
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dissipative particle dynamics parametrisation using infinite dilution activity coefficients: the impact of bonding†
Rachel L.
Hendrikse
 *a,
Carlos
Amador
b and
Mark R.
Wilson
a
*a,
Carlos
Amador
b and
Mark R.
Wilson
a
aDepartment of Chemistry, Durham University, Durham, DH1 3LE, UK. E-mail: rachel.hendrikse@durham.ac.uk
bProcter and Gamble, Newcastle Innovation Centre, Whitley Road, Newcastle upon Tyne, NE12 9BZ, UK
First published on 23rd December 2024
Abstract
Dissipative particle dynamics (DPD) simulations have proven to be a valuable coarse-grained simulation technique for studying complex systems such as surfactant and polymer solutions. However, the best method to use in parametrising DPD systems is not universally agreed. One common approach is to map infinite dilution activity coefficients to the DPD simulation ‘beads’ that represent molecular fragments. However, we show that here that this approach can lead to serious errors when bonding beads together to create molecules. We show errors arise from the verlaps between bonded beads, which alters their solubility. In this article, we demonstrate how these bonding errors can be accounted for when defining DPD force fields using simple theoretical methods to account for the overlapping volumes, and we demonstrate the validity of our approach by calculating the partition coefficients for a series of solutes into two immiscible solvents.
1 Introduction
The dissipative particle dynamics (DPD) simulation method was initially introduced by Hoogerbrugge and Koelman,1 and it has been continuously developed since then.2–5 DPD is a mesoscopic simulation technique in which groups of atoms are represented by single beads, which is intended to reduce the computational demand of simulating single atoms. Mesoscale modelling provides the bridge between the atomistic methods and the macroscale, retaining molecular details which are lost at a continuum scale, but making longer time and length scales more accessible than when using full atomistic modelling.When first introduced, DPD was primarily used for qualitative study, with DPD parameters chosen to investigate general behaviours of systems such as surfactants6,7 and polymer8–11 solutions. These molecules are typically simulated using a simple bead-and-spring model, allowing one to bond various ‘beads’ together to study the behaviour of larger molecules. This has proven to be a largely successful approach for studying these types of systems, including the prediction of liquid crystal phase formation6,8,10 and the nature of micellar solutions.7,10
A convenient method of relating atomistic simulations to mesoscopic simulation was presented by Groot and Warren2 who provided a mapping between DPD parameters and Flory–Huggins parameters. This mapping allows one to determine Flory–Huggins parameters using solubility parameters from molecular dynamics (or even experimental data) to determine how DPD beads should interact to model realistic systems. This approach has been widely used12–16 in the literature as a means of simulating ‘real molecules' using DPD, enabling quantitative predictions for various systems. This has made it possible to accurately calculate properties such as interfacial tension,12–14 critical micelle concentrations14 and other micelle properties14 for surfactant systems, as well as polymer properties such as radius of gyration15,17 and the end-to-end distance.18
While the mapping procedure provided by Groot and Warren2 has been widely used, there are limitations to its applicability for different systems. For example, the original expressions they provided for the mapping were determined on the basis of simulations which relied on the phase separation of beads, meaning this approach is difficult to use for determining the interaction between two soluble bead types. They also performed simulations over a limited range of DPD interaction parameters, choosing to focus on ranges where linear relationships can be fitted to their calculated values. Therefore, strictly, their expressions should only be used in the range in which they were intended. Although, they are often used outside of this range. Other methods that have since been used to determine realistic DPD parameters include mapping to infinite dilution activity coefficients,19–23 as an alternative to Flory–Huggins parameters. This has the benefit of being generally applicable to a greater range of system types, including bead types that are soluble in one another. Another approach is by mapping interactions to octanol–water partition coefficients,24–26 which has been shown to be successful for studying the behaviour of various surfactant systems.24–28 Alternatively, others have calculated DPD parameters by matching to experimental liquid densities of mixtures.29,30
It is of particular interest in this work that, no matter which parametrisation approach is used, most existing DPD studies neglect the impact that bonding has on the resulting solubility of molecules. However, this has been shown to be important,31 not least because the length of the bond can greatly influence the degree of overlap between the bonded beads. Bonding beads together has been shown to influence their solubility because of this overlapping bead volume. This oversight can lead to an incorrect determination of DPD parameters for bonded molecules, which is crucial for modelling the correct behaviour of systems. This is particularly true for systems involving shorter bond lengths, which typically result from a more finely coarse-grained system. Hence, dealing with this issue is essential for using DPD in quantitative studies of soft matter systems.
In this work, we show that DPD parameters can be accurately assigned based on their known infinite dilution activity coefficients, by using Widom insertion together with a simple approach to take into account the overlap of bonded beads. We validate our approach by simulating the partitioning of various solute molecules into two immiscible solvents, allowing us to determine partition coefficients as a means to check the predicted chemical potential of bonded beads and hence to check their infinite dilution activity coefficients. We suggest that this simple approach can make a major improvement to the parametrisation of DPD models.
2 Dissipative particle dynamics
2.1 Overview
In dissipative particle dynamics (DPD) atoms and molecules are modelled by simulating ‘DPD' beads, where a single bead can represent a number of atoms. The total force acting on a single unbonded DPD ‘bead’ is the sum of the conservative FCij, dissipative FDij, and random force FRij. All of these forces act within a defined cut-off range RC, beyond which the interaction force is zero. We choose to set the cut-off RC = 1, in line with existing literature. Additional bonding FBij and angle forces FAij contribute to longer molecules represented by a series of bonded beads.The conservative force takes the form
 | (1) |
![[r with combining circumflex]](https://www.rsc.org/images/entities/b_char_0072_0302.gif) ij = rij/|rij|. The conservative force assigns a chemical identity to the beads via varying the value of the aij parameter, which can be defined using various approaches which will be discussed in Section 2.2. The forces FDij and FRij are given by the following expressions.
