
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unusual mechanism of aziridine biosynthesis catalysed by the αKG-dependent non-heme enzyme TqaL†
Warispreet
Singh
*abc and
Meilan
Huang
 *a
*a
aDepartment of Chemistry & Chemical Engineering, Queen's University, Belfast BT9 5AG, UK. E-mail: m.huang@qub.ac.uk; w.singh@northumbria.ac.uk
bDepartment of Applied Sciences, Northumbria University, Newcastle upon Tyne, NE1 8ST, UK
cHub for Biotechnology in Build Environment, Newcastle upon Tyne, UK
First published on 2nd April 2025
Abstract
Aziridines are present in many synthetic pharmaceuticals. The synthesis of the aziridine ring remains challenging due to its highly strained three-membered ring structure. Recently, a non-heme αKG-dependent enzyme, TqaL, has been demonstrated to catalyze the synthesis of aziridines from L-Val. However, the detailed reaction mechanism of the enzyme remains elusive. Herein, we reported, for the first time, the mechanism of oxidative cyclisation for aziridine synthesis catalyzed by TqaL. Following the HAA step, the reaction proceeded via a unique concerted process with a single electron transfer from the isopropyl radical to the Fe(III)–OH motif, which was coupled with the electrophilic attack of the primary amine substrate on the tertiary isopropyl radical and simultaneous proton transfer from the substrate amine to the hydroxyl group of the Fe(III)–OH to give the aziridine. This research would provide a valuable structural basis for tailoring the non-heme αKG-dependent enzyme for the biosynthesis of highly active aziridine derivatives as pharmaceuticals.
1 Introduction
Asymmetric synthesis of three-membered nitrogen-containing aziridines is important for the pharmaceutical industry because it is a key building block for many therapeutic drugs with antimicrobial (azicemicins A and B1), antitumor and antimalarial activities. Notable examples include mitomycin C, a chemotherapy drug,2 and miraziridine A, a potent inhibitor of cathepsin B protease, which is overexpressed in various cancers.3 The synthesis of aziridines represents a challenge for organic chemists due to the high ring strain of the three-membered ring. The biosynthesis pathway of an aziridine natural product catalyzed by engineered heme P450 enzymes4,5 and non-heme iron alpha-ketoglutarate (αKG)-dependent enzymes6via bimolecular aziridination through a nitrene intermediate has been reported. The bimolecular aziridination catalyzed by iron-containing enzymes has been investigated by theoretical computations.7,8 However, to the best of our knowledge, the reaction mechanism for aziridine synthesis via unimolecular cyclisation catalyzed by αKG-dependent enzymes has never been reported, which has limited the ability to tailor the enzyme or discover other novel non-heme enzymes from nature for the synthesis of highly active aziridines.Recently, the biosynthesis of 2-aminoisobutyric acid by the oxidation of L-Val via an aziridine-containing intermediate, pleurocybellaziridine, was reported (Scheme 1). The intramolecular cyclisation is catalysed by the non-heme iron αKG-dependent TqaL enzyme.9 Traditionally, reactions catalysed by Fe(II)/αKG-dependent enzymes proceed through hydroxylation pathways;10,11 however, TqaL is a unique non-heme enzyme that catalyzes aziridination rather than hydroxylation. Mutagenesis studies suggested that Phe275 is critical for aziridination over hydroxylation, and mutation of Phe275 to Leu or Ala results in hydroxylated L-Valine as the major product.12 It was suggested that the aziridination is initiated by the abstraction of a hydrogen atom from the Cβ of L-Val to the Fe(IV)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O species.9,13 Transient kinetics, isotope experiments, combined with product analysis and mechanism probes, indicated that intramolecular C–N bond formation to install aziridine in L-Val occurs via a carbocation intermediate and excluded the possibility of obtaining the final aziridine product via a hydroxylated intermediate.11 These experimental hypotheses and evidence enable a detailed understanding of the reaction mechanism of the TqaL enzyme through computational modelling studies at the atomic level. However, in the reported crystal structure of TqaL (PDB: 7EEH), the Fe(IV)O motif, which is the main oxidant in many Fe(II)/αKG-dependent enzymes, is absent from the catalytic site, as is the L-Val substrate. The lack of information on how L-Val is positioned in relation to the Fe(IV)O oxidant has limited our understanding of the aziridination reaction catalysed by TqaL.
O species.9,13 Transient kinetics, isotope experiments, combined with product analysis and mechanism probes, indicated that intramolecular C–N bond formation to install aziridine in L-Val occurs via a carbocation intermediate and excluded the possibility of obtaining the final aziridine product via a hydroxylated intermediate.11 These experimental hypotheses and evidence enable a detailed understanding of the reaction mechanism of the TqaL enzyme through computational modelling studies at the atomic level. However, in the reported crystal structure of TqaL (PDB: 7EEH), the Fe(IV)O motif, which is the main oxidant in many Fe(II)/αKG-dependent enzymes, is absent from the catalytic site, as is the L-Val substrate. The lack of information on how L-Val is positioned in relation to the Fe(IV)O oxidant has limited our understanding of the aziridination reaction catalysed by TqaL.
 | ||
| Scheme 1 The biosynthesis of L-valine to 2-2-aminoisobutyric acid by the action of three enzymes (TqaL, TqaF and TqaM) as reported by Bunno et al.9 The focus of this study is the first step of the entire process: the oxidation of L-Val to pleurocybellaziridine by the non-heme iron αKG-dependent enzyme TqaL. | ||
Therefore, we employed molecular docking, molecular dynamic simulations (MD) and quantum mechanics and molecular mechanics (QM/MM) calculations to elucidate the configuration of Fe(IV)O motif and substrate orientation in the active site of the TqaL, and studied the reaction mechanism of aziridine synthesis. Our results show that the C–N bond formation reaction proceeds with the initial hydrogen abstraction from the activated Cβ of L-valine. Interestingly, following the C–H activation, unlike the mechanism of common αKG-dependent non-heme enzymes that catalyze the hydroxylation via the well-known OH rebound step, the reaction catalyzed by TqaL proceeds via nucleophilic attack of amine by the electronegative nitrogen and concerted proton transfer from amine to the Fe(III)–OH, giving the cyclized aziridine product with the release of a water molecule. The research provides structural insights to tailor TqaL for aziridine biosynthesis. The sequence relevance and substrate similarity with hydroxylase (both enzymes can catalyze L-leucine) would enable repurposing of the enzymes or expanding the αKG-dependent non-heme enzyme reservoir in nature.
2 Methods
2.1 Structure preparation
The 3D structure of the enzyme was taken from αKG-dependent dioxygenase TqaL from Neurospora crassa (PDB code: 7EEH).9 Chain A of the TqaL was used for the computational study. The missing loops of the structure (P97–V116, P172–L180 and G283–N291) were built using the loop modelling protocol (DOPE-HR method) with Modeller software14 implemented in USCF Chimera 1.15.15 The position of αKG was obtained from the αKG analogue N-oxalylglycine in the X-ray structure of the homologous enzyme prolyl hydrolase (PDB code: 6F0W),16 and then the nitrogen atom in N-oxalylglycine was changed to the methylene carbon to give αKG. The water molecule in the vicinity of active site was replaced with superoxo anion radical or oxo complex.The protonation states of the titratable residues were assigned at pH = 7 using the H++ server.17 His287 was doubly protonated with an overall charge of +1. The rest of the histidine residues carried an overall neutral charge. His190 and His237 were protonated at the epsilon nitrogen while His148, His184, His213, His239, and His253 were protonated at the delta nitrogen position. The side chain of all the aspartic and glutamic acids were deprotonated, while all the arginine and lysine residues were protonated. The parameters for L-Val were obtained from the general Amber force field (GAFF). The partial charges were computed using the RESP (restrained electrostatic potential) method by performing quantum mechanical (QM) calculations at the HF/6–31G* level of theory using Gaussian16.
