
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hydration-induced dynamical changes in lyophilised and weakly hydrated apoferritin: insights from molecular dynamics simulation†
Elisa
Bassotti
 a,
Gaio
Paradossi
a,
Ester
Chiessi
*a and
Mark
Telling
*bc
a,
Gaio
Paradossi
a,
Ester
Chiessi
*a and
Mark
Telling
*bc
aDepartment of Chemical Science and Technologies, University of Rome Tor Vergata, Via della Ricerca Scientifica I, 00133 Rome, Italy. E-mail: ester.chiessi@uniroma2.it
bSTFC, ISIS Facility, Rutherford Appleton Laboratory, Harwell Campus, OX11OQX, UK. E-mail: mark.telling@stfc.ac.uk
cDepartment of Materials, University of Oxford, Parks Road Oxford, OX1 3PH, UK
First published on 2nd January 2025
Abstract
The dynamics and functionality of proteins are significantly influenced by their interaction with water. For lyophilised (i.e. h ≤ 0.05 where h = g of H2O per g of protein) and weakly hydrated systems (i.e. h ≤ 0.38) hydration generally enhances protein mobility above the so-called ‘dynamical transition’ temperature (Td > 220 K). However, water-induced mobility hindrance at low temperatures (T < 175 K) has been reported in various proteins of varying secondary structure; namely green fluorescent protein (GFP), pig liver esterase, lysozyme, ribonuclease A (RNAse A) and apoferritin. By focussing on the dynamic behaviour of the apoferritin molecule, this study proposes mechanisms driving these hydration-induced mobility changes, particularly the less understood hindrance at low temperatures. Using atomistic molecular dynamics (MD) simulations of horse spleen apoferritin in the lyophilised (h = 0.05) and weakly hydrated (h = 0.31) states, we report here the impact of water on protein dynamics as a function of temperature. Through residue-specific mean squared displacement (MSD), radial distribution function (RDF), solvent accessible surface area (SASA), local hydration degree and hydrogen bonding analyses, we demonstrate that while water proximity directly correlates with mobility enhancement at high temperatures, the hydration-induced mobility reduction observed at temperatures below 175 K is primarily propagated through the protein backbone.
1. Introduction
The interaction between water and proteins is known to dictate protein dynamics and functionality.1–4 Indeed, the presence of water is usually essential for a protein to function.5 Complete recovery of biological activity is only observed above monolayer coverage, i.e. 2–3 water molecules per residue or h = 0.38 (where h = grams of H2O per gram of protein),4 although the hydration threshold for activity is commonly defined to be h = 0.2.6 Hydration water, the ensemble of water molecules directly exposed to the macromolecule surface, can play an active role in the function of proteins;1,2 for example, water molecules mimic the substrate present in the biotin binding site of streptavidin, inducing a conformational transition of a protein loop.7 In addition to lacking biological activity, dehydrated biomolecules exhibit significantly reduced dynamics;5 in general, water is expected to have a plasticizing effect on protein dynamics, i.e. hydration should cause the system to exhibit enhanced mobility compared to its dry state.8,9 While this is true above the so-called ‘dynamical transition’ temperature (Td),10–12 a thermal condition above about 200–250 K (depending on the system) where segmental motions in weakly hydrated proteins become anharmonic, water-induced dynamical hindrance at low temperature has been observed in various proteins, such as the green fluorescent protein (GFP),13 pig liver esterase,14 lysozyme,15,16 and ribonuclease A (RNAse A).17 This dichotomy presents a complex and intriguing challenge in understanding the role of water as related to protein dynamics. Several interpretations have been given to the dynamical transition of biomacromolecules.16 For some systems, a direct dynamical coupling between water and macromolecules has been proposed.18–21 Additionally, it has been observed that the thermal onset of interfacial water mobility occurs at a universal, surface-independent temperature.22 The relationship between hydration water and protein dynamics has been widely investigated.9 For proteins in solutions, dynamical correlations between the protein and solvation layer have been detected regionally,23,24 as well as correlation between the local structure of a water–protein interface and protein fluctuations in a protein dimerization interface.25 In this respect, the occurrence of a correlation between the structure and dynamics of water molecules in the hydration shell of biomacromolecules has also been shown.26,27 However, the detailed mechanisms underlying water-induced effects on protein mobility are still not fully understood, especially for low temperature phenomena. For example, the hindrance observed below 200 K for weakly hydrated GFP relative to its dry state was attributed to the high β-structure content of this protein.13 However, this phenomenon has also recently been observed, both from experimental quasi-elastic neutron scattering (QENS)28 data and comparative molecular dynamics (MD) simulations, in apoferritin;29 a multi subunit molecule which is virtually devoid of β-sheet structured regions.30 Such contrasting observations suggest that there must be other factors driving the low temperature mobility attenuation of proteins in the weakly hydrated state. The underlying mechanism needs further clarification.By focussing on the dynamical behaviour of the apoferritin molecule, this study aims to characterise the observed water-induced effects on protein mobility and propose a temperature-dependent mechanism which may explain the trends reported widely in the literature, particularly the less studied hindrance effect at low temperatures.
To tackle this issue, our team have performed atomistic molecular dynamics (MD) simulations on horse spleen apoferritin (PDB structure: 2W0O,30Fig. 1), using a protocol which proved to be effective in reproducing the experimental behaviour.29
 | ||
| Fig. 1 PDB structure 2W0O of horse spleen apoferritin. Each of the 24 subunits is represented in a different colour. | ||
Apoferritin (molecular weight ≈ 444 kDa,31 for horse spleen apoferritin) is the apo-form of the iron-regulatory protein ferritin, present in many living organisms,32 and consists of an ordered non-covalent assembly of 24 structurally equivalent subunits with a high content of α-helical regions, forming a hollow spherical nanoparticle permeable to water, ions and small molecules.30,33 These features confer to apoferritin potentialities in biomedicine.34,35 The native biological assembly is extremely stable,36,37 although fibrillation of apoferritin has been detected under denaturing conditions.38
In brief, the mobility of horse spleen apoferritin in the dry amorphous state and at low hydration has been investigated experimentally by means of quasi-elastic neutron scattering (QENS).39,40 To authenticate the experimental findings, in silico models of the lyophilised and weakly hydrated material were built, with the dynamical behaviour of the model assemblies being simulated using MD methods. Comparative analyses, via observation of the temperature dependence of the mean squared displacement (MSD) of the protein hydrogen atoms, validated the experimental results; the MSD parameter being accessible via both experimental QENS29 measurements and simulated scattering functions extracted from the MD trajectories.
Consistent with the expected plasticizing effect of water on protein dynamics,8 a dynamical transition, i.e. a significant mobility enhancement in the weakly hydrated protein with respect to the dry state, is observed above ∼220 K.40 However, in contrast, and relative to the dry state, the hydrated protein mobility is attenuated in the temperature range of 100–150 K.40
While similar behaviour is reported in other macromolecular systems,13–17 the unique pseudo-spherical symmetry of the apoferritin molecule, as compared to the aforementioned proteins, offers the opportunity to identify correlations between the hydration-induced dynamical effects experienced by individual residues relative to both their specific location within the oligomeric assembly and proximity to the solvation shell. As a result, the influence of hydration on protein dynamics can be determined via:
(i) Characterisation of water's effect on the mobility (mean squared displacement, MSD) of individual amino acids, providing a detailed analysis of local dynamic changes,
(ii) Topological characterisation, by means of radial distribution function (RDF) analysis, of residues experiencing significant hydration-induced changes in mobility allowing the effect of proximity to external and internal interfaces, and thus to water molecules, on dynamics to be better understood,
(iii) Local characterisation of the solvent accessible surface area (SASA) and degree of hydration at the single residue level, providing further insights into hydration-induced effects on protein mobility,
(iv) Characterisation of protein–water hydrogen bonds and of backbone vs. side-chain MSD contributions.
Our findings reveal that, for the apoferritin molecule at least, above Td water directly enhances protein mobility, consistent with existing theories of the dynamical transition.16 Conversely, at low temperatures, the water-induced hindrance of protein mobility turns out to be an indirect effect, propagated through the protein's backbone.
2. Methods
Building on our experience simulating polymers in solution,41 atomistic molecular dynamics simulations were carried out on lyophilised (h = 0.05 g of D2O per g of protein) and weakly hydrated (h = 0.31 g of D2O per g of protein) horse spleen apoferritin. The models were built following a specific route, going from the hydrated system to the lyophilised one; this procedure mimics the primary drying step of an in vitro freeze-drying process.29Such a simulation protocol was benchmarked against quasi-elastic neutron scattering data and proved to be effective in reproducing experimental trends and behaviour.29 The hydration level chosen for the lyophilised system is that expected after primary drying,42 while the h value of the hydrated model was set to 0.31 in order to match the hydration degree used in the neutron experiment.
