Unveiling the molecular mechanism of Mn and Zn-catalyzed Ullmann-type C–O cross-coupling reactions†
Received
13th July 2024
, Accepted 2nd December 2024
First published on 5th December 2024
Abstract
A detailed theoretical study delving into the molecular mechanisms of the Ullmann-type O-arylation reactions catalyzed by manganese and zinc metal ions has been investigated with the aid of the density functional theory (DFT) method. In contrast to the redox-active mechanisms proposed for classical Ullmann-type condensation reaction, a redox-neutral mechanism involving σ-bond metathesis emerged as the most appealing pathway for the investigated high-valent Mn(II) and Zn(II)-catalyzed O-arylation reactions. The mechanism remains invariant with respect to the nature of the central metal, ligand, base, etc. This unusuality in the mechanism has been dissected by considering three cases: ligand-free and ligand-assisted Mn(II)-catalyzed O-arylation reaction and ligand-assisted Zn(II)-catalyzed O-arylation reactions. In each class, a metal phenoxide species was identified as the active catalyst. The unusual mechanistic trends observed in these C–O cross-coupling reactions could be attributed to the stable electronic configurations (d5, d10) combined with the higher oxidation states of the catalytic metal centers (Mn(II), Zn(II)). The exploration into the electronic effects of functional groups in controlling the reaction feasibility within each metal variant disclosed a consistent trend irrespective of the transition metal catalyst involved in the reaction. It was found that the introduction of electron-withdrawing groups at the para-position of organic halide lowers the energy of their lowest unoccupied molecular orbitals (LUMO), thereby lowering the HOMO–LUMO gap between the coupling partners. This study thereby revealed a comprehensive understanding of the fundamental mechanisms exhibited by high-valent first-row transition metal catalysts that could foster the development of eco-friendly protocols for preparing biaryl ether moieties.
1. Introduction
Transition metal-catalyzed cross-coupling reactions in synthetic organic chemistry have attracted much attention owing to the construction of C–C and C–X (X = O, S, NH) bonds.1,2 More than a century ago, Ullmann reported the first copper-catalyzed coupling reaction between two aryl halides, which is known as the classical Ullmann reaction.3 A comparative methodology was taken on for C–O and C–N coupling by Ullmann after the disclosure of the classical Ullmann reaction.4,5 Then, at the beginning of the 19th century, Goldberg also pioneered the use of copper as a catalyst in the synthesis of aryl amides.6 These reactions came to be known as Ullmann-type condensation reactions. These synthetic responses made a major advance in organic chemistry because of the development of various carbon–heteroatom bond formation reactions. These synthetic methodologies have risen in prominence in recent decades as a result of their widespread use in the synthesis of biaryl ethers, which have a stronghold in pharmaceuticals and industry.7,8 However, these protocols have remained unexploited owing to the requirement for a stoichiometric amount of metal salts, long reaction times, harsh reaction conditions, such as high temperature and pressure, and intolerance to functional groups.9 Consequently, significant endeavors have been committed to the development of more appealing methods for Ullmann-type condensation reactions.
The exceptional ability exhibited by palladium metal has made it an excellent candidate for facilitating biaryl synthesis.10–12 Nonetheless, palladium-catalyzed C–O bond formation reactions suffer from a few impediments such as high cost, toxicity, etc. This has opened a resurgence in the Cu-catalyzed Ullmann reaction protocols by the introduction of ancillary ligands in the procedure, which came to be known as modified Ullmann condensation reactions. The incorporation of bidentate ancillary ligands could enhance the soluble nature of the Cu precursors so that mild reaction conditions are developed. However, recently numerous ventures have been advanced to clear the present circumstances with various metals such as nickel,13 iron,14 cobalt,15 manganese,16 zinc17etc.
Among the different 3d transition metal catalysts for cross-coupling reactions, the use of Mn and Zn as catalysts has been gaining ground nowadays, due to their non-toxic, earth-abundant, and eco-friendly characteristics. Within the realm of transition metals, manganese characterizes a distinctive position owing to its extra stable half-filled d5 electronic configuration and its versatile character to expand its oxidation state up to VII.18 Similarly, zinc as a catalyst remains unearthed in organic chemistry compared to other metals. One reason for this incomprehension is the “transition” position of zinc in the periodic table, between transition metals and main group elements. Hence, the chemistry of zinc is distinct from that of transition metals and is more closely related to main group chemistry because it is based on the [Ar] 3d10 4s2 electron configuration with filled d-shells. Because of this, compared to other transition metals, zinc does not have distinctive redox chemistry; the most well-known forms are Zn(0) and Zn(II) states. Henceforth, zinc is less well-known as a catalyst core for cross-coupling than other transition metals.