2.2 Molecular docking of L-Val
Flexible docking was performed to dock L-Val in the crystal structures of TqaL using AutoDock 4.2. The structure of the superoxo anion radical was built so that the non-heme iron binds to αKG in a bidentate fashion and to the superoxo anion radical. During the reaction, the superoxo anion radical is decarboxylated to give the Fe(IV)O species. To understand the location of substrate binding in the active site of TqaL during the reaction, the substrate was docked in both the Fe(III)-superoxo anion radical complex and the Fe(IV)O complex structures. The grid box was centred at one of the oxygen atoms of the superoxo complex (X, Y, Z: −29.58 Å × 79.286 Å × −8.907 Å) for the Fe(III)-superoxo anion radical complex structure, and the grid box was centred on the oxygen atom of the Fe(IV)O motif (X, Y, Z: −30.329 Å × 81.691 Å × −7.835 Å) for the Fe(IV)O complex. In both docking studies, the Lamarckian genetic algorithm (LGA) and the standard free energy scoring function were implemented and a total of 300 LGA runs were carried out for each ligand-protein complex.
2.3 MD simulations
To understand the substrate binding and conformational dynamics of L-Val in the active site of TqaL, MD simulations were performed using the compute unified device architecture (CUDA) version of particle-mesh Ewald molecular dynamics (PMEMD) in Amber2018 on graphics processing units (GPUs).19 The parameters for the Fe(IV)O complex were obtained from our recent research on αKG-dependent thebaine demethylases.11 The Amber ff19SB force field parameters were used for the protein modelling. The protein–substrate complex was then added to the TIP3P truncated octahedral water box, kept at a distance of 15 Å away from the water box edges. Four Na+ ions were added to neutralise the protein ligand complex and periodic boundary conditions were used in each simulation run. Particle mesh Ewald was used to compute the long-range electrostatic interactions with a cut-off value of 12 Å and the SHAKE algorithm was used to constrain the bonds involving hydrogen.
The entire system was subjected to two rounds of energy minimization using the steepest descent and conjugate gradient method. In the first energy minimisation, the solute molecules were restrained using a potential of 5 kcal mol−1 Å2 and the solvent and ions were subjected to 2000-step minimization. In the second round of energy minimisation, the entire system was subjected to 5000-step minimization using the steepest descent and conjugate gradient method. The entire system was then heated from 0 to 298.15 K for 50 ps at constant volume using a Langevin thermostat with a collision frequency of 1 ps−1 using a canonical ensemble. During the heating process, the non-hydrogen atoms of the solute molecules were restrained using a harmonic potential of 5 kcal mol−1 Å2. Then, another 2000-step energy minimization was conducted using the steepest descent and conjugate gradient methods. The equilibration was run at 298.15 K for 50 ps using a weak restraint of 0.1 kcal mol−1 Å2 on all the solute atoms in an NPT ensemble and a pressure of 1 bar was maintained using a Berendsen barostat. The 300-ns productive MD simulations were run in the NPT ensemble with a time step of 2 fs. The data was saved every 50 ps and the analysis of MD trajectories were conducted using CPPTRAJ.20
2.4 QM/MM calculations
The reaction mechanism of the of aziridine synthesis was studied by performing QM/MM calculations on the most populated snapshots obtained from the MD simulations using ChemShell 3.7.21,22 Prior to QM/MM calculations, 2500-step energy minimisation was performed for the selected MD snapshots using the steepest descend and conjugate gradient algorithms. The Fe(IV)O complex structure was used as the starting point of the reaction for all the QM/MM calculations. The QM region consisted of the side chain of His184, Asp186 and His253 truncated at the Cβ position, succinate, L-Val and the Fe(IV)O motif (Scheme 2). Since MD simulations revealed the important role of Arg74 in substrate binding, the effect of including Arg74 in the QM region is considered by additional QM/MM calculations where Arg74 was also added to the QM region.
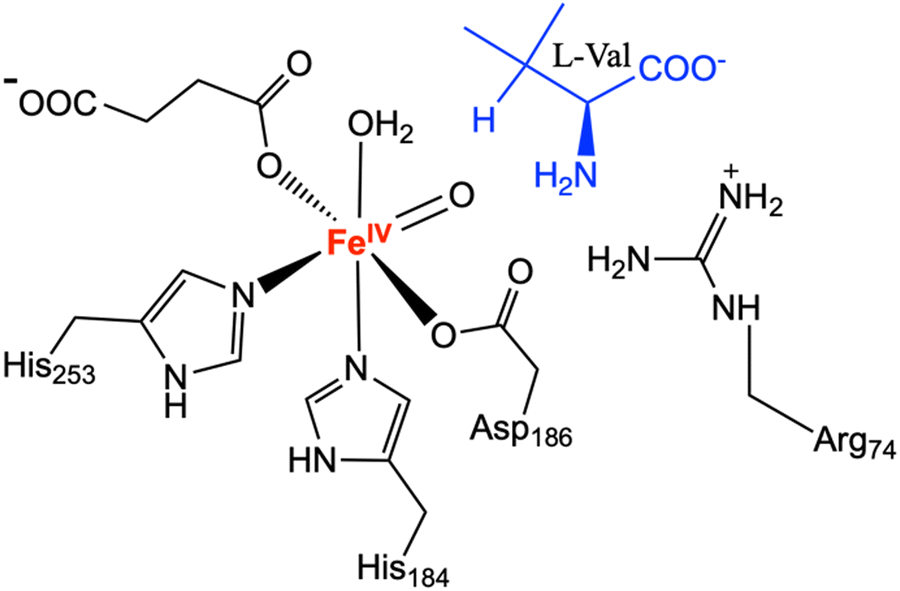 | ||
| Scheme 2 Quantum mechanics (QM) region in the quantum mechanics/molecular mechanics (QM/MM) calculations. | ||
To perform the QM/MM geometry optimisation, the protein system was split into the relaxed or frozen region. The relaxed region which constitutes protein residues and solvent molecules within 8 Å of iron were allowed to move freely during the geometry optimisation. The rest of the frozen region was kept fixed. The protein residues in the QM region were cut at the Cβ position and were statured using hydrogen link atoms. An electronic embedding scheme was used in all the QM/MM calculations to consider the effect of the electrostatic charges of the protein atoms in the MM region on the polarization of the electron density of the atoms in the QM region.