All details concerning the simulation protocol are reported in ref. 29, with the most salient aspects being summarised here. MD simulations were performed using the GROMACS software package43 (version 2020.6) and the OPLS-AA force field.44 The hydrated model was constructed using the 2W0O30 PDB entry, adding hydrogen atoms and then assembling the 24 subunits. A number of TIP3P45 water molecules equal to the number of D2O molecules in a sample with h = 0.31 was added after removal of crystallographic water molecules. The choice of OPLS-AA as the force field was based on its widespread use for the simulation of biomacromolecules; TIP3P being one of the water models compatible with the OPLS-AA energy landscape.46 The ionization state of the protein was that corresponding to pH 7, according to the experimental conditions before lyophilisation. Seven (7) Na+ ions per subunit were added for electroneutrality. The addition of each component was followed by energy minimization (steepest descent algorithm) with a 1000 kJ mol−1 nm−1 tolerance. To preserve structural integrity and minimize the loss of secondary structure, a preliminary 5 ns NPT simulation at 290 K was carried out on the weakly hydrated system using position restraints on protein heavy atoms. The final configuration was used as the starting structure for the NVT simulation at 290 K, run for a total of 150 ns.
The lyophilised model was obtained starting from the last configuration of the NVT simulation at 290 K of the hydrated system, removing water molecules in distinct steps until reaching a hydration level of h = 0.05. Each dehydration step was followed by a 10 or 20 ns NPT simulation at 100 K, to relax the system and mimic the low temperature lyophilisation conditions. Once a hydration state corresponding to h = 0.05 had been reached, the system was gradually brought from 100 to 290 K by performing four NPT simulations. A final NPT equilibration was carried out at 290 K for 400 ns; the structure obtained at the end of this simulation being used as the starting configuration for a subsequent NVT simulation at 290 K. The details of the in silico lyophilisation protocol are given in Section 1.1.2 of the ESI of ref. 29.
For both weakly hydrated and lyophilised apoferritin, after the simulation steps leading to the starting configuration at 290 K, NVT simulations were performed at 16 different temperatures, including 150 K. The simulation temperature scan was guided by the experimental data temperature points and performed on cooling from 290 to 10 K at 1 K ns−1. NVT isothermal runs of about 180 ns were collected, with a sampling frequency of 1 frame every 5 ps. The temperatures explored and the corresponding simulation times are reported in Table S1 of the ESI of ref. 29. An independent replica, simulated by following the above protocol, was also acquired to ensure reproducibility. The LINCS47 algorithm was used to constrain all bonds involving hydrogen atoms and allowed a simulation time step of 2 fs using the leapfrog integration algorithm.48 Periodic boundary conditions and the minimum image convention were applied. One series of simulations and one independent replica were acquired for both systems at all temperatures. Table 1 reports data on the systems’ composition and density, and on the size of the simulation box. Lyophilised and weakly hydrated systems are labelled as h = 0.05 and h = 0.31, respectively.
| Apoferritin | ||
|---|---|---|
| h = 0.05 | h = 0.31 | |
| a The box from the final frame of the NPT simulation is used for the subsequent NVT simulations. b Density in the NVT simulations. | ||
| Subunits | 24 | |
| Residues per subunit | 170 | |
| Biological assembly | 24-mer | |
| Water molecules | 1257 | 6844 |
| Sodium ions | 168 | |
| Total number of atoms | 69![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 051 051 |
85812 |
| Cubic box side before NPT simulation (nm) | 12.3 | 15 |
| Cubic box side after NPT simulationa (nm) | 10.4 | 12.3 |
| Densityb (g dm−3) | 722.6 | 532.5 |
The equilibration condition for the molecular structure in each trajectory was verified by monitoring the long-time behaviour of the root mean squared deviation (RMSD) of the protein's atoms (see Section SI.1.1 Root Mean Squared Deviation (RMSD) in the ESI†). The last 20 ns of the trajectories were typically considered for analysis; when not differently specified, the results presented are the mean values and associated error (standard deviations) from analysis of two individual production runs, i.e., the last 10 ns and the second-to-last 10 ns of the NVT trajectories. Trajectory analyses were conducted focusing solely on the dynamical behaviour of the hydrogen atoms since the simulations were benchmarked against neutron scattering experimental data,29 where the incoherent scattering from hydrogen atoms dominates the measured signal.28
The mobility of weakly hydrated and lyophilised apoferritin was investigated by analyzing the mean squared displacements (MSDs) of protein hydrogen atoms at 150 K and 290 K. The MSD parameter is calculated as a function of time using eqn (1):
| MSD(t) = 〈|r(t) − r0|2〉 | (1) |
Mobility was studied for each individual amino acid, averaging over the 24 subunits (see Section SI.1.7 Verification of the pseudo-symmetry assumption in residue-wise analyses in the ESI†). No correction for center of mass motion was applied (see Section SI.1.2 MSD per residue in the ESI†). To obtain the time independent MSD(t) value, for each 10 ns production run the MSD(t) curves were averaged over the interval 6–8 ns. In this range the MSD(t) response was seen to have broadly plateaued. The results presented in this work are the mean values and associated errors (standard deviations) from the two 10 ns production runs (for the effect of extending the length of the analysed trajectory see Section SI.1.2 MSD per residue in the ESI†). The residue-wise MSD was also selectively determined for backbone and side chain. All MSD calculations were performed considering hydrogen atoms only. Analyses of root mean squared fluctuation (RMSF) of all apoferritin hydrogen atoms detected that MSD and RMSF parameters give comparable information regarding protein mobility (see Section SI.1.8 in the ESI†).
The dynamics of protein–water hydrogen bonds in weakly hydrated apoferritin was evaluated by studying the normalized time autocorrelation function,
 | (2) |
SASA of selected apoferritin components has been calculated using a spherical probe with a radius of 0.14 nm and the values of van der Waals radii from the work of Bondi.50,51
Further details about trajectories analyses are given in Section SI.1 of the ESI.† The consistency between the original and independent replica simulations was verified for both lyophilised and weakly hydrated apoferritin (Section SI.1.2 in the ESI†). Graphic visualization was obtained using the molecular viewer software package VMD52 and the web-based open-source molecular viewer Mol* viewer.53
3. Results and discussion
As reported by us in ref. 29, the evolution of the mean square displacement as a function of temperature, MSD(T), for hydrated and lyophilised apoferritin, both experimentally detected from neutron scattering data and predicted from associated MD simulations, shows that hydration has two opposing effects on the overall average mobility of the protein: while there is a clear enhancement of the mobility upon hydration at temperatures greater than 175 K, the lyophilised protein is seen to be slightly more mobile than the hydrated system at low temperatures (T < 175 K).29To further this work, therefore, we now investigate the local mobility of apoferritin using MD tools to probe the simulations, better understand the trends observed on a molecular level and isolate the influence of water. Such analysis should provide critical insights into the different effects of hydration on protein's dynamics at low and high temperatures.
First, the mobility of apoferritin was studied on a local level by computing an MSD value for each individual amino acid, with an average over the 24 subunits being presented, at 150 and 290 K. In this way, the impact on local mobility relative to the chemical nature of the residues, as well as their position in the primary sequence, was explored. Next, topological characterisation, by means of RDF analysis, of residues experiencing significant hydration-induced changes in mobility allowed the effect of proximity to external and internal interfaces, and therefore to water molecules, on dynamics to be better understood. Then, the analysis of residue-resolved hydration and SASA focussed possible correlations between local hydration degree and hydration-induced effects on protein mobility. Finally, MSD per residue, also considering backbone and side-chain MSD components, and hydrogen bonding analyses allowed us to propose mechanisms for hydration-induced mobility changes at high and low temperatures.