ij = rij/|rij|. The conservative force assigns a chemical identity to the beads via varying the value of the aij parameter, which can be defined using various approaches which will be discussed in Section 2.2. The forces FDij and FRij are given by the following expressions.| FDij = −γωD(rij)(ij·vij)ij, | (2) |
| FRij = σωR(rij)ζijijΔt−1/2, | (3) |
In this work, we perform all of our simulations using the LAMMPS software, where the form of the weight function used is
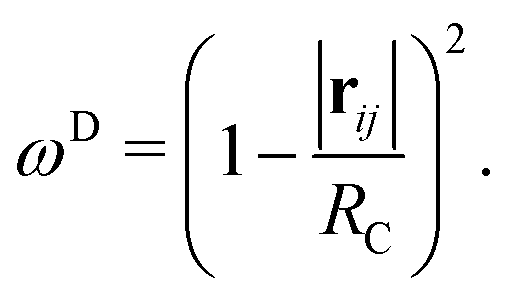 | (4) |
| ωD = [ωR]2 | (5) |
| σ2 = 2γkBT, | (6) |
For bonded molecules, we also impose ‘bonding’ and ‘angle’ potentials taking the form of a harmonic spring,
| UBij = KB(rij − l0)2, | (7) |
| UAijk = KA(θijk − θ0)2, | (8) |
It is worth noting that, by default, bonded beads do not directly interact with each other in LAMMPS (except via the bonded interaction). This differs from other DPD software available. One of the significant drawbacks of having bonded beads interacting is that the resulting bond length can be significantly larger than what is set using the equilibrium bond length l0 parameter. While bonded interactions can be switched on in LAMMPS, for the bulk of this work we choose to leave bonded beads as non-interacting for this reason. However, as discussed below, the resulting bond length is also slightly different from l0. We also note that it would be equally possible to leave bonded beads as interacting with each other, but this requires adjusting the setting of l0 accordingly to generate the correct effective distance between bonded beads. Given that this correction becomes dependent on a variety of factors, including the interaction aij parameter between the bonded beads themselves, this can be somewhat difficult in practice.
2.2 Determining aij values (cross interactions)
In the existing literature, the most common approach used to determine DPD interaction parameters aij is from their Flory–Huggins χ parameters, as first suggested by Groot and Warren.2 In Flory–Huggins theory the total free energy of mixing is given as | (9) |
| χ = 0.286Δa. | (10) |
Flory–Huggins parameters can be obtained from experimental measurement, but other developments have looked at alternative ways to calculate χ when it is not experimentally available. This includes using Hildebrand solubility parameters δ,
 | (11) |
While the original Flory–Huggins parameterisation approach has proven to be extremely popular across literature, alternative approaches are available. One example is that of Travis et al.33 where the conservative DPD interaction is based on regular solution theory (RST), which has the benefit of removing the restriction of equal repulsive interactions between like beads. Another is to determine aij using infinite dilution activity coefficients (IDAC),19,22 will will be the method of choice in our work.
The chemical potential  of a substance A in substance B is calculated as
of a substance A in substance B is calculated as
 | (12) |
 is the chemical potential of pure component A, γA is the activity coefficient, xA is the mole fraction of A in B, and the units of the chemical potential μ are energy per particle. The chemical potential can be calculated as a sum of two contributions
is the chemical potential of pure component A, γA is the activity coefficient, xA is the mole fraction of A in B, and the units of the chemical potential μ are energy per particle. The chemical potential can be calculated as a sum of two contributions| μ = μex + μid | (13) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) lnρΛ3 where Λ is the thermal de Broglie wavelength and ρ is the number density. μex is the excess chemical potential, calculated using the Widom insertion method.
lnρΛ3 where Λ is the thermal de Broglie wavelength and ρ is the number density. μex is the excess chemical potential, calculated using the Widom insertion method.
In the Widom insertion method, at each step, a ‘ghost’ particle is inserted at a random position in the simulation box. The energy difference ΔU between the system with and without this ghost particle is calculated, and the change in potential energy ΔU comes from the potential interactions of the ghost particle with the actual particles in the box. The excess chemical potential can then be derived from the probability of a particle insertion being favourable, which depends on the average Boltzmann-weighted energy cost of insertion. The excess chemical potential μex is therefore calculated as
 | (14) |
This approach has significant benefits over the method of relating Flory–Huggins parameters to their Δa value. Namely, the Groot and Warren2 relation was derived by performing a sweep in the range 8 ≤ Δa ≤ 25. Crucially, this relation was derived only for Δa values leading to phase separation, and therefore might not be reliable to determine interactions between components that are soluble within one another. The relation between Δa and χ also becomes non-linear at large values of Δa,35 which is not accounted for in eqn (10).
It was shown previously,36 that the Flory–Huggins χ-parameter which is calculated from simulated phase behaviour agrees very well with the free energy difference between a monomer surrounded by solvent particles, and a solvent particle surrounded by solvent particles (i.e. essentially an approximate IDAC), for particular cases. Another interesting study35 used the fact that, theoretically, one can relate the χ parameter in terms of excess chemical potentials36
 | (15) |
However, the approach of mapping to IDACs is reliant on the ability to correctly calculate the excess chemical potential μMex of a molecule, which is the main consideration of the current work. Furthermore, it is worth noting that the method relies on the average 〈exp(−ΔU/kBT)〉, which can be computationally sensitive. If ΔU varies widely (such as for molecules of bonded beads), the exponent can lead to a broad distribution, which can make accurate averaging difficult due to large variances. Therefore, a high number of insertions is required to converge to a reliable μex.
2.3 Effects of bonding
It is often assumed in the literature that one can determine the interactions of the beads individually (i.e. each bead represents a fragment of the molecule) and that bonding them together is a good representation of the molecule as a whole. From the point of view of using the Widom insertion technique to map to IDACs, this is equivalent to suggesting that the sum of the excess chemical potentials μSex of beads which are bonded into a chain is equivalent to the total excess chemical potential for the whole molecule μMex, i.e. | (16) |
While there are authors who have tried to quantify the effect that bonding has on the system properties and behaviours,37,38 to the best of our knowledge, one of the only studies in existing literature which attempts to take into account the impact of bonding on solubility is that of Saathoff.31 Saathoff31 argues that, due to the overlapping nature of DPD beads, eqn (16) does not hold and the resulting chemical potential of bonded molecules is lower than expected. Therefore, corrections must be applied to μex to produce the desired value of lnγ. Saathoff31 achieved this by performing a series of simulations in which μex was calculated for bonded pairs of beads in a solvent, while the length of the bond was systematically varied. The resultant data was fitted to determine expressions to apply corrections to μex. However, here we argue that the corrections required can be accounted for by simply calculating the expected volume of the beads, which are overlapping (as a function of bond length), rather than depending on fitted expressions.