The catalytic potential of zinc and manganese has been recently explored by several groups.19–22 Gopinathan Anilkumar et al. have reported novel manganese and zinc-catalyzed protocol for synthesizing biaryl ethers using aryl iodides and phenols as the substrates.17,23 Astonishingly the same group has also reported an eco-friendly ligand-free protocol for the Mn-catalyzed C–O cross-coupling reaction.23 These reaction protocols effectively tolerate the coupling of aryl iodides bearing electron-withdrawing substituents with phenols.
Despite the synthetic advancement in the Mn and Zn-catalyzed reaction protocols, a mechanistic understanding of the reactions is rarely found in the literature. Computational modeling using quantum mechanical techniques has become an excellent tool to understand the molecular mechanism of catalytic organic reactions, which may proceed either via radical or non-radical pathways. The radical mechanism generally involves single electron transfer (SET) and halogen atom transfer (HAT) mechanisms, whereas the non-radical pathway consists of the oxidative addition–reductive elimination (OA–RE) mechanism and σ-bond metathesis mechanism. The choice of the mechanism depends on several factors, like nature of the ligand, metal, oxidation state, etc. In this article, we report here a detailed mechanistic investigation of the manganese and zinc-catalyzed O-arylation reactions by making use of the density functional theory (DFT) method. To our surprise, a redox-neutral mechanism involving σ-bond metathesis was found to be the most promising mechanism for both the Mn(II) and Zn(II) catalyzed etherification reactions between the electron-deficient aryl halide and phenol irrespective of the nature of the metal, and presence or absence of an ancillary ligand. A redox-neutral mechanism in organo transition metal catalysis could be referred to those mechanisms in which the oxidation state of the central transition metal catalyst remains unchanged throughout the catalytic cycle. Such redox-neutral catalytic cycles have been reported for Cu(I), Co(II), and Zn(II)-catalyzed cross-coupling reactions, which proceed via either concerted oxidative addition–reductive elimination mechanisms, or nucleophilic substitution pathways or a σ-bond metathesis mechanism.24–27 Henceforth, probing the detailed mechanism of manganese and zinc-catalyzed cross-coupling reactions will provide insights for designing better catalysts and reaction protocols.
2. Computational methodology
All calculations were performed using DFT with no symmetry constraints using the Gaussian 09 suite of programs.28 The B3LYP functional with the D3 version of Grimme's dispersion correction was employed for geometry optimization.29,30 For Mn-catalyzed O-arylation reactions, the 6-31+G(d) basis set was used for all atoms except Mn, I, K and Cs. Whilst for Zn-catalyzed O-arylation, the 6-311+G(d) basis set was employed for all atoms except Zn, I, and Cs.31,32 It was found that both 6-31+G(d) and 6-311+G(d) provide results with comparable accuracy. The heavy atoms were described using effective core potentials in our study. For investigating the mechanism of Zn-catalyzed O-arylation reaction, the effective core potentials of Hay and Wadt with double ζ valence basis set LANL2DZ were used to describe I, Cs, and Zn atoms.33–35 For the Mn-catalyzed C–O cross-coupling reactions, effective core potentials involving Stuttgart–Dresden (SDD)36 basis sets employing triple zeta valence basis sets were found to be better than LANL2DZ for describing Mn, I, K, and Cs. The solvent effect was computed with the conductor-like polarizable continuum model (CPCM) solvation model using acetonitrile (ε = 35.69) as the solvent.37,38 Frequency calculations were used to confirm the located stationary points corresponding to the minima and saddle points and to obtain the thermal free energy correction. For the transition states, the intrinsic reaction coordinate calculation (IRC) was carried out to confirm that the obtained saddle point connects to the appropriate reactants and products in the potential energy surface39,40 (see ESI† for more details pertaining to the justification of the chosen computational methodology).
3. Results
To unravel the details governing the reaction mechanism of manganese and zinc-catalyzed O-arylation, the reaction between electron-deficient aryl halide and phenol was considered as the model reaction for our computational investigation. We have taken three distinctive circumstances, including the ligand-free and ligand-assisted manganese-catalyzed O-arylation reactions and ligand-assisted zinc-catalyzed O-arylation reactions. The model reactions chosen for the study were based on the experimental protocol developed by Anilkumar et al. which is shown in Scheme 1.17,23 The details garnered from the computational study on the mechanisms of these O-arylation reactions are provided in the succeeding sections.