3.1 MSD per residue analysis
By considering the mean squared displacement parameter per residue, heterogeneities in mobility along the primary apoferritin structure, both in the lyophilised and hydrated material, could be explored. The MSD parameter was considered and calculated for each individual amino acid at 290 K and 150 K. For the reason given earlier, only apoferritin hydrogen atoms were considered, and the result reported per residue was the average over the 24 subunits of the biological assembly. MSD per residue values along the primary structure at 290 and 150 K are shown in Fig. SI.4 and SI.5 (ESI†), respectively, comparing lyophilised and weakly hydrated protein. The corresponding distributions are reported in Fig. 2(a) and (b).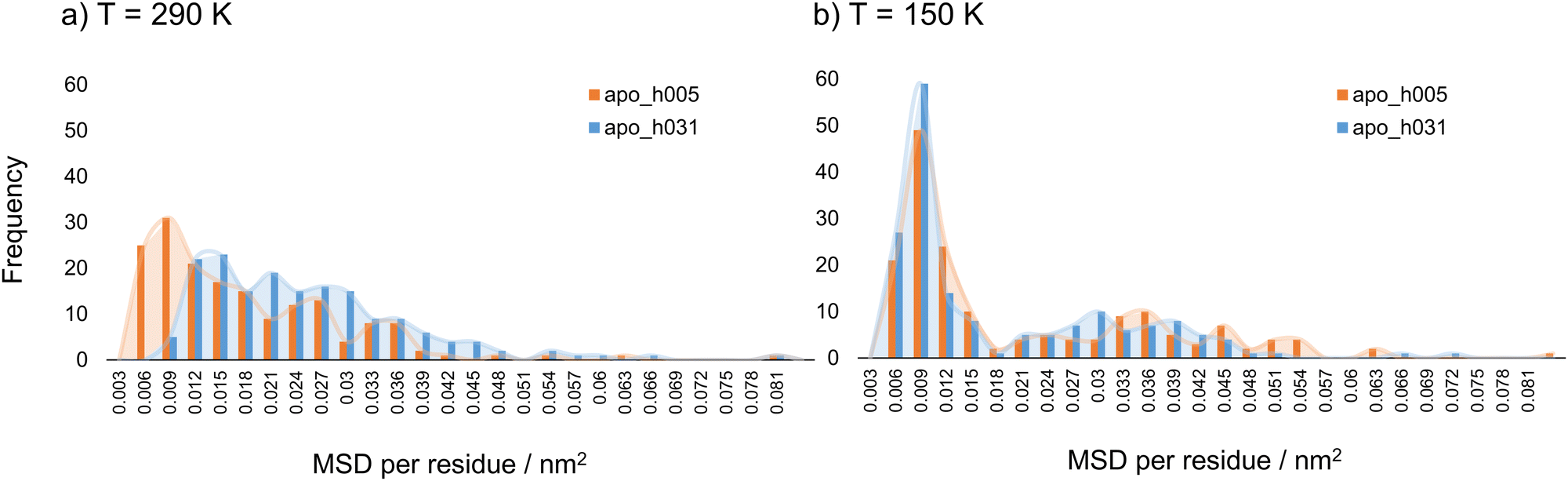 | ||
| Fig. 2 Distributions of MSD per residue values for apoferritin in the lyophilised (apo_h005, orange) and weakly hydrated (apo_h031, blue) states at (a) T = 290 K and (b) T = 150 K. | ||
Considering first the results at 290 K (Fig. 2(a)), multimodal distributions with fractions of comparable weight are detected at this temperature for both hydration states. The most populated fraction is associated with those residues that exhibit the lowest mobility; fractions with decreasing population being observed as a function of increasing residue mobility. For weakly hydrated apoferritin the whole distribution curve moves to higher MSD per residue values, as compared to the distribution of lyophilised material, highlighting the hydration-induced mobility enhancement at 290 K. At 150 K (Fig. 2(b)), however, the distributions of MSD per residue values are approximately bimodal, and this decrease in temperature leads to a drastic reduction in the population of residues with intermediate MSD values. An MSD of 0.009 nm2 is the highest frequency value for both lyophilised and weakly hydrated states at 150 K. Interestingly, this MSD value has the highest frequency also at 290 K for the lyophilised state. Fig. 2(b) illustrates the hydration-induced mobility hindering at 150 K at the single amino acid level: the distribution curve for lyophilised apoferritin exceeds the one of weakly hydrated protein starting from the MSD per residue value of 0.012 nm2 and the hindering effect is relevant for residues with higher mobility, for some of which the mobility in the hydrated state is almost suppressed. Although the overall effect of hydration on apoferritin mobility at 290 and 150 K is an enhancement and reduction, respectively, the influence on residue mobility at a given temperature appears to be non-uniform, with both increases and decreases in amino acids’ MSD (Fig. SI.4 and SI.5, ESI†). To better explore such changes, the residues were classified according to the variation in mobility upon hydration, relative to their dry state motion at the same temperature. The change in mobility (%) was defined using eqn (3):
 | (3) |
The residues were then classified according to change in mobility (%) upon hydration, as described in Table 2.
| Change in mobility | Effect of hydration |
|---|---|
| <−10% | Hindrance |
| From −10% to +10% | Negligible effect |
| From +10% to +60% | Moderate enhancement of mobility |
| >+60% | Great enhancement of mobility |
The rationale behind this classification system is discussed in Section SI.1.3 of the ESI.† Results obtained at 290 K are reported in Fig. SI.6 (ESI†) and summarized in Table 3.
| T = 290 K | Number of residues | Percentage of residues (%) |
|---|---|---|
| Hindrance | 12 ± 2 | 7 ± 1 |
| Negligible effect | 28 ± 3 | 17 ± 1 |
| Moderate enhancement | 53 ± 5 | 31 ± 3 |
| Great enhancement | 77 ± 9 | 45 ± 5 |
At 290 K the prevalent effect of hydration is an increase in mobility, with 45% of residues displaying a substantially enhanced mobility and 31% of residues showing a moderate rise in mobility. Despite this general trend in mobility enhancement upon hydration, there are also instances where certain residues show negligible changes or even decreases in mobility (Fig. SI.6, ESI†). This variability suggests that water's effect on residue mobility is not uniform across all residues and may depend on specific structural or chemical properties. The influence of structural or chemical properties was studied by evaluating the contribution of each amino acid class to the observed hydration-induced effects (Table 4).
| T = 290 K | Hindrance | Negligible effect | Moderate enhancement | Great enhancement |
|---|---|---|---|---|
| Methyl-containing | 9 ± 2 | 17 ± 2 | 30 ± 2 | 5 ± 2 |
| Charged | 0 | 2 | 10 ± 3 | 38 ± 3 |
| Polar neutral | 1 | 4 ± 1 | 8 ± 1 | 18 ± 1 |
| Aromatic | 2 | 5 | 2 ± 1 | 6 ± 1 |
| Aliphatic, non-CH3 | 0 | 0 | 3 ± 3 | 10 ± 3 |
Charged residues give the most significant contribution to the greatest mobility enhancement upon hydration, followed by neutral polar residues. Since these are hydrophilic amino acids, this finding suggests that water proximity and water–protein interaction likely play a role in high temperature hydration-induced phenomena, as discussed later. The results of the residue-resolved MSD analysis at 150 K are reported in Fig. SI.7 (ESI†) and summarized in Table 5.
| T = 150 K | Number of residues | Percentage of residues (%) |
|---|---|---|
| Hindrance | 91 ± 1 | 54 |
| Negligible effect | 54 ± 1 | 32 |
| Moderate enhancement | 23 | 13 |
| Great enhancement | 2 ± 1 | 1 |
The main effect of hydration at 150 K is hindrance, observed in 54% of residues. The possibility that the hindrance effect results from the local formation of ice or an uneven distribution of water molecules in the system was explored by analysing the distribution of the number of water–water hydrogen bonds per water molecule (Fig. SI.18, ESI†) and the water cluster size distribution (Fig. SI.19, ESI†) in weakly hydrated apoferritin at 290 K and 150 K. The maximum number of hydrogen bonds per water molecule at 150 K is much lower than 2 (Fig. SI.18, ESI†), the value expected for a tetracoordinated water molecule in ice. This finding rules out the formation and presence of ice nuclei. Similarly, the possibility of hindrance being caused by densely hydrated regions was excluded, as the majority of water molecules remain either single or form small clusters (Fig. SI.19, ESI†). While overall protein mobility shows a slowdown upon hydration, as observed experimentally and in simulated all-hydrogen-atoms MSD analysis29 and confirmed by these results, water affects residue mobility in a non-uniform manner, with some residues being either not influenced or showing an increased mobility (Fig. SI.7, ESI†). Hydration-induced effects are in fact dependent on the residues’ structural or chemical properties, with methyl-containing residues contributing most significantly to the hindered behaviour, followed by charged and neutral polar residues (Table 6).