ORCA 4.2.023 was used to run all the DFT calculations with U-B3LYP as the functional of choice.24 The def2-TZVP def2/J auxiliary basis set was used for the iron, while the rest of the atoms were treated with def2-SVP. The D3 dispersion correction and Becke–Johnson (BJ) damping were used in all the calculations. Keywords TightSCF, SlowConv, Grid4, and GridX4 were also used to ensure convergence. For the MM part of the calculations, DL_POLY25 was used with a FF19SB force field.26
To evaluate the ground state of the system, QM/MM geometry optimization was performed for four spin states: singlet (S = 0), triplet (S = 1), quintet (S = 2), and septet (S = 3). The QM/MM relaxed potential energy scan (PES) was run for the hydrogen atom abstraction (HAA) step by decreasing the distance (0.1 Å) between the oxygen atom of Fe(IV)O and the hydrogen atom to be abstracted from L-Val. Starting from the iron oxo oxidant, the reaction to form hydroxylated product via the rebound and the possible mechanisms to form the aziridine product include step-wise and concerted intramolecular cyclisation.
The rebound step in the hydroxylation mechanism was calculated by decreasing the distance between the hydroxyl group on the iron centre and the carbon radical on the L-Val with a decrement of 0.1 Å. For the aziridination mechanism, intramolecular attack of the amine on to the Cβ carbon was modelled by decreasing the distance between the nitrogen and the Cβ carbon of L-Val with a decrement of 0.1 Å. The transition state associated with each step was fully optimised using the dimer keyword and were then validated by the presence of one unique imaginary frequency. The final energies were also computed at the UB3LYP-D3BJ/def2-TZVPP level.
3 Results and discussion
Most of the reactions catalyzed by non-heme αKG-dependent enzymes (e.g., hydroxylation, demethylation, and epoxidation reactions) follows the common mechanism to first generate an iron oxo oxidant for the subsequent reactions (Fig. 1).10,11,27–30,31 The triplet molecular oxygen is coupled to the high spin ferrous iron in the active site of non-heme αKG-dependent enzymes, giving a Fe(III)-superoxo complex (1). The superoxol species is then converted to high-valent Fe(IV)O species (2) through oxidative decarboxylation of αKG. The Fe(IV)O motif would then flip to a favourable inline conformation (3) to initiate the oxidation reactions.9,32,33
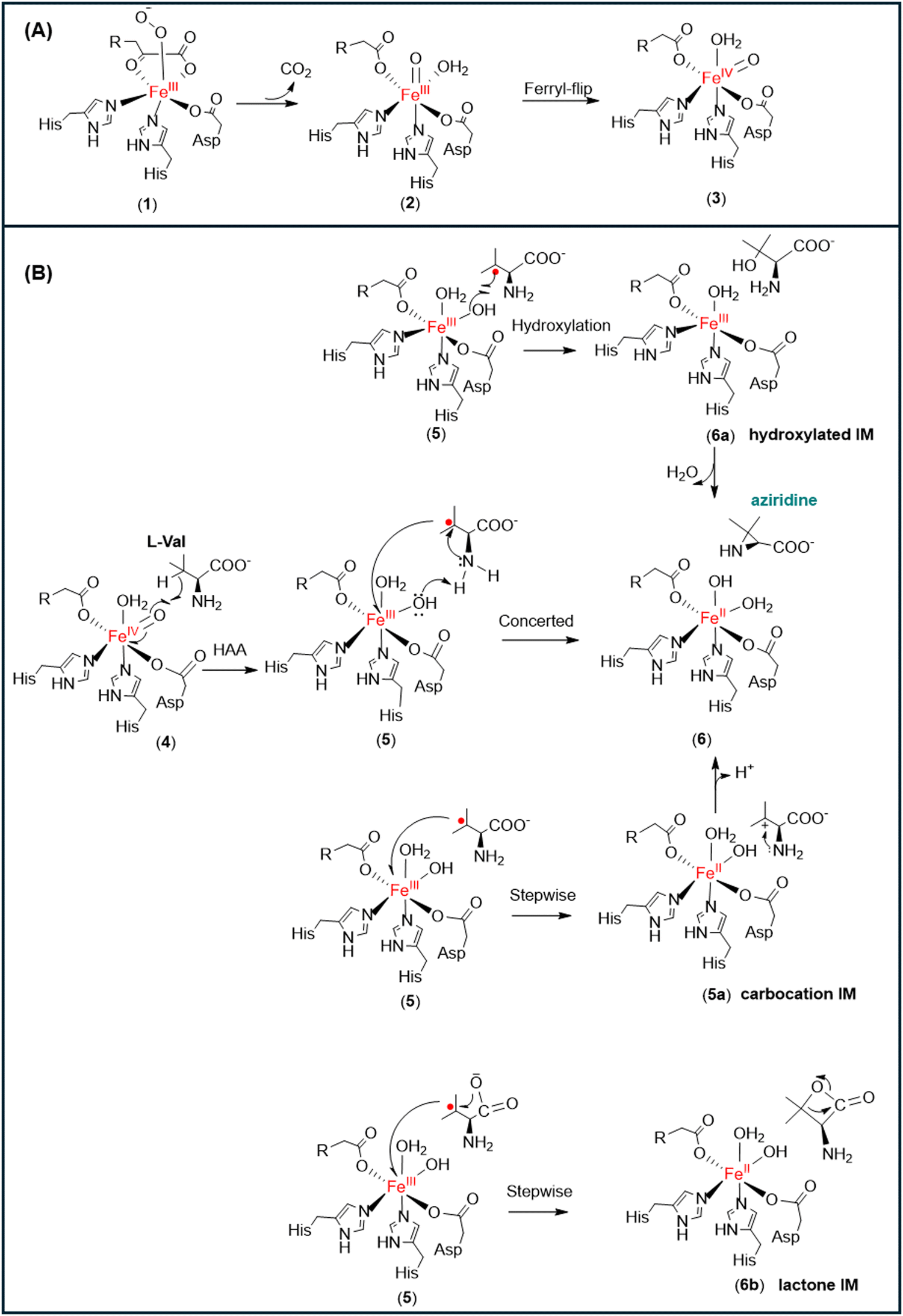 | ||
| Fig. 1 (A) Formation of the iron oxo oxidant (3) in non-heme αKG-dependent enzymes (R = −(CH2)2−COO−). (B) Possible reaction pathways for the formation of the three-membered aziridine from the Fe(III)–OH species. | ||
TqaL is a unique member of non-heme iron αKG-dependent enzymes, as it is the only known enzyme in the family that catalyses the aziridination reaction. In the presence of L-Val, the Fe(IV)O motif (4) abstracts a hydrogen atom from the Cβ atom of the substrate to form an isopropyl radical and Fe(III)–OH (5) (Fig. 1).12,13 However, unlike the other α-KG dependant non-heme enzymes that produce the hydroxylated product, TqaL catalyzes the aziridination of L-Val to give aziridine.
The formation of aziridine by intramolecular cyclisation may occur from the Fe(III)–OH intermediate (5) via three possible pathways (Fig. 1). One possibility is that the reaction proceeds with the rebound of the hydroxyl group to give hydroxylated L-Val (6a), which then cyclises to give the final product aziridine. Another possibility is the transfer of the single electron from the isopropyl radical to the Fe(III) species to generate a tertiary carbocation intermediate (5a), which is then attacked by the NH2 group of L-Val to form the aziridine product (6). Alternatively, the single electron transfer and the intramolecular cyclisation may occur simultaneously, which is accompanied by hydrogen atom abstraction (HAA) from the NH2 group of L-Val by the Fe(III)–OH species to give the final product (6).