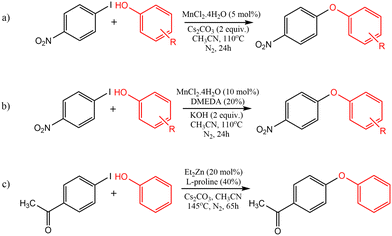 |
| | Scheme 1 The model reactions investigated in this study: (a) ligand-free Mn-catalysed etherification; (b) ligand-assisted Mn-catalysed etherification; (c) ligand-assisted Zn-catalysed etherification. | |
3.1. Possible active catalysts in acetonitrile solvent
The active catalyst involved is usually generated by the interaction of the precatalytic metal salts with the ligands. Meanwhile, in specific cases, the active catalyst also includes the coordination of the substrate molecule with the central metal atom. In order to ascertain the active catalysts participating in the O-arylation reactions under investigation, we first evaluated the relative Gibbs free energies associated with the formation of various metal catalytic species that could be generated within the reaction medium. The formation of these active catalytic species in each scenario is delineated below:
3.1.1. Identification of active catalysts in manganese-catalyzed O-arylation reaction.
3.1.1.1. Ligand-free variant.
Let us consider the model system shown in Scheme 1a. Here, the precatalyst MnCl2·4H2O initially loses four water molecules to form MnCl2. The phenol present in the solution gets deprotonated by the base Cs2CO3 to form caesium phenoxide. The formed caesium phenoxide undergoes a ligand exchange with one of the chloride ions of the precatalyst MnCl2 (Int 1a), resulting in the formation of another intermediate (Int 2a). Since an auxiliary ligand was absent in the catalytic system, the solvent molecule acetonitrile or the water molecules (displaced from MnCl2·4H2O) could explicitly take the unoccupied coordination sites on the Mn catalyst resulting in the formation of Int 3a and Int 4a, respectively. However, neither the coordination of a water molecule nor the coordination of acetonitrile is found to be favorable owing to the positive Gibbs free energy of formation. Thus, we have considered Int 2a as the active catalyst in our mechanistic study for the ligand-free Mn(II) catalyzed O-arylation reaction (Scheme 2).
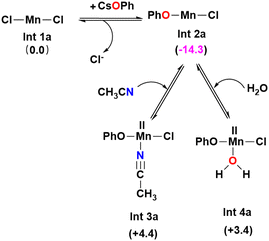 |
| | Scheme 2 Equilibrium between different Mn(II) complexes formed in acetonitrile solvent under the ligand-free condition (values in parentheses give the Gibbs free energy changes in (kcal mol−1)). | |
| | 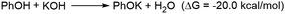 | (1) |
| |  | (2) |
| |  | (3) |
3.1.2. Identification of active catalysts in ligand-assisted zinc-catalyzed O-arylation reaction.
After identifying the active catalysts for ligand-assisted and ligand-free Mn-catalyzed O-arylation reaction, we next probed the active catalyst for the Zn-catalyzed O-arylation reaction. Initially, we investigated the catalytic activity of a Zn(0) proline complex, formed by the interaction of Zn metal with proline species, which was proposed to be the active catalytic species by the experimental group.17 The Zn metal was assumed to be formed in the reaction media via the thermal decomposition of Et2Zn as reported by Dumont et al.41 It was expected that the free proline ligand in the zwitterionic form, coordinates to the Zn(0) metal center through the two donor atoms, oxygen, and nitrogen. However, we could not trace out the expected Zn(0)-proline complex on the potential energy surface. This could be attributed to the highly stable 4s2 3d10 configuration of Zn metal, which prohibits it from receiving electron density from the ancillary ligands. Moreover, the very high temperature (>300 °C) required for the thermal decomposition of diethyl zinc coupled with the absence of strong reducing agents suggests the formation of Zn(0) intermediate species in the reaction medium to be highly unfavorable. Furthermore, the experimental studies also revealed that the product formation was not observed when Zn granules and Zn powder were used as the catalytic sources. This further manifests the unfavourability of Zn(0) complexes as pre-catalytic species. Thus, the catalytic cycle proposed by the experimental group for Zn(0) catalyzed etherification reaction proceeding via an oxidative addition–reductive elimination pathway was safely ruled out (Scheme 3).