| T = 150 K | Hindrance | Negligible effect | Moderate enhancement | Great enhancement |
|---|---|---|---|---|
| Methyl-containing | 34 | 17 ± 2 | 8 ± 2 | 2 ± 1 |
| Charged | 23 ± 1 | 22 ± 1 | 5 | 0 |
| Polar neutral | 18 ± 1 | 6 ± 3 | 7 ± 2 | 0 |
| Aromatic | 11 ± 1 | 2 ± 1 | 2 ± 1 | 0 |
| Aliphatic, non-CH3 | 5 ± 1 | 7 ± 1 | 1 | 0 |
It is noteworthy that, within the subset of hindered residues, separating the contributions of methyl and non-methyl residues to the change in mobility upon hydration (eqn (3)) reveals that methyl-containing residues exhibit, on average, a greater hindrance effect (−27 ± 2%) as compared to non-methylated residues (−19 ± 1%). This difference can be attributed to methyl groups’ more external position in the residue, allowing them to move more freely as compared to non-methylated moieties and making methyl-containing species intrinsically more sensitive to constraints. Considering that the extent of the rotational methyl motion is independent of the hydration level (Fig. 4a of ref. 29), its removal should increase the signal-to-noise ratio in the residue-resolved mobility analysis. Therefore, the MSD per residue parameters and changes in mobility (%) upon hydration (eqn (3)) were also calculated using trajectories in which the rotation of methyl groups was removed,29 at 150 and 290 K (see SI.2.3 MSD per residue analysis with methyl rotation removal, in the ESI†). In general, the outcome of such analysis makes the temperature-dependent effects of hydration on mobility more evident. At 290 K, the number of residues with greatly enhanced mobility increases (Table SI.1, ESI†), with methyl-containing residues now showing great mobility enhancement (Table SI.2, ESI†), as opposed to the case of trajectories with methyl rotations. In a similar way, the hindrance effect at 150 K is accentuated in the trajectories without methyl rotations (Table SI.3, ESI†), with the number of methyl-containing residues having hindered mobility increasing (Table SI.4, ESI†).
At both temperatures, some regions of the protein exhibit clusters of residues with similar changes in MSD, suggesting potential localized effects of hydration on protein dynamics (see Fig. SI.6 and SI.7, ESI†). Among these, it is worth mentioning that the cluster of hindered residues at 150 K formed by residues 104 to 118 (Fig. 3(a)) includes most of the residues belonging to the 3-fold channels of the protein (residues 109 to 120, Fig. 3(b)), which serve as the main entry and exit pathways for iron;54,55 the Fe(II) uptake is facilitated by an electrostatic gradient, allowing the ion to enter this funnel-shaped hydrophilic channel, whose conserved carboxy groups act as transient binding sites.55
 | ||
| Fig. 3 PDB 2W0O structure of apoferritin, highlighting (a) residues 104 to 118, showing suppressed mobility upon hydration at 150 K, in blue, and (b) residues 109 to 120, forming the 3-fold channels of the protein, in purple. (c) PDB 2W0O structure of apoferritin with residues colour-graded according to the number of water molecules within a 0.35 nm radius, using a blue-grey-red scale; residues 104 to 120 are highlighted. | ||
On the other hand, the cluster of residues with greatly enhanced mobility at 290 K formed by residues 145 to 153 (Fig. 4(a)) is located near the 4-fold channels of apoferritin (residues 160 to 171, Fig. 4(b)). These channels are hydrophobic and therefore not permeable to ions, but are thought to be permeable to oxygen.54,55 Additionally, they have smaller pore diameters56 compared to 3-fold channels, which inhibits uptake. In this context, the enhanced mobility observed upon hydration at 290 K may be correlated with the need for greater flexibility to allow molecules to pass through the channel.
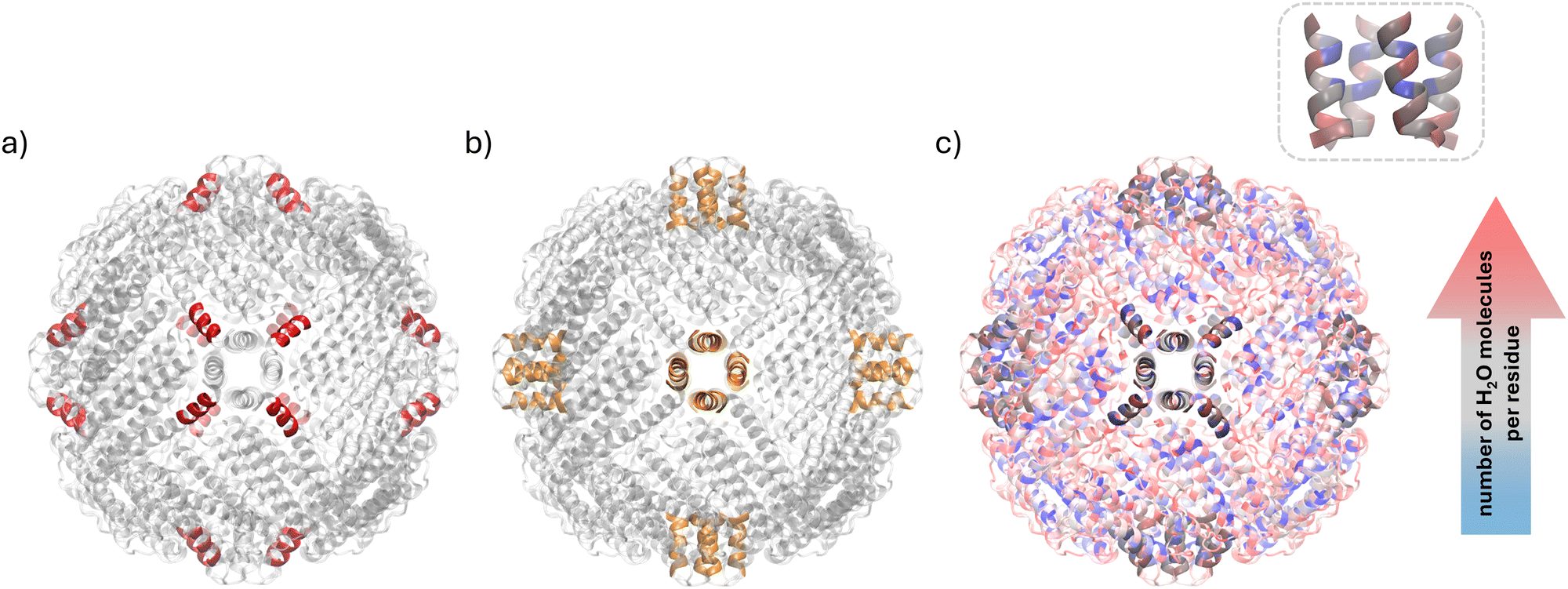 | ||
| Fig. 4 PDB 2W0O structure of apoferritin, highlighting (a) residues 145 to 153, having greatly enhanced mobility at 290 K, in red, and (b) residues 160 to 171, forming the 4-fold channels of the protein, in orange. (c) PDB 2W0O structure of apoferritin with residues colour-graded according to the number of water molecules within a 0.35 nm radius, using a blue-grey-red scale; residues 145 to 153 and 160 to 171 are highlighted. (c) Inset: Residues 160 to 171. | ||
The potential correlation between water proximity and hydration-induced effects on protein mobility was investigated by evaluating the local hydration degree of weakly hydrated apoferritin. The analysis consisted of calculating the number of water molecules within a 0.35 nm radius of each residue in the last and second-to-last 10 ns production runs at 290 and 150 K. The PDB 2W0O structure of apoferritin was then visualized, with residues colour-graded based on the average number of water molecules per residue (averaged over the 24 subunits) using a blue-grey-red scale (Fig. 3(c) and 4(c)). Significant (influenced and channel forming) residues were highlighted. Overall, most residues with greatly enhanced mobility at 290 K (Fig. 4(a)) appear to be medium to highly hydrated, with water mostly located near residues closer to the 4-fold channels (Fig. 4(b)). Residues forming the 4-fold channels also have mostly medium-high hydration levels (Fig. 4(c), inset). Conversely, residues with hindered mobility (Fig. 3(a)) and forming 3-fold channels (Fig. 3(b)) appear to be, in general, only weakly (medium-low) hydrated, with the only notable peak in the number of nearby water molecules being associated with ASP 112; the residue forming the highest number of hydrogen bonds at 150 K (see Section 3.4). While the colour-graded visualization provides a preliminary analysis, these observations were supported by radial distribution function analyses (see Section 3.2).