3.1 L-Val binding in the active site
The Fe(IV)O species is the main oxidant in non-heme αKG-dependent enzymes,10,27,28,30,34–36 therefore, the orientation of Fe(IV)O relative to the substrate in the active site is critical for the enzyme's catalytic capacity. The crystal structure of TqaL (PDB: 7EEH) was used in this study, where the missing loops were modelled using Modeller and then the entire crystal structure was relaxed by 300-ns MD simulations. The structure shows that the mononuclear non-heme iron is coordinated with a triad composed of His184, His253 and Asp186 (Fig. 2). The modelled AlphaFold2 structure is largely same as the crystal structure (Fig. S1, ESI†). Hence all the subsequent calculations and analysis are based on the MD refined crystal structure.
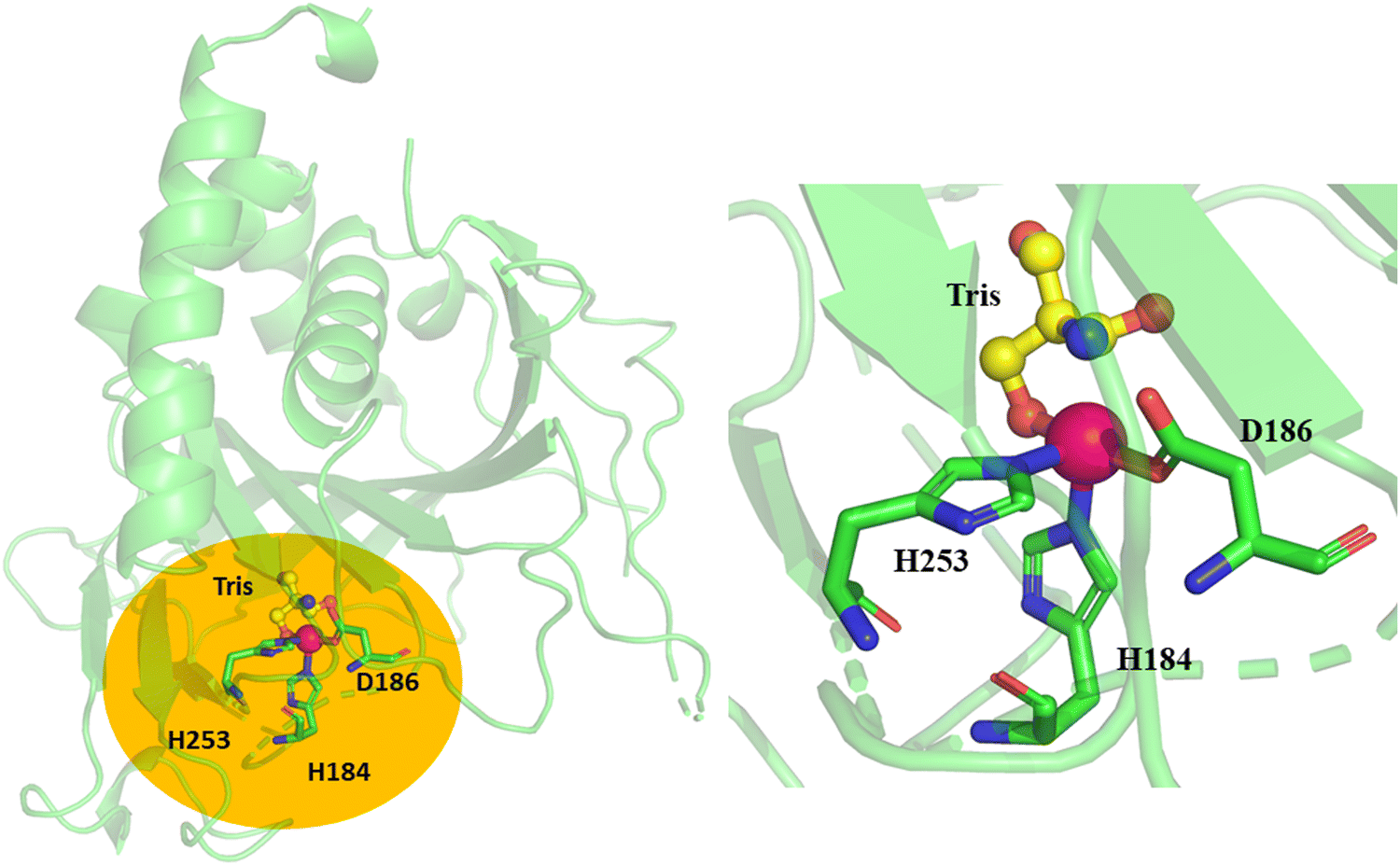 | ||
| Fig. 2 X-ray structure of chain A of TqaL (PDB code: 7EEH).9 The active site of TqaL consists of the mononuclear non-heme iron, shown as a pink sphere, coordinated to 2His/Asp protein residues shown in stick representation. The enlarged catalytic site is shown in the right panel. The Tris buffer is shown in ball-and-stick representations. The missing loops are shown in dotted representation. | ||
The location of L-Val substrate in the active site of TqaL was determined by docking the substrate in the Fe(III)-superoxo and Fe(IV)O complexes, which correspond to the pre- and post- decarboxylation states, respectively (Fig. 3). Molecular docking studies revealed that Arg74 stabilizes the carboxylate group of the L-Val substrate in the iron oxo complex, and the substrate binds trans to His253, indicating a ferryl flip during the oxidative decarboxylation process (Fig. 3B, see Table S1 (ESI†) for other docked poses energies). As a result, the Fe(IV)O motif rotates and aligns itself toward the Cβ carbon of the L-Val substrate (the distance between the Cβ hydrogen of L-Val and the iron oxo oxygen is 2.6 Å), facilitating the HAA to initiate the aziridination reaction. This type of ferryl flip is in agreement with the previous report on other αKG dependent non-heme enzymes.27,34,37
 | ||
| Fig. 3 Molecular docking of the L-Val substrate in the active site of the TqaL enzyme. The docking was performed in both the (A) pre-decarboxylation state (i.e. Fe(III)-superoxo complex) and (B) post-decarboxylation state (i.e. Fe(IV)O complex) to examine the substrate binding and the configuration of the Fe(IV)O species. | ||
To validate the docking pose and study the stabilization of L-Val in the active site, MD simulations of the Fe(IV)O state in complex with L-Val (Fig. S2 for the RMSD trajectory, ESI†) were performed. The octahedral active site geometry of the active site was maintained throughout the simulations and is consistent with previous literature.34,38 It has been established in non-heme and heme enzyme chemistry that the shorter the distance of the hydrogen atom to the oxo group of Fe(IV)O motif, the better it is for catalysis.39–41 Recent experimental evidence suggests that aziridination is initiated by the abstraction of hydrogen atoms from the Cβ of L-Val by the Fe(IV)O species.13 Therefore, sampling on the MD trajectory was performed based on the distance of the Cβ to the oxygen atom of the Fe(IV)O complex. Cluster analysis was conducted for the MD simulations and two dominant conformations were selected for further analysis based upon the distance between Fe(IV)O and the Cβ-hydrogen of L-Val (Fig. 4). In both conformations, the carboxylate group of L-Val makes electrostatic interactions with the side chain of Arg74 (Fig. S3, ESI†). Our MD simulations show that the isopropyl group of the L-Val is stabilized by the hydrophobic residues Phe164, Ile259 and Phe275, which is in accordance with the site-directed mutagenesis study, where Phe275 was suggested to be critical for the aziridination reaction and its mutations to Leu or Ala resulted in a hydroxylated major product.12 MD simulations also showed that the amine group of L-Val is well hydrated and forms water-mediated hydrogen bonds with Glu181. The Cβ hydrogen atom of the substrate is 2.6 Å from the iron oxo complex, which is an ideal distance to initiate the aziridination reaction by HAA, and agrees with the previous mechanism hypothesis that aziridination is initiated by hydrogen atom abstraction from Cβ of V-Val.13 However, in the other dominant conformation, the amine group of the substrate moves slightly outwards such that the water-mediated hydrogen bonds with Glu181 breaks (Fig. 4). This results in an increased distance between the Cβ hydrogen atom and the iron oxo atom (2.6 Å vs 3.0 Å).