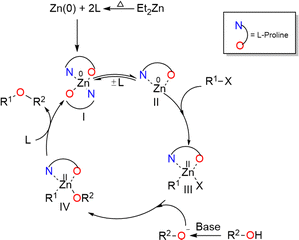 |
| | Scheme 3 The mechanistic pathway proposed for Zn-catalyzed O-arylation reaction by the experimental group. | |
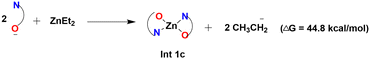
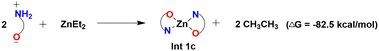
The experimental study has shown a good catalytic activity for a diethyl zinc source for the Zn-catalyzed O-arylation reaction.17 In the reaction system, the diethyl zinc could interact with the supporting ligand L-proline to form a tetrahedral zinc–proline complex (Int 1c). Zn(II) ions are generally expected to form tetrahedral complexes with the ancillary ligands.42,43 Recently, several studies have also demonstrated the catalytic efficacy of Zn(II)(L-proline)2 complexes for a number of chemical transformations.44,45 The L-proline ligand in our reaction system, in its zwitterionic state, is expected to interact with the diethyl zinc in two ways: (i) deprotonation of the proline ligand by the base Cs2CO3 forming a proline anion, followed by ligand exchange with diethyl zinc liberating two ethyl anions and Int 1c. Our calculations demonstrated that the former process is highly unfavorable owing to the positive Gibbs free energy of formation (44.8 kcal mol−1) (eqn (4)). (ii) The second pathway entails the interaction of diethyl zinc with the neutral proline ligand (zwitterion) resulting in the formation of Int 1c liberating two ethane molecules. The above process is found to be highly favorable owing to the favorable Gibbs free energy change (−82.5 kcal mol−1) (eqn (5)). (More information regarding the selection of metal salt is given in the ESI†).
| |  | (4) |
| |  | (5) |
In the reaction media, the Cs2CO3 could react with phenol to form caesium phenoxide by deprotonation (eqn (6)). This step proceeds with a favorable Gibbs free energy of formation of −12.0 kcal mol−1, thereby making the formation of caesium phenoxide a favorable one. The caesium phenoxide could then interact with the Int 1c to form a three-coordinate proline ligated Zn-phenoxide complex (Int 2c), by the dissociation of one proline ligand. Several studies have reported the favourability of the formation of such Zn(II)-phenoxide complexes.46,47 Though the formation of Int 2c from Int 1c involves a positive Gibbs free energy of formation (10.8 kcal mol−1) (eqn (7)), the overall free energy of formation of Int 2c from Int 1c is found to have a slightly favorable Gibbs free energy of formation (−1.2 kcal mol−1) (eqn (8)). Hence, it becomes obvious that the formation of PhOCs makes the overall reaction an exergonic one. In other words, the counter ion, Cs, plays a relevant role in determining the favorability of formation of Int 2c. As Int 2c is coordinatively unsaturated, a molecule of the solvent acetonitrile can explicitly coordinate to generate a four-coordinate Int 3c species (Fig. 1). However, the Gibbs free energy of formation of Int 3c from Int 2c is +5.1 kcal mol−1, which predicates that the solvent coordination by acetonitrile was found to be unfavorable for proline ligated-Zn(II) phenoxide species. On the grounds of the above results, the Int 2c is taken as the active catalyst for investigating the mechanism of Zn(II) catalyzed O-arylation reaction in our study. Consequently, Int 1c can be considered as an off-cycle species.
| | 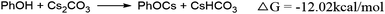 | (6) |
| |  | (7) |
| |  | (8) |
 |
| | Fig. 1 Solvent coordinated proline ligated zinc phenoxide complex (Int 3c). | |
3.2. Mechanism of Zn(II) vs. Mn(II) C–O cross-coupling reactions
In general, four common mechanisms have been proposed for the transition metal-catalyzed C–X cross-coupling reactions. These comprise of oxidative addition–reductive elimination48–52 and σ-bond metathesis as the non-radical pathways and single electron transfer (SET)53–55 and halogen atom transfer mechanism (HAT)56 as the radical pathways. A detailed investigation of these mechanisms with the aid of DFT studies is discussed in the following sections.
3.2.1. Oxidative addition–reductive elimination (OA–RE) mechanism.
The oxidative addition–reductive elimination pathway has been identified as a plausible mechanism for numerous C–X cross-coupling reactions.57,58 The investigation of the Mn(II) catalyzed etherification reaction revealed that the formation of an oxidative addition product for the ligand-free as well as ligand-assisted Mn-catalyzed etherification reaction is highly unfavorable owing to the positive Gibbs free energy of formation of 34.3 kcal mol−1 and 30.3 kcal mol−1, respectively. These results indicate the reluctance of the Mn(II) metal center to facilitate the oxidative addition of aryl halide on the active catalysts, owing to the instability of Mn in the higher oxidation state (IV). As a result, the classical mechanism of oxidative addition–reductive elimination could be ruled out for the ligand-free and ligand-assisted Mn-catalyzed C–O cross-coupling reactions.