3 Calculating the chemical potential
In this section, we first consider how one might determine the excess chemical potential via Widom insertion for single, unbonded beads. Then we discuss our theoretical approach for how one might calculate the chemical potential of a series of bonded beads, taking into account their overlapping volumes as a result of bonding.3.1 Single beads
To find a general relationship between aij and the excess chemical potential μex for single beads, we perform a parameter sweep in the range 10 ≤ aij ≤ 100, with an interval of Δaij = 5. Following the standard approach,2 we set all self-interactions to aii = 25. These simulations are conducted in the const-NVT ensemble, in a cubic box with periodic boundaries and edge length 20RC. The beads are initialised with a random initial configuration using n = 24000 simulation beads (box density ρ = 3RC−3).
From performing Widom insertions of single beads into a bath of non-bonded beads we determine how μex depends on the choice of cross-interaction parameter aij, which is shown in Fig. 1. We find that at higher values of aij the relation between aij and μex can be described linearly, while at lower values it cannot.
 | ||
| Fig. 1 Calculated relationship between the excess chemical potential μex and interaction parameter aij for the Widom insertion of single beads, for a solvent which interacts with aii. | ||
Therefore for aij < 70, we fit an expression of the form
| μex = Aaij3 + Baij2 + Caij | (17) |
It is worth noting that the fit shown in Fig. 1 is strictly speaking only valid for when aii = 25, and will vary slightly for different values of the solvent interaction. While intuitively one might expect eqn (17) to hold for all aii (due to the bulk density being set as ρ = 3RC−3 in all cases), a change in μex results from slight differences in the radial distribution function at different aii. Examples of other solvent aii interactions can be found in the ESI.† However, in the partitioning calculations presented in this article, we only use aii = 25 for single beads. We will show in Section 4.3 that sometimes it is necessary to use a different self-interaction value for the solvent when the beads are bonded, however, we will also show that eqn (17) still holds for the cases studied in this article.
3.2 Bonded beads
Suppose you have two overlapping spheres each with radius R which are separated by d, the volume of the overlapping portion is | (18) |
 | (19) |
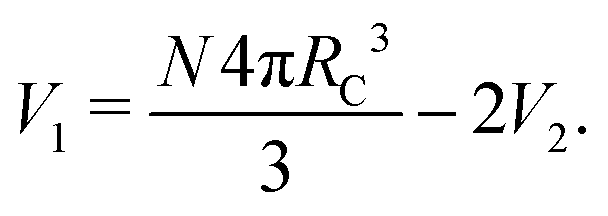 | (20) |
 | (21) |
If l0 < RC, then for molecules of length N ≥ 3, there is the possibility of three overlapping spheres, as illustrated in Fig. 2.
 | ||
| Fig. 2 Overlapping beads when l0 < RC, for cases where N = 2 (a) and N = 3 (b). | ||
The volume of the region where three beads overlap can be calculated as
 | (22) |
 | (23) |
 | (24) |
 | (25) |
One simplification we make in our approach is that we use the bond length setting in eqn (7) to determine the distance between beads, and hence calculate the overlap. In practice, over the simulation period the bond length will fluctuate. In dissipative particle dynamics, there is normally a relatively broad distribution of bond lengths, depending on the strength chosen for the bonding. This means that the volume of overlap fluctuates also. Given that the average volume of overlap is not exactly the same as that determined from the average bond length (since volume scales with l03), our approach is a slight approximation. However, we show in this article that simply using l0 for the volume calculation is still an effective method for correcting the chemical potential of the beads.
One further simplification that we have made is to derive eqn (25) under the assumption that conformational effects can be neglected for relatively small molecules. However, it is worth noting that as the molecule grows in size, conformational effects may become more important. This is likely to be true for longer polymers where parts of the solute molecule have the capacity to fold in on themselves and overlap.
4 Partition coefficients
In this section, we test our expressions for the excess chemical potential by applying them in the prediction of partition coefficients and comparing with simulated values. This approach stems from the fact that we can relate the excess chemical potential (predicted via Widom insertion) with the resulting partition coefficient of a solute into two immiscible liquids, a relation which will be presented in this section.We simulate a box containing two immiscible phases and insert a small amount of solute into the system. This allows us to vary the interaction parameters between the solute and the two solvent phases to extensively test eqn (21) and (25). For solute partitioning into two solvents α and β, the partition coefficient is defined as
 | (26) |
At equilibrium, the chemical potentials of the solute in the two solvents are equal. By making use of this fact, as well as decomposing the chemical potential into its excess and ideal parts (eqn (13)), this allows us to express the partition coefficient K at infinite dilution in terms of the difference between the excess chemical potentials of the two phases39
 | (27) |
In this section, we compare the partition coefficients of single bead solutes (Section 4.1), with those for bonded molecules (Section 4.2). Following this, we also investigate the effects of bonding the solvent molecules on K (Section 4.3). In all cases, we perform simulations consisting of two-phase separating solvents (type A and type B beads), defining the partition coefficient as K = cA/cB. To ensure their phase separation we set their interaction to be aAB = 100 which results in a negligible solubility of type A beads in type B and vice versa. We set all self-interactions to be aii = 25, unless specified otherwise. All simulations are conducted in boxes of size 60 × 20 × 20 with a bead density of ρ = 3RC−3. We refer to our solute beads as type C. The phase separation produced by types A and B (in the absence of solute beads) is illustrated in Fig. 3.