3.2 Radial distribution function analysis
Having identified those species whose mobility is most influenced by water at both low and high temperatures, radial distribution function (RDF) analysis was performed to investigate how hydration affects mobility of hydrogen atoms depending on their position relative to the protein's internal and external interfaces (see Section SI.1.4, ESI†). The centre of mass of the whole biological assembly was used as the reference position of these RDFs, that, due to the pseudo-spherical symmetry of apoferritin, provide an overall picture of the distribution of the different species throughout the protein shell, allowing the relationship between hydration-induced mobility effects and water proximity to be explored. RDFs of weakly hydrated apoferritin were calculated for: (i) hydrogen atoms associated with residues having greatly enhanced mobility at 290 K, (ii) hydrogen atoms associated with residues with hindered mobility at 150 K, and (iii) methyl and non-methyl hydrogen atoms at 150 K; radial distributions being compared to the RDF of water molecules at the same temperature.The RDF of hydrogen atoms that experience a great enhancement of mobility at 290 K (Fig. 5) shows two peaks (red curve) that broadly correlate with water molecule distribution (blue curve), suggesting a direct effect of water proximity on the mobility enhancement observed at high temperatures. Conversely, the radial distribution of hindered hydrogen atoms at 150 K (Fig. 6(a)) and that of methyl hydrogens (Fig. 6(b)) suggest the existence of an indirect mechanism for hydration-induced hindrance at 150 K. In this respect, the double peak shape of the RDFs associated with water molecules in Fig. 5 and 6 shows, irrespective of temperature, a higher local concentration of water at the internal and external surfaces of the pseudo-spherical biological assembly. The double peak shape of the RDF associated with those hydrogen atoms belonging to residues with greatly enhanced mobility in Fig. 5 is a consequence of the uneven distribution of water molecules and indicates the proximity of such residues to water at the temperature of 290 K.
 | ||
| Fig. 5 RDFs of water molecules (blue), hydrogens belonging to residues with greatly enhanced mobility (red, as defined by column 4 Table 4), and hydrogens belonging to all other residues (grey, as defined by columns 1–3 Table 4). T = 290 K. The protein's COM was chosen as the point of reference for RDF calculation. Inset: Equatorial section of hydrated apoferritin. Protein atoms are coloured in blue, water oxygen atoms are red. | ||
 | ||
| Fig. 6 (a) RDFs of water molecules (blue), hydrogens belonging to hindered residues (purple, as defined in column 1 of Table 6), and hydrogens belonging to all other residues (grey, as defined in columns 2–4 in Table 6). (b) RDF of hindered residues (i.e. the purple curve in (a)) split into CH3 (orange) and non CH3 (green), and RDFs of water molecules (blue). T = 150 K. The protein's COM was chosen as the point of reference for RDF calculation. | ||
3.3 Residue-resolved analysis of solvent accessible surface area and hydration
To support the findings suggested by RDF analyses, the exposure and extent of hydration in weakly hydrated apoferritin were explored at a local level. This was done by computing, for each of the 170 amino acids of the subunit, the SASA and the number of water molecules located within 0.35 nm of any atom of that residue. This cutoff distance approximates the thickness of the water first coordination shell surrounding a hydrophilic moiety. The results obtained at 290 and 150 K, with an average over the 24 subunits of the biological assembly, are reported in Fig. 7 and 8, respectively. | ||
| Fig. 7 Number of H2O molecules within a 0.35 nm radius of each atom of a residue as a function of the SASA of that residue at 290 K for weakly hydrated apoferritin. Average values over the 24 subunits and time. (a) Data for all residues. Surface- and water-exposed residues belonging to class (i) are those falling in the top-right part of the plot (shaded box area). The dotted line represents the correlation between SASA and a residue's degree of hydration, which is expected for proteins in solution but only holds at low SASA values in a weakly hydrated system. To aid visualization, only representative error bars (showing the minimum and maximum size of the error bar for each observable) are included; errors are standard deviations over the last 10 ns of the trajectory. (b) Data for residues with greatly enhanced mobility (red) and not influenced residues (grey) with respect to the dry system. (c) Data for charged residues, polar neutral residues, and all other residues. | ||
 | ||
| Fig. 8 Number of H2O molecules within a 0.35 nm radius of each atom of a residue as a function of the SASA of that residue at 150 K for weakly hydrated apoferritin. Average values over the 24 subunits and time. (a) Data for all residues. Surface- and water-exposed residues belonging to class (i) are those falling in the top-right part of the plot (shaded box area). The dotted line represents the correlation between SASA and a residue's degree of hydration, which is expected for proteins in solution but only holds at low SASA values in a weakly hydrated system. To aid visualization, only representative error bars (showing the minimum and maximum size of the error bar for each observable) are included; errors are standard deviations over the last 10 ns of the trajectory. (b) Data for residues with hindered mobility (blue) and not influenced residues (grey) with respect to the dry system. (c) Data for methyl-containing residues, charged residues, and all other residues. | ||
Typically, for a protein in solution, SASA and a residue's degree of hydration correlate i.e. the larger the SASA the higher the hydration. However, in this system such correlation only holds in the range of low SASA values (see the dotted lines in Fig. 7(a) and 8(a)), because of the weak hydration level (h = 0.31 g D2O per g of protein). In the correlation range, the slope of the linear fit line is greater at 150 K, as compared to 290 K, because of the higher surface density of water molecules at low temperature, as proved by the higher hydrogen bond connectivity between them (see Section SI.7.1 of the ESI†).
The amino acids, ALA119 and VAL101, with hydrophobic side-chains located on the external surface of the apoferritin spherical assembly are examples of residues where the SASA/hydration degree correlation breaks down. As shown in Fig. 7(a), these two residues have quite large SASA values of ≈0.9 nm2 but very low hydration (less than 1 water molecule). As Fig. 7(a) and 8(a) demonstrate, for weakly hydrated apoferritin, SASA-only is not a useful metric with which to probe the local extent of hydration.
However, such analysis does highlight that at 290 K (Fig. 7(b)) most residues with greatly enhanced mobility (according to the classification of eqn (3)) are highly hydrated. In contrast, those not influenced have a lower hydration degree, confirming the result shown by the RDFs in Fig. 5.
Moreover, Fig. 7(c) shows that charged and polar amino acids, i.e. those categories exhibiting the highest hydration-induced mobility enhancement at 290 K (see Table 4), are significantly hydrated. On the other hand, the results at 150 K (Fig. 8(b)) suggest no direct correlation between the extent of local hydration and hindered mobility, as shown by the overall view provided by the RDFs in Fig. 6(a). In this respect, Fig. 8(c) does confirm that methyl-containing amino acids, i.e. the category undergoing the highest hydration-induced mobility suppression at 150 K (see Table 5), largely exhibit a low hydration level.
Introducing a two-parameter criterion based on the results in Fig. 7(a) and 8(a), it is possible to discriminate between surface- and water-exposed residues (class (i)) and buried OR scarcely hydrated residues (class (ii)). The aim of this classification is to quantitatively detect effects on mobility coming from a direct contact with water molecules. The findings of the MSD per residue analysis have been then elaborated by distinguishing between the above defined classes of residues in terms of, first, hydration-induced mobility effect (Tables SI.6 and SI.8, ESI†) and, subsequently, hydration-induced mobility effect and chemical characteristics (Tables SI.7 and SI.9, ESI†). A consistent picture of water-induced effect on mobility, water- and surface-exposure, and amino acid class is obtained, where the direct influence of water proximity on protein mobility is clearly shown at 290 K, but missing at 150 K (see Section SI.6, ESI†).