 | ||
| Fig. 4 The most populated conformations (A) and (B) obtained from the MD simulations of Fe(IV)O in complex with L-Val. The clustering was based on the distance between Fe(IV)O and the Cβ of L-Val. | ||
To study the mechanism of L-Val aziridination, QM/MM calculations were performed for both conformations using the unrestricted B3LYP (U-B3LYP) functional method, which is widely adopted for studying non-heme iron enzymes. A recent study by the Siegbahn group indicates the reliability of B3LYP in capturing the essential electronic properties of metalloenzymes.24 Employing the def2-TZVP basis set for iron ensures a high level of accuracy in describing the metal center's electronic environment. For the surrounding lighter atoms, the def2-SVP basis set offers a computationally efficient, yet reliable representation. This combination has been extensively utilized in theoretical studies in exploring the mechanisms of non-heme iron enzymes because it provides a balance between computational cost and accuracy. Incorporating D3 corrections effectively improves the description of intermolecular forces in biological systems; hence, it would give more reliable mechanistic insights. The computational settings used in this study are consistent with methodologies employed in recent QM/MM studies of non-heme enzymes.34,42,43
3.2 Aziridination mechanism disclosed by QM/MM calculations
The ground state of the reactant complex was determined by optimising the representative starting structures (Fig. 4) obtained from MD simulations. Four possible spin states of non-heme iron were considered: low-spin open-shell singlet, intermediate spin triplet, quintet, and high-spin septet. Our results show that the spin state order was quintet (S = 2) < triplet (S = 1) < septet (S = 3) < singlet (S = 0) (Table S2, ESI†). The quintet is the ground state and is more stable than the triplet spin state by 8.01 kcal mol−1. The singlet and septet spin states are 15–17 kcal mol−1 higher in energy compared to the quintet spin state. This is in agreement with the reports for other 2-OG dependent non-heme enzymes for which the quintet is the ground state for the Fe(IV)O complex.34,38,44–46 In the electronic configuration of the reactant complex at the quintet state, the spin density of the Fe(IV)O unit is 3.71 and can be described as π*xy↑ π*xz↑ π*yz↑ σ*x2−y2↑ σ*z2. σ*z2 is the virtual orbital, and π*xy, π*xz, π*yz and σ*x2−y2 orbitals are singly occupied with four alpha electrons (Fig. 5A). The electronic structure of Fe(IV)O at the quintet state is consistent with previous theoretical studies on other non-heme αKG-dependent enzymes.34,37,47
 | ||
| Fig. 5 (A) The electronic configuration obtained during the aziridination reaction catalysed by the TqaL enzyme at the quintet state (S = 2). (B) QM/MM reaction profile obtained for aziridine formation via C–N bond formation by intramolecular cyclisation catalysed by the TqaL enzyme at the quintet state (S = 2) for both dominant conformations obtained from MD simulations. | ||
3.3 Hydrogen atom abstraction by Fe(IV)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) O
O
The HAA by the Fe(IV)O unit is well established in various reactions catalysed by non-heme αKG-dependent enzymes such as hydroxylation,10,28,37,48 epoxidation49 or demethylation,34,38 however, the mechanism of formation of the cyclised product aziridine initiated through HAA by Fe(IV)O is unreported. In the reactant complexes for structures A (colour coded green) and B (colour coded cyan) (RC) at the ground state (quintet state), the Cβ hydrogen of the isopropyl group of L-Val is 2.86 Å and 2.40 Å away from the oxygen atom of the Fe(IV)O unit, respectively (Fig. 5). The HAA step was also calculated at the triplet (S = 1) spin potential energy surface for structure A; however, the high potential energy barrier of 37.2 kcal mol−1 indicates that it is not feasible for the HAA reaction to occur at the triplet surface (Fig. S4, ESI†). Therefore, all the subsequent analysis are for the quintet spin state surface for both A and B structures.
The HAA for structure A and B went through an activation free energy barrier of 12.7 and 10.7 kcal mol−1, respectively and results in the formation of exothermic intermediate IM1 at −7.2 and −9.0 kcal mol−1, respectively (Fig. 5B). In the TS1 structures obtained from the two starting structures (A and B), the distances between the oxygen atom and the Cβ hydrogen are 1.36 Å and the distances between the β carbon and the Cβ hydrogen are 1.25 Å (Fig. 6). The spin density of the Fe(IV)O unit increased from 3.71 in RC to 4.01 in TS1 and the spin density of Cβ decreased from 0 in RC to −0.33 in TS1 (Fig. 6). The change in the spin population and orbital analysis indicates the transfer of the alpha electron from Cβ of L-Val to the Fe(IV)O unit and development of the radical character on Cβ. The electronic configuration of IM1, where the Fe(III)–OH motif has the spin density of 4.55, can be described as π*xy↑ π*xz↑ π*yz↑ σ*x2−y2↑ σ*z2↑ σC–H↓, and the spin density of −0.96 for Cβ of L-Val indicates the radical of beta spin on the Cβ (Fig. 5A). The electron transfer to the Fe(IV)O unit can either follow a σ or π channel depending on the angle of the Cβ hydrogen atom facing the Fe(IV)O motif in the active site. The transfer of an alpha electron to the dz2 orbital indicates the σ pathway and the transfer of beta electron to the dπ* orbital known as the π channel. Here, it was observed that the alpha electron was transferred to the σ*z2 orbital, indicating that HAA follows a σ pathway. This is in accordance with the HAA mechanism of other non-heme enzymes reported in previous work.47
 | ||
| Fig. 6 Stationary and saddle points obtained during the QM/MM calculations of the reaction pathways for the formation of aziridine catalyzed by the TqaL enzyme at the quintet state (S = 2) for structure (A) (green) and structure (B) (cyan). The distances are labelled in black text (in Å), and the spin densities (ρ) for each complex are shown in red. | ||
For most non-heme αKG-dependent enzymes, the hydrogen atom is transferred to the iron-oxo motif, followed by the transfer of the hydroxyl group to the substrate radical to give a hydroxylated product via a rebound mechanism. However, recent isotope and quantitative production analysis excluded the possibility that the formation of the final aziridine product goes through the hydroxylated L-Val intermediate.13 Nevertheless, we still studied the possibility of forming the aziridine product by the rebound mechanism and found that the rebound step goes through the activation energy barrier of 9.6 kcal mol−1, giving hydroxylated L-Val exothermically (Fig. S5, ESI†). To obtain the desired product aziridine, the reaction would proceed by transferring the proton from NH2 to the hydroxyl group of L-Val; however, this step is prohibited with a high potential energy barrier (Fig. S6, ESI†). Thus, following the hydrogen abstraction, there must be an alternative pathway, other than the OH rebound mechanism, that the Fe(III)–OH species must follow for the intramolecular cyclisation of L-Val to produce aziridine. Therefore, we investigated other possible reaction mechanisms (via a carbocation or lactone intermediate) to form aziridine from L-Val (Fig. 1), as shown in the below section.