 | ||
| Fig. 3 Phase separation of beads of type A (red) and type B (blue) when their DPD interaction values are set as: aAB = 100, aAA = 25 and aBB = 25. | ||
4.1 Non-bonded solute
In order to first test the applicability of eqn (27), we perform a set of simulations in which DPD beads are unbonded, single beads. We examine the effects of varying the strength of the interaction with the solute beads (type C) and examine the partitioning for varying choices of aAC and aBC. We find that the value of K calculated is weakly dependent on the solute concentration, as shown in Fig. 4 (which is to be expected as we have assumed infinite dilution). Therefore, we find the value of K at infinite dilution by extrapolating to a concentration of 0, in order to directly compare with that predicted using eqn (27). The results of this study are shown in Table 1, showing good agreement between the theoretically predicted value and the simulated values. This also provides confidence in the calculated relationship between aij and the chemical potential which is shown in Fig. 1. | ||
| Fig. 4 The partitioning (K) of unbonded solute beads (C type) in unbonded solvent beads (A and B type). There is a weak dependence of lnK on the concentration of the solute. | ||
| a AC | a BC | lnK (calculated using eqn (27)) |
lnK (simulated) |
|---|---|---|---|
| 25 | 35 | 3.22 | 3.244 (0.016) |
| 25 | 40 | 4.56 | 4.697 (0.007) |
| 30 | 35 | 1.71 | 1.528 (0.008) |
| 30 | 40 | 2.86 | 2.938 (0.026) |
| 40 | 60 | 4.08 | 4.267 (0.058) |
| 60 | 70 | 1.56 | 1.638 (0.052) |
4.2 Bonded solute
We next investigate the effects of bonding the solute molecules together, while keeping the solvent as single beads. We investigate for solute molecules with various lengths (N = 2, 3, 4). The bond length l0 is also expected to play a role in solubility, as this significantly influences the degree of overlap between bonded beads. Therefore we test three bond lengths l0 = 0.4RC, l0 = 0.6RC and l0 = 1.0RC. Similarly to in Section 4.1, we simulate various solute concentrations and extrapolate to zero solute concentration to determine a value for lnK.
If we assume that the chemical potential for the molecules can be calculated using eqn (16) (i.e. the overlap is not taken into account), Fig. 5a shows the relationship between the simulated, and predicted values for lnK. We observe that the deviation between the predicted and simulated values is largest when the bond length is shorter, and also increases as the value of K increases. This is hypothesised to be due to the increasing volume of overlap between the bonded beads at shorter bond lengths.
 | ||
| Fig. 5 Relationship between the predicted and simulated values of lnK. The predicted values are calculated assuming that the excess chemical potential of bonded molecules can be calculated using (a) eqn (16) (i.e. uncorrected) and (b) eqn (21) (i.e. corrected). The symbol represents the number of bonded beads in the molecule: 2 (+), 3 (○) and 4 (Δ), while the colour represents the bond length l0: 0.4RC (green), 0.6RC (red) and 1.0RC (black). The unbonded single-bead solute is represented by the blue (*) points. Note that error bars are smaller than the symbol size, but values can be found in the ESI.† | ||
However, if we calculate the predicted value of lnK using our volume-correction equations (eqn (21) and (25)) the relationship between the simulated and predicted values greatly improves, as shown in Fig. 5b. To quantify the degree of improvement, we calculate mean squared error values for the two different bond lengths before and after applying the correction, which are shown in Table 2. We note the significant improvement for the shorter bond lengths of l0 = 0.4RC and l0 = 0.6RC, while there is minimal change when l0 = 1RC. Note that all values shown in Fig. 5a and b can be found in the ESI.†
K between the predicted and simulated values when using the uncorrected form of the chemical potential vs. the corrected form. Note that bond length is in units of RC
| K prediction | Bond length | MSE |
|---|---|---|
| Uncorrected | 0.4 | 2.35 |
| Corrected | 0.4 | 0.030 |
| Uncorrected | 0.6 | 1.19 |
| Corrected | 0.6 | 0.070 |
| Uncorrected | 1.0 | 0.072 |
| Corrected | 1.0 | 0.112 |
4.3 Bonding the solvent
In this section, we investigate the effect of bonding the solvent. We perform simulations in which we determine lnK for a single bead solute (type C), which partitions into two solvents where one of those solvents consists of bonded beads (type A) and the other solvent remains unbonded (type B). Similarly to before, we investigate the effects of the number of bonded beads in the molecule, as well as the bond length.
Fig. 6a shows the predicted lnK vs. the simulated values for a variety of choices for aAB and aAC, where all self-interactions are kept as aii = 25. We see a small difference between the simulated and the predicted values. We hypothesise that, due to the bonding of the solvent, there is a difference in K which results from the change in density that bonding produces. The act of bonding molecules together decreases the pressure of the system, therefore increasing the equilibrium density, resulting in larger excess chemical potentials than those determined in Section 3.1 (eqn (17)). This pressure decrease results from the fact that bonding reduces the number of interacting particles in the system.
 | ||
| Fig. 6 Comparison between the predicted and simulated lnK values for varying aAB and aAC choices. Fig (a) shows the partitioning when all self-interactions are set to aii = 25.0, while (b) shows the partitioning behaviour when the self-interaction of the bonded beads (aAA) is corrected based on the pressure. Note that error bars are smaller than the symbol size, but values can be found in the ESI.† | ||
We aim to compensate for this by altering the self-interaction value for the bonded solvent. The relationship between the number of bonded beads and the pressure is shown in Fig. 7. We calculate this pressure by performing simulations in a cubic domain of length L = 20RC with a bead density of 3RC−3. For a given bonded solvent, we choose to set the self-interaction aAA to reproduce the same pressure as that of an unbonded solvent (for which we find a pressure of P = 23.7). This is to ensure that in all cases the pure solvent produces a bead density of ≈3RC−3, when simulated in coexistence with the unbonded solvent. We find that this approach works well for cases where a bond length of l0 = 0.6RC and l0 = 1RC are used, however, is not a perfect approach for correcting the density when l0 = 0.4RC. In this case, we suspect that other factors need to be taken into account to perfectly correct the density upon bonding. Therefore in this section, we focus on bond lengths l0 = 0.6RC and l0 = 1RC. The corrected aAA values used for the simulations in this section are presented in Table 3. Fig. 8 provides an example of how the solvent density changes due to this updated self-interaction value.