3.4 Protein–water hydrogen bonding analysis
Considering the results of RDF and residue-resolved hydration analyses, we investigated the mechanisms underlying the influence of water on protein's dynamics by studying protein–water hydrogen bonds (HBs).First, the total number of protein–water hydrogen bonds was calculated (see Section SI.1.5, ESI†) as a function of temperature in the range 100–290 K (Fig. 9). The number of hydrogen bonds formed shows a non-linear decrease with increasing temperature, as expected.57
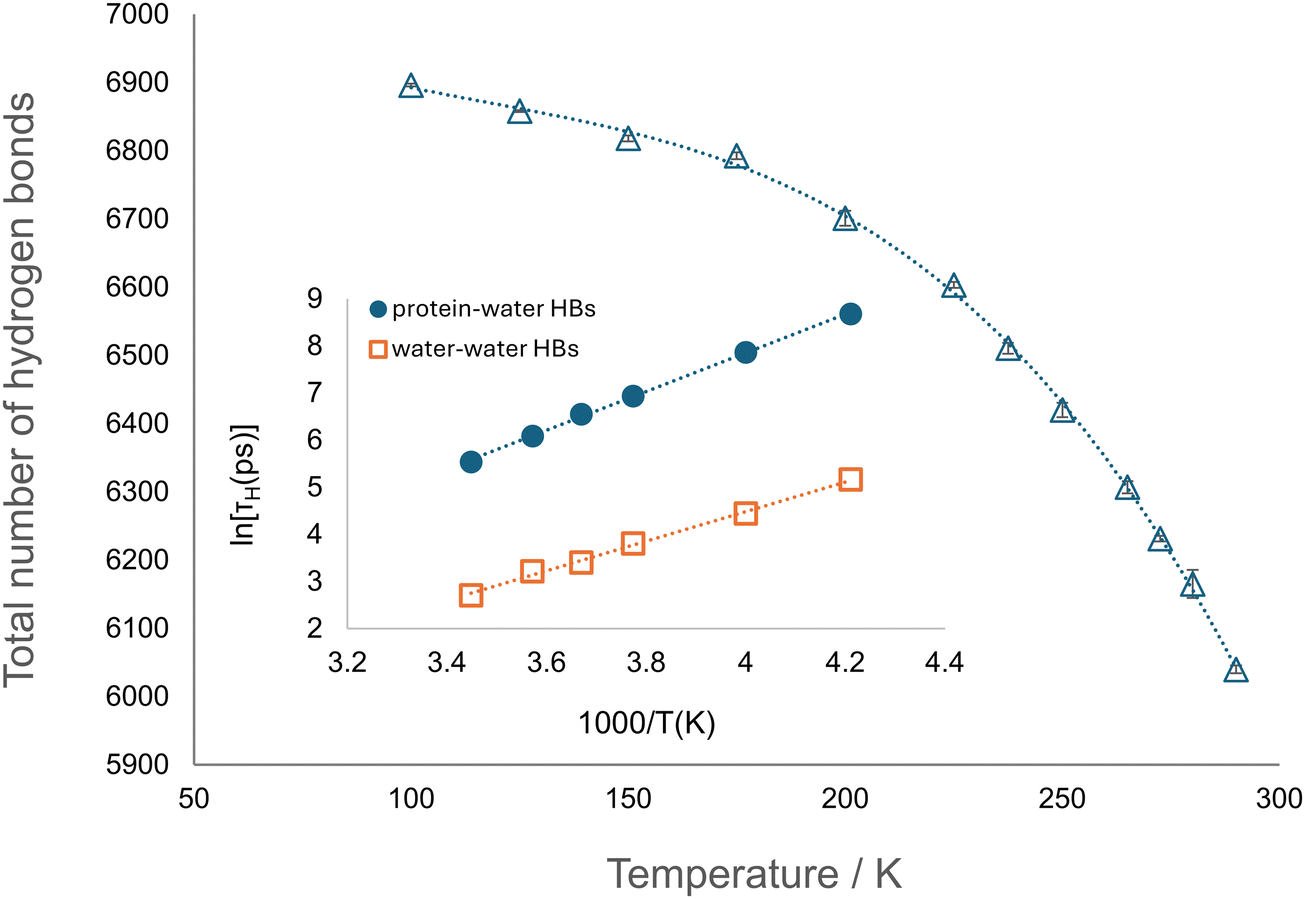 | ||
| Fig. 9 Total number of protein–water hydrogen bonds as a function of temperature (T = 100–290 K). The results and errors are averages and standard deviations as determined by analysing the last and second-to-last 20 ns intervals of each trajectory, respectively. Dotted line is a guide to the eye. Inset: Characteristic relaxation time of protein–water (blue) and water–water (orange, data from Commun. Chem., 2024, 7, 83) hydrogen bonds in hydrated apoferritin as a function of temperature. The results and errors are averages and standard deviations from analysis of the last and second-to-last 20 ns intervals of each trajectory, respectively. Error bars, if not visible, are within the symbol size. | ||
To further characterise the protein–water interaction, the hydrogen bond autocorrelation function CH(t) was calculated. The time decay of this function is due to the exchange of water molecules hydrogen bonded to protein, occurring with a characteristic relaxation time, τH, defined as the time when CH(t) reaches the value e−1. By fitting an Arrhenius form for ln(τH) as a function of T−1, an activation energy of 34.1 ± 0.5 kJ mol−1 is obtained (Fig. 9 (inset)). This is in agreement with the finding of a system independent protein–water hydrogen bond relaxation process with an energy barrier of ∼30–35 kJ mol−1.22 It is worth noting that this barrier has a common value to that observed for water–water hydrogen bond relaxation in apoferritin29 and other macromolecular systems.22
To assess how the interaction with water influences residue mobility, the presence of protein–water hydrogen bonds formed for more than 20% of the 290 and 150 K 10 ns trajectories, even if only intermittently (i.e., not continuously present in each 5 ps sampled frame of the 10 ns trajectory but instead making and breaking at various times), was considered.
At 290 K there is a direct correlation between enhanced mobility and hydrogen bonding with water: residues that exhibit a greater increase in mobility upon hydration tend to have stronger bond interactions with water molecules (Fig. 10).
 | ||
| Fig. 10 Number of hydrogen bonds formed for at least 20% of the trajectory (even non-continuously) at 290 K by amino acids experiencing (a) hindrance, (b) no effect, (c) moderate enhancement of the mobility, and (d) great enhancement of the mobility upon hydration. Each panel displays results specific to a particular class of amino acids, with x-ticks representing only residues within that class. Note that x-ticks without corresponding bars indicate amino acids that did not form hydrogen bonds. Results and errors are averages and standard deviations over the last and second-to-last 10 ns intervals of each trajectory, respectively. Individual versions of each panel, with expanded x-axes, are reported in Fig. SI.21 (ESI†). | ||
This correlation is absent at 150 K (Fig. 11). At this temperature, the distribution of the number of HBs among residues with different hydration-induced effects is more uniform. However, notable peaks (i.e. >80 HBs) are observed, with 8 out of these 13 peaks corresponding to residues that experience hindrance upon hydration. These 8 residues include charged amino acids (GLU or ASP) and a terminal leucine (LEU171). Most neighbouring residues (11 out of 15) are also hindered (see Fig. SI.7, ESI†). These results show that only a small (∼5%) fraction of amino acids characterised by both high water exposure and strong residue–water interaction (i.e., a significantly high number of HBs) are directly hindered by water proximity. For instance, residue ASP 112, which forms the highest number of persistent hydrogen bonds with water (Fig. 11(a)) and is located in the 3-fold channel (Fig. 3(b)), was found to participate in a hydrogen bond/dipolar interaction network. This network includes water molecules and atoms of ASP 112 and neighbouring residues, connected by hydrogen bonds and dipolar interactions (Fig. SI.20, ESI†). Such connectivity is probably involved in the water-induced hindering effect of this region. Since this is not a common feature of the system, it appears that the generalised (i.e., observed in 54% of residues) mobility attenuation observed at 150 K arises from propagation of a hindrance effect along the biopolymer chain, as discussed in Section 3.5.
 | ||
| Fig. 11 Number of hydrogen bonds formed for at least 20% of the trajectory (even non-continuously) at 150 K by amino acids experiencing (a) hindrance, (b) no effect, (c) moderate enhancement of the mobility, and (d) great enhancement of the mobility upon hydration. Each panel displays results specific to a particular class of amino acids, with x-ticks representing only residues within that class. Note that x-ticks without corresponding bars indicate amino acids that did not form hydrogen bonds. Results and errors are averages and standard deviations over the last and second-to-last 10 ns intervals of each trajectory, respectively. Individual versions of each panel, with expanded x-axes, are reported in Fig. SI.22 (ESI†). | ||
3.5 Backbone vs. side-chain contributions to water-induced mobility changes
Finally, to further investigate the temperature-dependent mechanism driving the hydration-induced effects on mobility, we computed mean square displacement parameters for each individual amino acid at 150 K and 290 K, separating out backbone and side-chain hydrogen contributions to the total hydrogen atom MSD. First, for each residue, the MSD was computed including only the backbone hydrogen atoms, which account for about 24% of the total hydrogen atoms. Next, the MSD parameter was computed including only side-chain hydrogen atoms of each amino acid. The values of the residue-wise MSD for the backbone and side-chain of dry and weakly hydrated apoferritin are reported in Fig. SI.10 and SI.11 (ESI†) for the temperatures of 290 and 150 K, respectively. At 290 K both systems show that the residue-wise MSD of the backbone is lower than that of the side chain for almost all residues. This result confirms the direct role of water in affecting the protein local mobility at this temperature, since the SASA and degree of hydration of side-chains are greater than those of the backbone (Fig. SI.13 and SI.14, ESI†). Comparing the mobility of dry and weakly hydrated apoferritin at 290 K, the residue-wise MSD for the backbone is always greater in the solvated system. This trend generally extends to the side-chains as well, although there are some exceptions occurring for amino acids with hydrophobic side-chains and thus lower water affinity. At 150 K (Fig. SI.11, ESI†), in contrast to 290 K (Fig. SI.10, ESI†), there are several amino acids for which the MSDs of the backbone and side chains are comparable. This result suggests a dynamical coupling between these residue components. The hydration-induced hindering, as shown by the greater MSD values of the dry system, in some hydrophobic residues is more accentuated for side chains, as compared to the backbone (see residues ALA14, PHE50, LEU93, and ALA109 in Fig. SI.11, ESI†).The change in mobility upon hydration (%) for backbone and side-chain components was then calculated using eqn (3). Finally, to assess the potential influence of backbone and side-chain contributions relative to each other, the average change in mobility across all residues was calculated separately for backbone and side-chain hydrogen atoms. The results are reported in Table 7.
| Average % change backbone | Average % change side-chain | |
|---|---|---|
| 150 K | −7.2 ± 0.1 | −7.5 ± 0.7 |
| 290 K | 88 ± 1 | 72 ± 1 |
At 150 K the backbone and side-chain residue mobilities show, within error, the same hindrance upon hydration. This is further evidenced by strong agreement (Fig. SI.12, ESI†) between the distribution of backbone and side-chain hydrogen atom mobility changes. This clear correlation between backbone and side-chain hydration-induced mobility changes at 150 K suggests that the low temperature water-induced hindrance observed in apoferritin occurs via an indirect mechanism, where the slowing of backbone motion propagates to affect side-chain motion.