3.4 Intramolecular cyclisation via a concerted mechanism
It was previously hypothesized that the formation of aziridine goes through a carbocation intermediate rather than the lactone intermediate.13 The lactone-based pathway was tested for structure A and it gave the barrier of HAA in excess of 19 kcal mol−1 (Fig. S6, ESI†); therefore, it was discarded for future calculations. In the IM1 for structure B (green colour) (Fig. 6), the hydrogen atom of the NH2 is only 1.99 Å from the hydroxyl group of Fe(III)–OH, so the HAA from the NH2 group of IM1 by Fe(III)–OH was also studied. However, the potential energy scan shows a high barrier of 24.3 kcal mol−1 (Fig. S7, ESI†), indicating that it is unlikely for the reaction to proceed with HAA from the amine group. We also considered the possible stepwise mechanism that includes a single electron transfer from the isopropyl radical to form the potential carbocation 5a (Fig. 2B), which would then undergo intramolecular cyclisation to give the aziridine product. However, all the optimisation attempts to obtain 5a reverted back to 5, further underpinning that the reaction proceeds via an alternative mechanism.During the HAA, the angle of the isopropyl group increased from 111.6° in the initial reactant complex (RC) to 120.1° in the IM1 (Fig. 6), which corresponds to a trigonal planar geometry of the isopropyl group and resembles the conformation of a tertiary carbon radical. The trigonal planar geometry of IM1 is crucial, as it allows for IM1 to act as a Lewis acid, with its partially occupied p orbital on the Cβ to orient itself perpendicular to the plane of the isopropyl group, and therefore enable the intramolecular cyclisation to form aziridine. The NH2 in IM1 is only 2.48 Å from the Cβ atom of L-Val, which facilitates nucleophilic attack of the planar Cβ of L-Val and the intramolecular cyclisation of L-Val. This process is coupled with a single electron transfer of beta spin localised on the Cβ atom to the central iron atom, going through the transition state TS2 with an activation energy barrier of 12.2 kcal mol−1 (Fig. 5B).
The spin density of Fe(III)–OH decreases from 4.52 in IM1 to 4.15 in the transition state TS2 and that of Cβ of L-Val increases from −0.96 to −0.48. In the TS2 structure, the distance of NH2 to the Cβ atom of L-Val is 2.20 Å and the hydroxy group of Fe(III)–OH is 1.95 Å away from the proton of NH2 group. The intramolecular cyclisation occurs via a concerted mechanism with nucleophilic attack of NH2 of the L-Val to the Cβ atom and the transfer of the proton from NH2 to the hydroxy group of Fe(III)–OH. During the concerted process, a water molecule leaves to exothermically yield an aziridine product at −36.7 kcal mol−1 (Fig. 5B).
4 Conclusions
TqaL offers an attractive biosynthetic route for the incorporation of three-membered aziridine ring structures into the core of biologically active molecules. It is critical to decide the binding of the substrate in the enzyme for rational engineering to improve the substrate scope to potentiate its use for the pharmaceutical industry. However, the substrate is missing in the crystal structure of the enzyme and the reaction mechanism of aziridine synthesis via unimolecular cyclisation catalyzed by a non-heme enzyme has never been reported.Here, we reported that the binding of L-Val in the active site of TqaL is stabilised by Arg74, Phe164, and Phe275. These residues enable the Cβ hydrogen of the L-Val substrate to be abstracted by the Fe(IV)O motif. The resulting Fe(III)–OH species then accepts an electron from the Cβ radical of the substrate. The single electron transfer is accompanied by the attack of a polar amine to the carbon radical and simultaneous transfer of a proton from the amine group of the substrate to the Fe(III)–OH species to form the final aziridine product. The active conformation for substrate binding and the elucidation of the uni-molecular cyclisation for aziridine synthesis sets the basis for engineering non-heme enzymes to exploit their use in the biosynthesis of aziridine-derived active therapeutics.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the computing resources from the QUB high performance computing Tier2 resource, funded by EPSRC (EP/T022175). W. S. acknowledges the support of Research England's Expanding Excellence in England (E3) Fund.References
- J. Foulke-Abel, H. Agbo, H. Zhang, S. Mori and C. M. H. Watanabe, Mode of action and biosynthesis of the azabicycle-containing natural products azinomycin and ficellomycin, Nat. Prod. Rep., 2011, 28, 693–704 RSC.
- G. S. Singh, Synthetic Aziridines in Medicinal Chemistry: A Mini-Review, Mini-Rev. Med. Chem., 2016, 16, 892–904 CrossRef CAS PubMed.
- Y. Nakao, M. Fujita, K. Warabi, S. Matsunaga and N. Fusetani, Miraziridine A, a Novel Cysteine Protease Inhibitor from the Marine Sponge Theonella aff. mirabilis1, J. Am. Chem. Soc., 2000, 122, 10462–10463 CrossRef CAS.
- H. Tsutsumi, Y. Katsuyama, M. Izumikawa, M. Takagi, M. Fujie, N. Satoh, K. Shin-ya and Y. Ohnishi, Unprecedented Cyclization Catalyzed by a Cytochrome P450 in Benzastatin Biosynthesis, J. Am. Chem. Soc., 2018, 140, 6631–6639 CrossRef CAS PubMed.
- C. C. Farwell, R. K. Zhang, J. A. McIntosh, T. K. Hyster and F. H. Arnold, Enantioselective Enzyme-Catalyzed Aziridination Enabled by Active-Site Evolution of a Cytochrome P450, ACS Cent. Sci., 2015, 1, 89–93 CrossRef CAS PubMed.
- N. W. Goldberg, A. M. Knight, R. K. Zhang and F. H. Arnold, Nitrene Transfer Catalyzed by a Non-Heme Iron Enzyme and Enhanced by Non-Native Small-Molecule Ligands, J. Am. Chem. Soc., 2019, 141, 19585–19588 CrossRef CAS PubMed.
- G. Mukherjee, F. G. C. Reinhard, U. K. Bagha, C. V. Sastri and S. P. De Visser, Sluggish reactivity by a nonheme iron(IV)-tosylimido complex as compared to its oxo analogue, Dalton Trans., 2020, 49, 5921–5931 RSC.