 | ||
| Fig. 7 Bulk pressure in simulations with number density 3, where the bond length l0 and the number of bonded beads N is varied. Also shown (black) is the bulk pressure of unbonded beads at the same number density. | ||
| N | Bond length | a AA |
|---|---|---|
| 2 | 0.6 | 27.9 |
| 3 | 0.6 | 28.9 |
| 4 | 0.6 | 29.4 |
| 2 | 1.0 | 26.6 |
| 3 | 1.0 | 27.2 |
| 4 | 1.0 | 27.5 |
 | ||
| Fig. 8 Example of the improvement of the bead density when the self-interaction for bonded beads is altered. Example for when four A-type molecules are bonded together with equilibrium bond length l0 = 0.6RC. Type-B beads are left unbonded and aAB = 100. In the original case aAA = aBB = 25, while in the corrected case aAA = 29.4 and aBB = 25.0. | ||
We note here that eqn (17) was calculated by performing Widom insertions into a solvent of unbonded beads with aii = 25. We expect the relation between μex and aij to alter slightly depending on the solvent parameters i.e. if aii ≠ 25, or if the solvent is bonded (or not). However, we find a negligible difference between the fit presented in eqn (17), and the fit for when the solvent is bonded and has the aij values we use in this article. Further information supporting this is presented in the ESI.†
Fig. 6b shows the new predicted vs simulated relationship for lnK. We see that the updated calculated values more closely match the simulated results once the self-interaction for the bonded solvent is updated. Similarly to previously, we calculate the mean squared error (MSE) for the uncorrected and corrected cases, which is shown in Table 4, where we observe that the MSE increases with more bonds in the solvent and that the correction greatly decreases the MSE between the simulated and predicted values of lnK. Note that all values shown in Fig. 6 can be found in the ESI.†
K (compared with the calculated value using eqn (25)) when the self-interaction of the solvent is corrected to account for the density change of the solvent upon bonding
| N (solvent) | Bond length | MSE (uncorrected) | MSE (corrected) |
|---|---|---|---|
| 2 | 0.6 | 0.344 | 0.010 |
| 2 | 1.0 | 0.378 | 0.131 |
| 4 | 0.6 | 0.857 | 0.024 |
| 4 | 1.0 | 0.900 | 0.176 |
It may seem counter-intuitive that the effects of bonding the solute molecules appear more significant than the effects of bonding the solvent molecules. However, this conclusion is expected from a theoretical point of view. In the case of the solvent, the effect of bonding beads does not significantly alter the degree of overlap, as in a bulk solvent of single beads there is expected to be a degree of overlap anyway. This contrasts with what is discussed in Section 4.2, for solutes, where the correction applied in calculating μMex accounts for a difference between beads which have no overlap at all, and those which overlap because they are bonded.
This point can be illustrated by considering the effect that bonding has on the radial distribution function of the fluid. We simulate a selection of pure systems in cubic simulation boxes, for which the radial distribution function g(r) is shown in Fig. 9. The simulations are conducted in boxes with edge length L = 20RC and density ρ = 3RC−3. Therefore, the only differences between the three cases shown is in the choice of bonding, and the repulsion parameter aii. As previously, we choose to set this so that the pressure is the same in all simulations (the values used in Fig. 9 can be found in Table 3).
 | ||
| Fig. 9 Radial distribution function g(r) for a variety of simple test cases. In the cases of bonded simulations, we use an equilibrium bond length of l0 = 0.6RC for molecules of two different lengths N. Also illustrated vertically are integer values of the equilibrium bond length. | ||
For a system consisting of single beads, if our beads were non-interacting, the average distance d between beads could be calculated as
 | (28) |
The process of bonding leads to the appearance of peaks at approximately integer values of the equilibrium bond lengths. However, bonding does not significantly alter the average overlap volume in the systems. Upon bonding, there is effectively a narrowing of the distribution of bead distances. The system loses some of its closest beads (reflected by only a significant decrease in g(r) for r < 0.5RC) but also some that would otherwise have been slightly further apart (g(r) decrease in the range 0.7RC < r < 0.9RC).
Finally, we also test systems with both bonded solute and bonded solvent, to test combining the bonding effects discussed thus far. Exact details of this study can be found in the ESI,† however, we find that we can combine the corrections discussed thus far to make good predictions for the expected chemical potential, and hence partitioning.
4.4 Solute–solute interactions
Until now, the examples studied considered solute molecules consisting of a single type of bead. However, a solute molecule could potentially be made up of various bead types. In this section, we briefly discuss the bonding overlap effects which would need to be considered if one wishes to define a full set of interactions for more complex systems.If we consider an example system where the solute molecule is made up of three beads and a fourth solvent bead (Fig. 10), there are 6 cross interaction aij values to determine. Three of those interactions are related to how the solute interacts with the solvent (aAD, aBD and aCD). In this case, the values are best determined by aiming to reproduce the correct activity coefficients of each of the solute beads at infinite dilution in the solvent (assuming that the system to be simulated consists of significantly more solvent molecules than solute). There are, however, three other cross-interaction values that are between the solute molecules themselves (aAB, aAC and aBC). In this case, one could choose to determine the cross-interaction in two different ways depending on which bead is chosen for the Widom insertion. For example, aAB could be determined by matching to the IDAC of bead A in bead B, or vice versa.
 | ||
| Fig. 10 Simple example system of a solute consisting of three bead types (A)–(C) in a solvent (D). | ||
Upon bonding of the solute beads, bead overlaps lead to an effective change in the IDAC coefficient, as discussed previously. However, unlike in Section 4.2, the effective interaction (as a result of bonding) has changed for both beads involved in the interaction. Therefore, we now consider the effect that bonding has on the solute–solute interactions.