In contrast, at 290 K, the hydration-induced mobility increases experienced by the backbone and side-chain hydrogen atoms are different, suggesting that the backbone propagation mechanism proposed is exclusive to low temperature phenomena. Note that, at 290 K, backbone hydrogen atoms show greater mobility enhancement on average compared to side-chain hydrogen atoms because their mobility changes more uniformly upon hydration across all protein residues, leading to a narrower distribution of values (see Fig. SI.12, ESI†). While the only effect of hydration on the backbone is that of mobility enhancement, side-chain hydrogens experience a wider range of effects, including hindered or unaffected mobility (Fig. SI.10, ESI†). This heterogeneity leads to a lower average change in mobility (%) for side-chains, as reported in Table 7.
To better understand the role of water in hydration-induced changes to backbone and side-chain mobility, the hydration of apoferritin's backbone and side-chain atoms was studied by separately calculating the SASA, degree of hydration (number of water molecules within a 0.35 nm radius), and protein–water hydrogen bonding for each atom group at 290 and 150 K. Irrespective of temperature and hydration state, the SASA of the side chains is much larger than that of the backbone (Fig. SI.13, ESI†). However, the relative hydration of side chains vs. backbone is not so accentuated (Fig. SI.14, ESI†), since the water content is not sufficient to saturate the protein surface; a surface populated also by hydrophobic side chains with a lower water affinity as compared to the hydrophilic backbone. The relative hydrogen bonding capability of side chains and backbones (Fig. SI.15, ESI†) is similar to the corresponding relative hydration. In terms of distribution of water molecules surrounding the side chains and backbone, no significant temperature dependant difference is detected. This finding shows that the different effect of hydration on protein mobility at 290 and 150 K does not depend on a different partition of water molecules between the two (side group and backbone) amino acid components.
4. Conclusions
Experimental observations and molecular dynamics simulations of horse spleen apoferritin reveal that while protein mobility is enhanced upon hydration above the dynamical transition temperature, the presence of water hinders protein internal motion at low temperatures (T < 175 K).29,40 This study focuses on elucidating the temperature-dependent mechanisms underlying the interplay between hydration and protein dynamics. The high temperature regime is characterised by significant enhancement in residue mobility which can be directly correlated with water proximity, as evidenced by radial distribution function, SASA, local hydration degree, and hydrogen bonding analyses. While water directly facilitates mobility enhancement above the dynamical transition temperature (Td ∼ 220 K), it imposes a more complex, indirect hindrance at low temperatures, suggesting a mechanistic shift from direct to indirect effects of hydration. At temperatures below 175 K water proximity directly hinders motion only for a small fraction of residues (about 5%), with the overall reduction in mobility, involving more than 50% of residues, primarily arising from backbone propagation effects which indirectly mute side-chain dynamics.Considering the structure of proteins for which a water-induced attenuation of mobility has been detected at low temperature using neutron spectroscopy (see Table SI.10, ESI†), the presence of a specific secondary structure does not seem to be the determining factor for this effect. However, these proteins share the feature to have relatively long polypeptide chains, with 124 or more residues. Vice versa, insulin, a small protein of 51 amino acids,29 and shorter chain homopolypeptides,58 do not display water-induced reduction of mobility below 175 K. While the level of hydration, h, is likely to play a role, we hypothesise that the propagation of mobility hindering could be amplified in long chain weakly hydrated polypeptides.
Finally, concerning the role of the backbone in this low temperature phenomenon, backbone-specific vibrational motions of myoglobin were found to be damped in the presence of hydration water at 150 K, while a similar damping was undetected for a hydrated amino acids mixture with the chemical composition of myoglobin but lacking the polypeptide chain.59
In this work we propose a mechanism for the influence of water on apoferritin dynamics. Our contribution provides working hypotheses to be explored in future experiments and simulations of other biomacromolecules and biomimetic polymers, to increase the overall knowledge on how the interplay between water and complex molecules determines the system behaviour.
Author contributions
M. Telling: conceptualization, supervision, investigation, methodology, validation, visualization, writing – original draft, writing – review & editing. E. Chiessi: supervision, data curation, methodology, resources, validation, visualization, writing – original draft, writing – review & editing. E. Bassotti: data curation, formal analysis, methodology, resources, validation, visualization, writing – original draft, writing – review & editing. G. Paradossi: resources, validation, visualization, writing – review & editing.Data availability
All the data presented in support of the findings and conclusions of this study, and present in the paper and/or the ESI,† have been uploaded as an appendix to the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the CINECA award, under the ISCRA initiative, for the availability of high-performance computing resources and support, and the UK's Science and Technology Facilities Council's (STFC) SCARF (Scientific Computing Application Resource for Facilities) High Performance Computing cluster for computing support. We also thank STFC's ISIS Pulsed Neutron and Muon facility, UK, for access to the neutron instrumentation referred to in this work. EB acknowledges the Italian Ministry of University and Research for PhD support. EC and EB acknowledge financial support from the PRIN2022 project “Exploiting double thermoresponsivity for a new class of biocompatible Composite MicroGELS (Co-MGELS)” number 2022PAYLXW (Italian Ministry of University and Research, MUR). The authors acknowledge, and appreciate, UK Research and Innovation (UKRI) open access publication funding.References
- M. Tarek and D. J. Tobias, Biophys. J., 2000, 79, 3244–3257 CrossRef CAS PubMed.
- F. Mallamace, C. Corsaro, D. Mallamace, S. Vasi, C. Vasi and G. Dugo, Comput. Struct. Biotechnol. J., 2015, 13, 33–37 CrossRef CAS PubMed.
- M.-C. Bellissent-Funel, A. Hassanali, M. Havenith, R. Henchman, P. Pohl, F. Sterpone, D. van der Spoel, Y. Xu and A. E. Garcia, Chem. Rev., 2016, 116, 7673–7697 CrossRef CAS PubMed.
- J. A. Rupley and G. Careri, Advances in Protein Chemistry, Elsevier, 1991, vol. 41, pp. 37–172 Search PubMed.
- S. Khodadadi, J. H. Roh, A. Kisliuk, E. Mamontov, M. Tyagi, S. A. Woodson, R. M. Briber and A. P. Sokolov, Biophys. J., 2010, 98, 1321–1326 CrossRef CAS PubMed.
- V. Kurkal, R. M. Daniel, J. L. Finney, M. Tehei, R. V. Dunn and J. C. Smith, Biophys. J., 2005, 89, 1282–1287 CrossRef CAS.
- E. Ayan, B. Yuksel, E. Destan, F. B. Ertem, G. Yildirim, M. Eren, O. M. Yefanov, A. Barty, A. Tolstikova, G. K. Ketawala, S. Botha, E. H. Dao, B. Hayes, M. Liang, M. H. Seaberg, M. S. Hunter, A. Batyuk, V. Mariani, Z. Su, F. Poitevin, C. H. Yoon, C. Kupitz, A. Cohen, T. Doukov, R. G. Sierra, Ç. Dağ and H. DeMirci, Commun. Biol., 2022, 5, 1–13 CrossRef.
- R. B. Gregory, Protein-solvent interactions, Routledge/CRC Press, 1995 Search PubMed.
- G. Schirò and M. Weik, J. Phys.: Condens. Matter, 2019, 31, 463002 CrossRef PubMed.
- W. Doster, S. Cusack and W. Petry, Nature, 1989, 337, 754–756 CrossRef CAS PubMed.
- J. H. Roh, J. E. Curtis, S. Azzam, V. N. Novikov, I. Peral, Z. Chowdhuri, R. B. Gregory and A. P. Sokolov, Biophys. J., 2006, 91, 2573–2588 CrossRef CAS PubMed.
- W. Doster, Eur. Biophys. J., 2008, 37, 591–602 CrossRef CAS PubMed.
- J. D. Nickels, H. O’Neill, L. Hong, M. Tyagi, G. Ehlers, K. L. Weiss, Q. Zhang, Z. Yi, E. Mamontov, J. C. Smith and A. P. Sokolov, Biophys. J., 2012, 103, 1566–1575 CrossRef CAS PubMed.