- G. Coin, R. Patra, S. Rana, J. P. Biswas, P. Dubourdeaux, M. Clémancey, S. P. de Visser, D. Maiti, P. Maldivi and J.-M. Latour, Fe-Catalyzed Aziridination Is Governed by the Electron Affinity of the Active Imido-Iron Species, ACS Catal., 2020, 10, 10010–10020 CrossRef CAS.
- I. Abe, R. Bunno, T. Awakawa and T. Mori, Aziridine formation by a Fe(II)/α-ketoglutarate dependent oxygenase and 2-aminoisobutyrate biosynthesis in fungi, Angew. Chem., Int. Ed., 2021, 60, 15827–15831 CrossRef PubMed.
- E. I. Solomon, S. Goudarzi and K. D. Sutherlin, O2 Activation by Non-Heme Iron Enzymes, Biochemistry, 2016, 55, 6363–6374 CrossRef CAS PubMed.
- W. Singh, C. Hui, C. Li and M. Huang, Thebaine is Selectively Demethylated by Thebaine 6-O-Demethylase and Codeine-3-O-demethylase at Distinct Binding Sites: A Computational Study, Inorg. Chem., 2021, 60, 10199–10214 CrossRef CAS PubMed.
- H. Tao, R. Ushimaru, T. Awakawa, T. Mori, M. Uchiyama and I. Abe, Stereoselectivity and Substrate Specificity of the Fe(II)/α-Ketoglutarate-Dependent Oxygenase TqaL, J. Am. Chem. Soc., 2022, 144, 21512–21520 CrossRef CAS PubMed.
- L. Cha, J. C. Paris, B. Zanella, M. Spletzer, A. Yao, Y. Guo and W.-C. Chang, Mechanistic Studies of Aziridine Formation Catalyzed by Mononuclear Non-Heme Iron Enzymes, J. Am. Chem. Soc., 2023, 145, 6240–6246 CrossRef CAS PubMed.
- B. Webb and A. Sali, Comparative Protein Structure Modeling Using MODELLER, Curr. Protoc. Bioinformatics, 2016, 54, 5.6.1–5.6.37 Search PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, UCSF Chimera--a visualization system for exploratory research and analysis, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- K. Lippl, A. Boleininger, M. McDonough, M. I. Abboud, H. Tarhonskaya, R. Chowdhury, C. Loenarz and C. J. Schofield, Born to sense: biophysical analyses of the oxygen sensing prolyl hydroxylase from the simplest animal Trichoplax adhaerens, Hypoxia, 2018, 6, 57–71 CrossRef PubMed.
- J. C. Gordon, J. B. Myers, T. Folta, V. Shoja, L. S. Heath and A. Onufriev, H++: a server for estimating pKas and adding missing hydrogens to macromolecules, Nucleic Acids Res., 2005, 33, W368–W371 CrossRef CAS PubMed.
- H. M. A. D. A. Case, K. Belfon, I. Y. Ben-Shalom, S. R. Brozell, D. S. Cerutti, T. E. Cheatham, III, G. A. Cisneros, V. W. D. Cruzeiro, T. A. Darden, R. E. Duke, G. Giambasu, M. K. Gilson, H. Gohlke, A. W. Goetz, R. Harris, S. Izadi, S. A. Izmailov, C. Jin, K. Kasavajhala, M. C. Kaymak, E. King, A. Kovalenko, T. Kurtzman, T. S. Lee, S. LeGrand, P. Li, C. Lin, J. Liu, T. Luchko, R. Luo, M. Machado, V. Man, M. Manathunga, K. M. Merz, Y. Miao, O. Mikhailovskii, G. Monard, H. Nguyen, K. A. O’Hearn, A. Onufriev, F. Pan, S. Pantano, R. Qi, A. Rahnamoun, D. R. Roe, A. Roitberg, C. Sagui, S. Schott-Verdugo, J. Shen, C. L. Simmerling, N. R. Skrynnikov, J. Smith, J. Swails, R. C. Walker, J. Wang, H. Wei, R. M. Wolf, X. Wu, Y. Xue, D. M. York, S. Zhao and P. A. Kollman, Amber 2021, University of California, San Francisco, 2021 Search PubMed.
- R. Salomon-Ferrer, A. W. Götz, D. Poole, S. Le Grand and R. C. Walker, Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald, J. Chem. Theory Comput., 2013, 9, 3878–3888 CrossRef CAS PubMed.
- D. R. Roe and T. E. Cheatham, PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data, J. Chem. Theory Comput., 2013, 9, 3084–3095 CrossRef CAS PubMed.
- P. Sherwood, A. H. de Vries, M. F. Guest, G. Schreckenbach, C. R. A. Catlow, S. A. French, A. A. Sokol, S. T. Bromley, W. Thiel, A. J. Turner, S. Billeter, F. Terstegen, S. Thiel, J. Kendrick, S. C. Rogers, J. Casci, M. Watson, F. King, E. Karlsen, M. Sjøvoll, A. Fahmi, A. Schäfer and C. Lennartz, QUASI: A general purpose implementation of the QM/MM approach and its application to problems in catalysis, J. Mol. Struct. THEOCHEM, 2003, 632, 1–28 CrossRef CAS.
- Y. Lu, K. Sen, C. Yong, D. S. D. Gunn, J. A. Purton, J. Guan, A. Desmoutier, J. Abdul Nasir, X. Zhang, L. Zhu, Q. Hou, J. Jackson-Masters, S. Watts, R. Hanson, H. N. Thomas, O. Jayawardena, A. J. Logsdail, S. M. Woodley, H. M. Senn, P. Sherwood, C. R. A. Catlow, A. A. Sokol and T. W. Keal, Multiscale QM/MM modelling of catalytic systems with ChemShell, Phys. Chem. Chem. Phys., 2023, 25, 21816–21835 RSC.
- F. Neese, The ORCA program system, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- P. E. M. Siegbahn and M. R. A. Blomberg, A Systematic DFT Approach for Studying Mechanisms of Redox Active Enzymes, Front. Chem., 2018, 6, 644 CrossRef CAS PubMed.
- W. Smith and T. R. Forester, DL_POLY_2.0: A general-purpose parallel molecular dynamics simulation package, J. Mol. Graphics, 1996, 14, 136–141 CrossRef CAS PubMed.
- C. Tian, K. Kasavajhala, K. A. A. Belfon, L. Raguette, H. Huang, A. N. Migues, J. Bickel, Y. Wang, J. Pincay, Q. Wu and C. Simmerling, ff19SB: Amino-acid-specific protein backbone parameters trained against quantum mechanics energy surfaces in solution, J. Chem. Theory Comput., 2020, 16, 528–552 CrossRef PubMed.
- W. Singh, D. Quinn, T. S. Moody and M. Huang, Reaction mechanism of histone demethylation in αKG-dependent non-heme iron enzymes, J. Phys. Chem. B, 2019, 123, 7801–7811 CrossRef CAS PubMed.
- A. Timmins, M. Saint-André and S. P. de Visser, Understanding How Prolyl-4-hydroxylase Structure Steers a Ferryl Oxidant toward Scission of a Strong C–H Bond, J. Am. Chem. Soc., 2017, 139, 9855–9866 CrossRef CAS PubMed.