Fig. 11 summarises the bonding effects addressed in Section 4.2 (interactions between a bonded solute and the solvent) and Section 4.3 (interactions between a solute and a bonded solvent). In this figure, we also illustrate the effects addressed in this section between the bonded solute molecules.
 | ||
| Fig. 11 Effect of bonding (a) the solute on its interaction with the solvent (Section 4.2); (b) the solvent on its interaction with the solute (Section 4.3); (c) the solute on its interaction with other solute molecules (Section 4.4). | ||
If, for example, we consider the interaction between bead A (left-hand molecule) and bead B (right-hand molecule) in Fig. 11c, this could be determined via matching to the IDAC of bead A in a bath of bead B. In this case, then the effect of bonding on the first molecule can partially be taken into account in the same way as described in Section 4.3, by altering the interaction to account for its overlap with its bonded neighbour. However, since bead B in the second molecule is also overlapping with its neighbours there is an extra impact of bonding.
However, the effects of bonding on the second molecule cannot necessarily be accounted for in the same manner as the first. As the interaction was determined via the insertion of a single bead into a bath of beads, there was already a degree of overlap of the B-type beads in the bath. Now that the molecule has been bonded to its neighbours, the degree of overlap has changed very little, the impact being the type of bead it is overlapping with. This is similar to the effects of bonding the solvent discussed in the previous section.
For example, if we refer again to our example case, we can determine the effect the overlap of the second solute molecule will have on the excess chemical potential μex. If we assume that the average volume of overlap VOL between a pair of beads is unchanged before and after bonding, then
 | (29) |
 which is relatively small.
which is relatively small.
However, we also note that for the cross-interaction values that are between the solute molecules, it may be best to determine the aij value for each pair as an average of the two IDAC values from one bead in another. For example, one will determine a different value calculating aAB depending on if one matches to the IDAC of bead A in bead B, or visa versa. Therefore, a potential choice is to determine both and take the average value i.e. . This is likely to be an effective approach if the two approaches yield similar interaction values (that is aA→B and aB→A are fairly similar).
. This is likely to be an effective approach if the two approaches yield similar interaction values (that is aA→B and aB→A are fairly similar).
5 Altering intramolecular interactions
As briefly mentioned in Section 2.1, by default LAMMPS does not allow beads which are directly bonded to interact. However, this behaviour can be switched on using the ‘special bonds’ function, and here we investigate the impact that switching these interactions on has on our results. This is motivated by the fact that many popular DPD codes, such as DL_MESO,40 choose to allow this interaction.We repeat a small subset of our calculations from Section 4.2 (a bonded solute in an unbonded solvent), but allowing the bonded beads to also interact with each other via the usual DPD repulsive force. The first noticeable difference between the two cases is the average bond length. In our original simulations, although the equilibrium bond length is set as l0 = 0.6RC, the average resulting bond length in the simulation is 0.58RC. This decrease, compared to the l0, is due to the pressure of the system. However, if desired, we could force the bond length to be closer to the value set in eqn (7) by increasing the strength of the bonding potential parameter KB. In our repeated simulations with bonded interactions turned on, we find that the average bond length in the system increases to 0.61RC. This increase is due to the added interparticle repulsion between two bonded beads. Details of these calculations of apparent bond length can be found in the ESI.†
A comparison of the partitioning lnK for the two cases is shown in Fig. 12, where the simulation data is summarised in the ESI.† Due to the slight change in bond length, one might expect to see a difference in the resulting partitioning due to the change in the degree of overlap. However, despite the slight change in average bond length, there is no significant change in partitioning within the error of the simulated value of lnK. If we consider eqn (19) we can determine that the change in the overlap between two beads would only vary by ≈4% upon the bond length changing from 0.58RC to 0.61RC, which would not result in a significant change in the chemical potential, likely indicating why we see very little variation between the two simulation cases in Fig. 12.
 | ||
| Fig. 12 A comparison of the partitioning lnK which results when the bonded interactions are turned on and turned off. The symbol represents the number of beads in the bonded solvent: 2 (+) and 3 (○). The equilibrium bond length in all cases is set as l0 = 0.6. Note that error bars are smaller than the symbol size, but values can be found in the ESI.† | ||
We also now consider the effect that including bonded interactions has on a bonded solvent, repeating the pressure calculation originally shown in Fig. 7. Due to the increase in the number of interactions in the fluid, turning the bonded interactions on alters the relationship between the pressure and the self-interaction value. The new aii value required to produce the same pressure as a non-bonded solvent is shown in Table 5 (full figure included in the ESI†). Comparing Table 3 and, we see that altering the nature of bonded bead interactions alters the pressure when the bond length is shorter, but not when it is longer. This is because the longer bond length of l0 = 1RC is the same as the cut-off, RC, and so beads have negligible interaction with each other even when bonded interactions are switched on. However, the difference in pressure for shorter bond lengths can be accounted for when setting the self-interactions (as discussed above). Therefore, we conclude that the results presented in this paper are applicable to systems with bonded beads with excluded bead interactions turned on or off.
| N | Bond length | a AA |
|---|---|---|
| 2 | 0.6 | 27.5 |
| 3 | 0.6 | 28.3 |
| 4 | 0.6 | 28.6 |
| 2 | 1.0 | 26.6 |
| 3 | 1.0 | 27.2 |
| 4 | 1.0 | 27.5 |
6 Application to real systems
In this section, we briefly demonstrate the applicability of our model to real systems by studying the partitioning of three different molecules into a water/dodecane system. The coarse-graining of the solutes, along with the representation for dodecane, are shown in Fig. 13. The coarse-grained modelling for these three systems is very different, highlighting the versatility of the method for real molecules. | ||
| Fig. 13 Coarse-graining used for various real molecules simulated in this section. Note that the beads are for illustrative purposes only and do not indicate the shape or size of the beads nor the degree of the overlap. | ||
For each solute we calculate the interaction parameters aij using our equations to correct μex due to their overlap. We then determine the resulting partition coefficient K, and compare with experimental data. In each case, we also calculate another set of interaction parameters aij for if we were to ignore the overlap of the beads, to show the improvement that our method has over this approach.