- V. Kurkal, R. M. Daniel, J. L. Finney, M. Tehei, R. V. Dunn and J. C. Smith, Chem. Phys., 2005, 317, 267–273 CrossRef CAS.
- J. H. Roh, V. N. Novikov, R. B. Gregory, J. E. Curtis, Z. Chowdhuri and A. P. Sokolov, Phys. Rev. Lett., 2005, 95, 038101 CrossRef CAS PubMed.
- S. Magazù, F. Migliardo and A. Benedetto, J. Phys. Chem. B, 2011, 115, 7736–7743 CrossRef.
- K. Wood, C. Caronna, P. Fouquet, W. Haussler, F. Natali, J. Ollivier, A. Orecchini, M. Plazanet and G. Zaccai, Chem. Phys., 2008, 345, 305–314 CrossRef CAS.
- L. Tavagnacco, E. Chiessi, M. Zanatta, A. Orecchini and E. Zaccarelli, J. Phys. Chem. Lett., 2019, 10, 870–876 CrossRef CAS.
- K. Wood, A. Frölich, A. Paciaroni, M. Moulin, M. Härtlein, G. Zaccai, D. J. Tobias and M. Weik, J. Am. Chem. Soc., 2008, 130(14), 4586–4587 CrossRef CAS PubMed.
- G. Schirò, Y. Fichou, F.-X. Gallat, K. Wood, F. Gabel, M. Moulin, M. Härtlein, M. Heyden, J.-P. Colletier, A. Orecchini, A. Paciaroni, J. Wuttke, D. J. Tobias and M. Weik, Nat. Commun., 2015, 6, 6490 CrossRef PubMed.
- L. Tavagnacco, M. Zanatta, E. Buratti, M. Bertoldo, E. Chiessi, M. Appel, F. Natali, A. Orecchini and E. Zaccarelli, Chem. Sci., 2024, 15, 9249–9257 RSC.
- L. Zheng, Z. Liu, Q. Zhang, S. Li, J. Huang, L. Zhang, B. Zan, M. Tyagi, H. Cheng, T. Zuo, V. G. Sakai, T. Yamada, C. Yang, P. Tan, F. Jiang, H. Chen, W. Zhuang and L. Hong, Chem. Sci., 2022, 13, 4341–4351 RSC.
- J. T. King and K. J. Kubarych, J. Am. Chem. Soc., 2012, 134, 18705–18712 CrossRef CAS PubMed.
- J. N. Dahanayake and K. R. Mitchell-Koch, Front. Mol. Biosci., 2018, 5, 65 CrossRef PubMed.
- D. El Ahdab, L. Lagardère, T. J. Inizan, F. Célerse, C. Liu, O. Adjoua, L.-H. Jolly, N. Gresh, Z. Hobaika, P. Ren, R. G. Maroun and J.-P. Piquemal, J. Phys. Chem. Lett., 2021, 12, 6218–6226 CrossRef CAS PubMed.
- J. N. Dahanayake and K. R. Mitchell-Koch, Phys. Chem. Chem. Phys., 2018, 20, 14765–14777 RSC.
- S. Jiao, D. M. Rivera Mirabal, A. J. DeStefano, R. A. Segalman, S. Han and M. S. Shell, Biomacromolecules, 2022, 23, 1745–1756 CrossRef CAS.
- M. Telling, A Practical Guide to Quasi-elastic Neutron Scattering, Royal Society of Chemistry, 2020 Search PubMed.
- E. Bassotti, S. Gabrielli, G. Paradossi, E. Chiessi and M. Telling, Commun. Chem., 2024, 7, 83 CrossRef CAS PubMed.
- N. de Val, J.-P. Declercq, C. K. Lim and R. R. Crichton, J. Inorg. Biochem., 2012, 112, 77–84 CrossRef CAS PubMed.
- R. R. Crichton, R. Eason, A. Barclay and C. F. Bryce, Biochem. J., 1973, 131(4), 855–857 CrossRef CAS.
- R. Laghaei, W. Kowallis, D. G. Evans and R. D. Coalson, J. Phys. Chem. A, 2014, 118, 7442–7453 CrossRef CAS.
- R. Laghaei, D. G. Evans and R. D. Coalson, Proteins, 2013, 81, 1042–1050 CrossRef CAS.
- H. Veroniaina, X. Pan, Z. Wu and X. Qi, Expert Rev. Anticancer Ther., 2021, 21, 901–913 CrossRef CAS PubMed.
- A. I. Kuruppu, L. Turyanska, T. D. Bradshaw, S. Manickam, B. P. Galhena, P. Paranagama and R. De Silva, Biochim. Biophys. Acta, Gen. Subj., 2022, 1866, 130067 CrossRef CAS PubMed.
- K. Yoshizawa, Y. Mishima, S.-Y. Park, J. G. Heddle, J. R. H. Tame, K. Iwahori, M. Kobayashi and I. Yamashita, J. Biochem., 2007, 142, 707–713 CrossRef CAS PubMed.
- B. Maity, Z. Li, K. Niwase, C. Ganser, T. Furuta, T. Uchihashi, D. Lu and T. Ueno, Phys. Chem. Chem. Phys., 2020, 22, 18562–18572 RSC.
- R. Jurado, J. Adamcik, A. Sánchez-Ferrer, S. Bolisetty, R. Mezzenga and N. Gálvez, Biomacromolecules, 2021, 22, 2057–2066 CrossRef CAS PubMed.
- M. T. F. Telling, L. Clifton, J. Combet, B. Frick, S. Howells and V. G. Sakai, Soft Matter, 2012, 8, 9529–9532 RSC.
- M. T. F. Telling, S. Howells, J. Combet, L. A. Clifton and V. García Sakai, Chem. Phys., 2013, 424, 32–36 CrossRef CAS.
- G. Paradossi and E. Chiessi, Phys. Chem. Chem. Phys., 2017, 19, 11892–11903 RSC.
- A. Matejčíková, E. Tichý and P. Rajniak, J. Drug Delivery Sci. Technol., 2022, 74, 103550 CrossRef.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, J. Am. Chem. Soc., 1996, 118, 11225–11236 CrossRef CAS.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- S. P. Kadaoluwa Pathirannahalage, N. Meftahi, A. Elbourne, A. C. G. Weiss, C. F. McConville, A. Padua, D. A. Winkler, M. Costa Gomes, T. L. Greaves, T. C. Le, Q. A. Besford and A. J. Christofferson, J. Chem. Inf. Model., 2021, 61, 4521–4536 CrossRef CAS PubMed.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- R. W. Hockney, Methods Comput. Phys., 1970, 9, 135–211 Search PubMed.
- A. Luzar and D. Chandler, Nature, 1996, 379, 55–57 CrossRef CAS.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- F. Eisenhaber, P. Lijnzaad, P. Argos, C. Sander and M. Scharf, J. Comput. Chem., 1995, 16, 273–284 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS.
- D. Sehnal, S. Bittrich, M. Deshpande, R. Svobodová, K. Berka, V. Bazgier, S. Velankar, S. K. Burley, J. Koča and A. S. Rose, Nucleic Acids Res., 2021, 49, W431–W437 CrossRef CAS PubMed.
- V. V. Sudarev, S. M. Dolotova, S. M. Bukhalovich, S. V. Bazhenov, Y. L. Ryzhykau, V. N. Uversky, N. A. Bondarev, S. D. Osipov, A. E. Mikhailov, D. D. Kuklina, T. N. Murugova, I. V. Manukhov, A. V. Rogachev, V. I. Gordeliy, I. Yu Gushchin, A. I. Kuklin and A. V. Vlasov, Int. J. Biol. Macromol., 2023, 224, 319–343 CrossRef CAS PubMed.
- P. Arosio, F. Carmona, R. Gozzelino, F. Maccarinelli and M. Poli, Biochem. J., 2015, 472, 1–15 CrossRef CAS PubMed.
- X. Peng, C. Lu, Z. Liu and D. Lu, Phys. Chem. Chem. Phys., 2021, 23, 17158–17165 RSC.
- P. K. Nandi, N. J. English, Z. Futera and A. Benedetto, Phys. Chem. Chem. Phys., 2016, 19, 318–329 RSC.
- G. Schirò, C. Caronna, F. Natali and A. Cupane, Phys. Chem. Chem. Phys., 2010, 12, 10215–10220 RSC.
- G. Schirò, C. Caronna, F. Natali, M. M. Koza and A. Cupane, J. Phys. Chem. Lett., 2011, 2, 2275–2279 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp03481c |
| This journal is © the Owner Societies 2025 |