- J. Lu, B. Wang, S. Shaik and W. Lai, QM/MM Calculations Reveal Important Role of α-Heteroatom Substituents in Controlling Selectivity of Mononuclear Nonheme HppE-Catalyzed Reactions, ACS Catal., 2020, 10, 9521–9532 CrossRef CAS.
- M. Srnec, S. R. Iyer, L. M. K. Dassama, K. Park, S. D. Wong, K. D. Sutherlin, Y. Yoda, Y. Kobayashi, M. Kurokuzu, M. Saito, M. Seto, C. Krebs, J. M. Bollinger and E. I. Solomon, Nuclear Resonance Vibrational Spectroscopic Definition of the Facial Triad FeIVO Intermediate in Taurine Dioxygenase: Evaluation of Structural Contributions to Hydrogen Atom Abstraction, J. Am. Chem. Soc., 2020, 142, 18886–18896 CrossRef CAS PubMed.
- S. Martinez, M. Fellner, C. Q. Herr, A. Ritchie, J. Hu and R. P. Hausinger, Structures and Mechanisms of the Non-Heme Fe(II)- and 2-Oxoglutarate-Dependent Ethylene-Forming Enzyme: Substrate Binding Creates a Twist, J. Am. Chem. Soc., 2017, 139, 11980–11988 CrossRef CAS PubMed.
- T. Borowski, A. Bassan and P. E. M. Siegbahn, Mechanism of Dioxygen Activation in 2-Oxoglutarate-Dependent Enzymes: A Hybrid DFT Study, Chem. – Eur. J., 2004, 10, 1031–1041 CrossRef CAS PubMed.
- A. Timmins and S. P. De Visser, A Comparative Review on the Catalytic Mechanism of Nonheme Iron Hydroxylases and Halogenases, Catalysts, 2018, 8, 314 CrossRef.
- W. Singh, C. Hui, C. Li and M. Huang, Thebaine is Selectively Demethylated by Thebaine 6-O-Demethylase and Codeine-3-O-demethylase at Distinct Binding Sites: A Computational Study, Inorg. Chem., 2021, 60, 10199–10214 CrossRef CAS PubMed.
- S. Sinnecker, N. Svensen, E. W. Barr, S. Ye, J. M. Bollinger, F. Neese and C. Krebs, Spectroscopic and Computational Evaluation of the Structure of the High-Spin Fe(IV)-Oxo Intermediates in Taurine: α-Ketoglutarate Dioxygenase from Escherichia coli and Its His99Ala Ligand Variant, J. Am. Chem. Soc., 2007, 129, 6168–6179 CrossRef CAS PubMed.
- L. M. Hoffart, E. W. Barr, R. B. Guyer, J. M. Bollinger and C. Krebs, Direct spectroscopic detection of a C-H-cleaving high-spin Fe(IV) complex in a prolyl-4-hydroxylase, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 14738–14743 CrossRef CAS PubMed.
- M. G. Quesne, R. Latifi, L. E. Gonzalez-Ovalle, D. Kumar and S. P. de Visser, Quantum Mechanics/Molecular Mechanics Study on the Oxygen Binding and Substrate Hydroxylation Step in AlkB Repair Enzymes, Chem. – Eur. J., 2014, 20, 435–446 CrossRef CAS PubMed.
- W. Singh, D. Quinn, T. S. Moody and M. Huang, Reaction Mechanism of Histone Demethylation in alphaKG-dependent Non-Heme Iron Enzymes, J. Phys. Chem. B, 2019, 123, 7801–7811 CrossRef CAS PubMed.
- S. Shaik, D. Kumar, S. P. De Visser, A. Altun and W. Thiel, Theoretical Perspective on the Structure and Mechanism of Cytochrome P450 Enzymes, Chem. Rev., 2005, 105, 2279–2328 CrossRef CAS PubMed.
- C. Hui, W. Singh, D. Quinn, C. Li, T. Moody and M. Huang, Regio- and Stereoselectivity of CYP450BM3-Catalyzed Oxidation of Complex Terpenoids: A QM/MM study, Phys. Chem. Chem. Phys., 2020, 22, 21696–21706 RSC.
- R. Lonsdale, K. T. Houghton, J. Żurek, C. M. Bathelt, N. Foloppe, M. J. De Groot, J. N. Harvey and A. J. Mulholland, Quantum Mechanics/Molecular Mechanics Modeling of Regioselectivity of Drug Metabolism in Cytochrome P450 2C9, J. Am. Chem. Soc., 2013, 135, 8001–8015 CrossRef CAS PubMed.
- H. Li, W. Zhu and Y. Liu, Mechanism of Uncoupled Carbocyclization and Epimerization Catalyzed by Two Non-Heme Iron/α-Ketoglutarate Dependent Enzymes, J. Chem. Inf. Model., 2019, 59, 5086–5098 CrossRef CAS PubMed.
- C. Hui, W. Singh, D. Quinn, C. Li, T. S. Moody and M. Huang, Regio-and stereoselectivity in the CYP450 BM3-catalyzed hydroxylation of complex terpenoids: a QM/MM study, Phys. Chem. Chem. Phys., 2020, 22, 21696–21706 RSC.
- H. Li, W. Zhu and Y. Liu, Mechanism of Uncoupled Carbocyclization and Epimerization Catalyzed by Two Non-Heme Iron/alpha-Ketoglutarate Dependent Enzymes, J. Chem. Inf. Model., 2019, 59, 5086–5098 CrossRef CAS PubMed.
- J. Lu and W. Lai, Mechanistic Insights into a Stibene Cleavage Oxygenase NOV1 from Quantum Mechanical/Molecular Mechanical Calculations, ChemistryOpen, 2019, 8, 228–235 CrossRef CAS PubMed.
- S. Zhang and Y. Liu, Mechanism of fatty acid decarboxylation catalyzed by a non-heme iron oxidase (UndA): a QM/MM study, Org. Biomol. Chem., 2019, 17, 9808–9818 RSC.
- Y. Liu and Y. Liu, Computational Study of Aromatic Hydroxylation Catalyzed by the Iron-Dependent Hydroxylase PqqB Involved in the Biosynthesis of Redox Cofactor Pyrroloquinoline Quinone, Inorg. Chem., 2022, 61, 5943–5956 CrossRef CAS PubMed.
- X. Zhang, Z. Wang, J. Gao and W. Liu, Chlorination versus hydroxylation selectivity mediated by the non-heme iron halogenase WelO5, Phys. Chem. Chem. Phys., 2020, 22, 8699–8712 RSC.
- J. Li, H.-J. Liao, Y. Tang, J.-L. Huang, L. Cha, T.-S. Lin, J. L. Lee, I. V. Kurnikov, M. G. Kurnikova, W.-C. Chang, N.-L. Chan and Y. Guo, Epoxidation Catalyzed by the Nonheme Iron(II)- and 2-Oxoglutarate-Dependent Oxygenase, AsqJ: Mechanistic Elucidation of Oxygen Atom Transfer by a Ferryl Intermediate, J. Am. Chem. Soc., 2020, 142, 6268–6284 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp03708a |
| This journal is © the Owner Societies 2025 |