6.1 Dodecane and water solvents
We use a single water bead to represent multiple water molecules, and dodecane is modelled by four bonded beads of the same type. In our simulations, we use a constant bead density of ρ = 3RC−3. Therefore we can find a value RC (and a means for conversation into real units) by matching to the experimentally known density of dodecane at room temperature. This approach leads us to determine RC = 6.56 Å. From this, we define the coarse-graining of water NW by also matching to the density of water at room temperature, finding that 1 water bead represents NW ≈ 3.14 water molecules.To determine an appropriate bond length for the dodecane molecule, we use the fact the C–C–C angles are approximately 109.5°, and that the experimental C–C bond is 1.543 Å.41 The separation between three carbon atoms is therefore approximately 3.77 Å, meaning an equilibrium bond length of 0.6RC is a good choice.
6.2 Solutes
As shown in Fig. 13, diethyl carbonate can be represented by two beads of the same type, due to its symmetry. This means that eqn (19), (20), and (21) can be directly applied to account for the overlap. For diethyl carbonate, we use an equilibrium bond length of 0.6RC, which converts to a realistic bond length for this molecule.For simulating heptanol, we need to use two different types of beads. This means that we need to calculate the volume of the overlapping portions separately, rather than using eqn (19) and (20). Then we correct each bead's μex individually, due to the overlapping portions. However, practically this is simple and not much more complicated than our previous equations (which assume each bonded bead interacts with the solvent with the same aij parameter). Once again, we use an equilibrium bond length of 0.6RC, since this is representative of the real bond length.
Finally, we model the benzene as a triangle of bonded beads, where an angle constraint is imposed at every corner, and the equilibrium angle is set to 60°. Using that the average C–C bond in a benzene molecule is 1.39 Å, we determine the separation between bonded beads should be 2.41 Å, meaning an equilibrium bond length of 0.4RC is a good choice.
The choice to model benzene using three beads in a triangle formation has been used in other DPD models24 and makes sense from a geometric point of view. However, this formation of beads has a very large degree of overlap and so we expect that in most previous author's models, the chemical potential of rings is not fully reflected. Using our method to calculate μex requires us to know the degree of overlap each bead has with one another. Our previous expressions for volume are based on a linear construction of the molecule, therefore we need another approach. The analytical expression for the volumes of overlap in a ring is quite complex compared to the cases used in the article so far. However, the volume of overlap can very easily be calculated using a Monte Carlo simulation. Therefore eqn (25) can still be applied to calculate the excess chemical potential, once these volumes have been found. Details of this calculation, including the volumes V1, V2 and V3 for different bond lengths, can be found in the ESI.†
The target values for excess chemical potential μ are obtained from experimental data. The exact aij values used in each simulation case can be found in the ESI,† along with details of the experimental data used and additional information about these simulations.
6.3 Partitioning results
Table 6 shows the results of our partitioning study for each molecule. We show the values of lnK obtained when the overlap of beads is not taken into account (uncorrected) when calculating the aij interaction parameters for the molecule, compared to when we use our approach to account for the overlap (corrected) when determining suitable aij values.
K in the cases where the volume of overlap is not accounted for (uncorrected) and it is accounted for using our method (corrected). Experimental lnK are obtained from ref. 42, where K is defined as the ratio of the concentration in the dodecane to the water
| Solute | Simulated lnK (uncorrected) |
Simulated lnK (corrected) |
Experimental lnK |
|---|---|---|---|
| Diethyl carbonate | 0.817 | 1.168 | 1.3 |
| Heptanol | 0.484 | 1.965 | 1.9 |
| Benzene | 2.542 | 4.475 | 5.1 |
We observe a significant improvement in our corrected parameters compared to the uncorrected ones, and we see this for all solute cases tested. This is particularly notable for our benzene representation. For all solutes, the use of uncorrected parameters leads the solute molecules to behave with significantly less hydrophobicity than they should. However, correcting the parameters for the overlap generates a much more realistic behaviour. Generating the correct behaviour, and in particular the correct hydrophobicity, is crucial for many types of systems, but in particular for self-assembling systems such as surfactants.
7. Conclusions and discussion
In this article, we have shown that there are effects resulting from the bonding of beads, which are generally not considered in the parameterisation of DPD models. However, we show that these bonding effects can be compensated for by considering the degree of overlap between bonded beads. The degree of overlap depends on the chosen bond length, and shorter bond lengths lead to the need to consider the effects of bonding more carefully. Although, as highlighted in Section 5, the average bond length is slightly different to its harmonic potential setting (i.e. that set by eqn (7) using parameter l0), we show that this deviation is small enough that we can use l0 to estimate the volume of overlap of the beads, and hence correct the chemical potential.We have also considered more complex cases, where molecules can be made up of a combination of fragment types. While we have only considered linearly bonded molecules in this work with a single bond length, the methodology presented could easily be extended to consider more complex molecules, including those containing rings or branching etc.
Finally, it is worth considering the general applicability of the results presented in this work. In recent years, DPD has become a very versatile tool for the simulation of soft matter systems. Accordingly, we expect the method presented here to find uses in the simulation of surfactants, where accurate parametrization is required to obtain predictions of interfacial tensions between fluids20,43 and for predicting critical micelle concentrations;21,25 for model membranes where partitioning of small molecules through the membrane is strongly dependent on the accurate parametrization of the interactions with lipid molecules;44 for DPD studies of peptides and proteins45 where folding depends critically on accurate monomer–monomer aij values; in addition to a wide variety of other problems.
Author contributions
RH: conceptualization, data curation, formal analysis, investigation, methodology, software, validation, visualization, writing – original draft. CA: project administration, writing – review and editing. MW: funding acquisition, project administration, supervision, writing – review and editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the support of Durham University for the use of its HPC facility, Hamilton. RH would like to acknowledge support through an EPSRC/P&G funded Prosperity Partnership grant EP/V056891/1.Notes and references
- P. J. Hoogerbrugge and J. M. V. A. Koelman, Europhys. Lett., 1992, 19, 155–160 CrossRef.
- R. D. Groot and P. B. Warren, J. Chem. Phys., 1997, 107, 4423–4435 CrossRef CAS.
- P. Español and P. Warren, Europhys. Lett., 1995, 30, 191–196 CrossRef.
- R. D. Groot, J. Chem. Phys., 2003, 118, 11265–11277 CrossRef CAS.
