, 132, 141–146 CrossRef.
- A. J. Arduengo, S. F. Gamper, M. Tamm, J. C. Calabrese, F. Davidson and H. A. Craig, J. Am. Chem. Soc., 1995, 117, 572–573 CrossRef CAS.
- I. Alkorta and J. Elguero, J. Phys. Chem., 1996, 100, 19367–19370 CrossRef CAS.
- M. Jabłoński and M. Palusiak, Phys. Chem. Chem. Phys., 2009, 11, 5711–5719 RSC.
- P. Costa and W. Sander, Angew. Chem., Int. Ed., 2014, 5, 5122–5125 CrossRef.
- J. M. Standard, J. Phys. Chem. A, 2017, 121, 381–393 CrossRef CAS.
- J. E. Del Bene, I. Alkorta and J. Elguero, Chem. Phys. Lett., 2017, 675, 46–50 CrossRef CAS.
- M. Jabłoński, Molecules, 2022, 27, 5712 CrossRef PubMed.
- C. H. Bauschlicher, H. F. Schaefer III and P. S. Bagus, J. Am. Chem. Soc., 1977, 99, 7106–7110 CrossRef CAS.
- C.-H. Hu, Chem. Phys. Lett., 1999, 309, 81–89 CrossRef CAS.
- P. Politzer, J. S. Murray, T. Clark and G. Resnati, Phys. Chem. Chem. Phys., 2017, 19, 32166–32178 RSC.
- A. C. Legon, Phys. Chem. Chem. Phys., 2014, 16, 12415–12421 ( See also Phys. Chem. Chem. Phys. , 2014 , 16 , 25199 ) RSC , for correction.
- I. Alkorta and A. C. Legon, Molecules, 2017, 22, 1786–1799 CrossRef PubMed.
- I. Alkorta and A. C. Legon, Chem. Phys. Lett., 2023, 833, 140929 CrossRef CAS.
- I. Alkorta and A. C. Legon, ChemPlusChem, 2024, 89, e202400314 CrossRef CAS PubMed.
- I. Alkorta and A. C. Legon, ChemPlusChem, 2021, 86, 778–784 CrossRef CAS PubMed.
- I. Alkorta and A. C. Legon, J. Phys. Chem. A, 2024, 128, 5963–5968 CrossRef CAS PubMed.
- I. Alkorta and A. C. Legon, Phys. Chem. Chem. Phys., 2022, 24, 25822–25833 RSC.
- I. Alkorta and A. C. Legon, J. Phys. Chem. A, 2023, 127, 4715–4723 CrossRef CAS PubMed.
- C. Hättig, D. P. Tew and A. Köhn, J. Chem. Phys., 2010, 132, 231102 CrossRef.
- C. Hättig, W. Klopper, A. Köhn and D. P. Tew, Chem. Rev., 2012, 112, 4–74 CrossRef.
- J. G. Hill and K. A. Peterson, J. Chem. Phys., 2014, 141, 094106 CrossRef.
- T. B. Alder, G. Knizia and H. J. Werner, J. Chem. Phys., 2007, 127, 221106 CrossRef.
- M. K. Kesharwani, N. Sylvetsky, A. Köhn, D. P. Tew and J. M. L. Martin, J. Chem. Phys., 2018, 149, 154109 CrossRef PubMed.
- H. Kruse, R. Szabia and J. Šponer, J. Chem. Phys., 2020, 152, 214104 CrossRef CAS PubMed.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby and M. Schütz, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 242–253 CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson and H. Nakatsuji, et al., Gaussian 16, Revision, A.03, Gaussian, Inc., Wallingford, CT, USA, 2016 Search PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS.
- Jmol: an open-source Java viewer for chemical structures in 3D, https://www.jmol.org.
- I. Alkorta and A. C. Legon, Phys. Chem. Chem. Phys., 2022, 24, 12804–12807 RSC.
- M. S. Schuurman, J. Giegerich, D. Lang, B. Kiendl, K. Pachner, R. J. MacDonell, A. Krüger and I. Fischer, Chem. – Eur. J., 2015, 21, 14486–14495 CrossRef CAS.
- J. M. Vrtilek, C. A. Gottlieb and P. Thaddeus, Astrophys. J., 1987, 314, 716–725 CrossRef CAS.
- M. C. McCarthy and P. Thaddeus, J. Mol. Spectrosc., 2002, 211, 235–240 CrossRef CAS.
- M. C. McCarthy and P. Thaddeus, J. Mol. Spectrosc., 2002, 211, 228–234 CrossRef CAS.
- I. Alkorta and A. C. Legon, ChemPlusChem, 2023, 88, e202300032 CrossRef CAS PubMed.
- N-Heterocyclic Carbenes, ed. S. P. Nolan, 2014, Wiley-VCH, Verlag GmbH & Co Search PubMed.
- M. Haaf, T. A. Schmedake and R. West, Acc. Chem. Res., 2000, 33, 704–714 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp03894k |
| This journal is © the Owner Societies 2025 |