- C. Marsh, G. Backx and M. Ernst, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1997, 56, 1676–1691 CrossRef CAS.
- T. L. Rodgers, O. Mihailova and F. R. Siperstein, J. Phys. Chem. B, 2011, 115, 10218–10227 CrossRef CAS PubMed.
- Z. Li and E. E. Dormidontova, Macromolecules, 2010, 43, 3521–3531 CrossRef CAS.
- M. Lísal and J. K. Brennan, Langmuir, 2007, 23, 4809–4818 CrossRef PubMed.
- A. G. Schlijper, P. J. Hoogerbrugge and C. W. Manke, J. Rheol., 1995, 39, 567–579 CrossRef CAS.
- A. A. Gavrilov, Y. V. Kudryavtsev and A. V. Chertovich, J. Chem. Phys., 2013, 139, 224901 CrossRef PubMed.
- K. Zhang and C. W. Manke, Mol. Simul., 2000, 25, 157–166 CrossRef.
- S. Chen, N. Phan-Thien, X.-J. Fan and B. C. Khoo, J. Non-Newtonian Fluid Mech., 2004, 118, 65–81 CrossRef.
- S. Wang, S. Yang, R. Wang, R. Tian, X. Zhang, Q. Sun and L. Liu, J. Pet. Sci. Eng., 2018, 169, 81–95 CrossRef.
- E. Mayoral and A. G. Goicochea, J. Chem. Phys., 2013, 138, 094703 CrossRef.
- Y.-D. He, Y.-H. Tang and X.-L. Wang, J. Membr. Sci., 2011, 368, 78–85 CrossRef.
- R. Groot and K. Rabone, Biophys. J., 2001, 81, 725–736 CrossRef PubMed.
- S. Sengupta and A. Lyulin, Polymers, 2020, 12, 907 CrossRef.
- T. Ni, G.-S. Huang, P. Gao, Y.-T. Xu and M.-Z. Yang, Polym. J., 2011, 43, 635–641 CrossRef.
- A. Vishnyakov, M.-T. Lee and A. V. Neimark, J. Phys. Chem. Lett., 2013, 4, 797–802 CrossRef PubMed.
- X. Wang, K. P. Santo and A. V. Neimark, Langmuir, 2020, 36, 14686–14698 CrossRef.
- M.-T. Lee, R. Mao, A. Vishnyakov and A. V. Neimark, J. Phys. Chem. B, 2016, 120, 4980–4991 CrossRef.
- R. L. Hendrikse, C. Amador and M. R. Wilson, Soft Matter, 2023, 19, 3590–3604 RSC.
- R. L. Hendrikse, C. Amador and M. R. Wilson, Soft Matter, 2024, 20, 6044–6058 RSC.
- R. L. Anderson, D. J. Bray, A. S. Ferrante, M. G. Noro, I. P. Stott and P. B. Warren, J. Chem. Phys., 2017, 147, 094503 CrossRef.
- R. L. Anderson, D. S. D. Gunn, T. Taddese, E. Lavagnini, P. B. Warren and D. J. Bray, J. Phys. Chem. B, 2023, 127, 1674–1687 CrossRef PubMed.
- R. L. Anderson, D. J. Bray, A. Del Regno, M. A. Seaton, A. S. Ferrante and P. B. Warren, J. Chem. Theory Comput., 2018, 14, 2633–2643 CrossRef PubMed.
- M. Panoukidou, C. R. Wand, A. Del Regno, R. L. Anderson and P. Carbone, J. Colloid Interface Sci., 2019, 557, 34–44 CrossRef.
- R. L. Hendrikse, A. E. Bayly and P. K. Jimack, J. Phys. Chem. B, 2022, 126, 8058–8071 CrossRef PubMed.
- D. J. Bray, R. L. Anderson, P. B. Warren and K. Lewtas, J. Chem. Theory Comput., 2020, 16, 7109–7122 CrossRef PubMed.
- D. J. Bray, R. L. Anderson, P. B. Warren and K. Lewtas, J. Phys. Chem. B, 2022, 126, 5351–5361 CrossRef PubMed.
- J. Saathoff, J. Chem. Phys., 2018, 148, 154102 CrossRef PubMed.
- H. Rezaei and H. Modarress, Chem. Phys. Lett., 2015, 620, 114–122 CrossRef.
- K. P. Travis, M. Bankhead, K. Good and S. L. Owens, J. Chem. Phys., 2007, 127, 014109 CrossRef.
- E. Mayoral and E. Nahmad-Achar, J. Chem. Phys., 2012, 137, 194701 CrossRef PubMed.
- T. P. Liyana-Arachchi, S. N. Jamadagni, D. Eike, P. H. Koenig and J. I. Siepmann, J. Chem. Phys., 2015, 142, 044902 CrossRef PubMed.
- C. M. Wijmans, B. Smit and R. D. Groot, J. Chem. Phys., 2001, 114, 7644–7654 CrossRef.
- M. R.-B. A. Gama Goicochea and R. López-Rendón, Mol. Phys., 2007, 105, 2375–2381 CrossRef.
- M.-T. Lee, A. Vishnyakov and A. V. Neimark, J. Phys. Chem. B, 2013, 117, 10304–10310 CrossRef.
- A. S. Paluch, S. Parameswaran, S. Liu, A. Kolavennu and D. L. Mobley, J. Chem. Phys., 2015, 142, 044508 CrossRef.
- S. M. Michael, A. Seaton, R. L. Anderson and W. Smith, Mol. Simul., 2013, 39, 796–821 CrossRef.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1–S19 RSC.
- M. H. Abraham and W. E. Acree Jr., New J. Chem., 2004, 28, 1538–1543 RSC.
- H. Alasiri and W. G. Chapman, J. Mol. Liq., 2017, 246, 131–139 CrossRef.
- H.-M. Ding, W.-D. Tian and Y.-Q. Ma, ACS Nano, 2012, 6, 1230–1238 CrossRef PubMed.
- A. Vishnyakov, D. S. Talaga and A. V. Neimark, J. Phys. Chem. Lett., 2012, 3, 3081–3087 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp03791j |
| This journal is © the Owner Societies 2025 |