We next explored the possibility of an oxidative addition–reductive elimination mechanism in the zinc-catalyzed O-arylation reaction by expecting the reaction to proceed through a Zn(II)/Zn(IV) catalytic cycle. However, our attempts to locate a stable oxidative addition product Zn(IV) on the potential energy surface (PES) were unsuccessful owing to the poor stability of Zn complexes in higher oxidation states (IV). This observation indicates that the oxidative addition–reductive elimination pathway is an unfavorable pathway for the Zn(II) catalyzed C–O cross-coupling reactions also.
3.2.2. Single electron transfer (SET) mechanism.
We next probed the possibility of a single electron transfer mechanism for the Mn/Zn-catalyzed O-arylation reaction, a single electron transfer from the M(II) center (M = Mn/Zn) of the active catalyst to aryl halide, resulting in the formation of an aryl iodide radical anion and a M(III) radical cation. The aryl radical anion in addition gets dissociated into an iodide ion and an aryl radical. The generated aryl radical eventually reacts with the M(III) radical cation to yield the coupled product biphenyl ether. The free energies of the entirely separated M(III) complex and the aryl radical were used to calculate the barrier height for the SET mechanism. Since the accurate calculation of the activation barrier is complicated, we have roughly estimated the activation barrier for the SET mechanism using the Marcus–Hush theory56 (see ESI†). The calculated free energy barrier for the SET mechanism was prohibitively large (ligand-free variant: 45.6 kcal mol−1, ligand-assisted variant: 46.6 kcal mol−1) for the ligand-free and ligand-assisted Mn-catalyzed O-arylation reactions. Consequently, the SET mechanism for the manganese-catalyzed coupling has been excluded in our study.
Similarly, in the case of the zinc-catalyzed O-arylation reaction, the activation barrier obtained for SET using the Marcus Hush model and Saveants model are 88.7 kcal mol−1 and 51.0 kcal mol−1, respectively (see ESI†). Both the activation barriers were found to be exceptionally high for the model reaction considered for our study. Thus, it was revealed that the SET pathway is not an optimal pathway for both the investigated classes of Zn(II) and Mn(II) catalyzed O-arylation reactions.
3.2.3. Halogen atom transfer (HAT) mechanism.
We have also explored the possibility of the halogen atom transfer pathway for each model reaction system. The HAT has been proposed as the favourable mechanistic route for the O- and S-arylation Ullmann-type reactions.2,59 This mechanism involves the simultaneous cleavage of the Ph–I bond with the creation of the M–I bond (M = Mn/Zn). In this mechanism, an iodide ion is transferred from the aryl iodide to the metal center, which leads to the formation of an M(III) intermediate and an aryl radical. The aryl radical generated further attacks the oxygen atom of the phenoxide ligand and affords the coupled product, biphenyl ether. Due to the complexity in estimating the accurate activation barrier, we have estimated the activation barriers roughly using the Saveants model60 (see ESI†). Unfortunately, the free energy barrier obtained under this mechanism for both the Mn and Zn-catalyzed reactions was found to be very high (ligand-free variant: 42.2 kcal mol−1; ligand-assisted variant: 40.4 kcal mol−1, Zn catalyzed O-arylation: 41.4 kcal mol−1). Therefore, this mechanism is kinetically forbidden under the given sets of experimental conditions for the model systems used in the study.
3.2.4. Binuclear mechanism.
Next, our investigation delved into the potential occurrence of a dimer formation especially in the case of the ligand-free variant of the Mn-catalyzed O-arylation reaction. On a thorough study, we successfully optimized a dimeric species (Int 6a) that could have been formed from Int 2a (Fig. 3). Next, we explored the feasibility of oxidative addition of Int 6a occurring at one of the manganese atoms. Despite successfully optimizing the oxidative addition product (Int 6b) on the potential energy surface, the substantial energy of formation (+39.3 kcal mol−1) led us to dismiss the mechanism reliant on dimeric intermediates (see ESI,† Scheme S4 for more details).
3.2.5. σ-bond metathesis mechanism.
Our study revealed that the three commonly proposed mechanisms for the transition metal-catalyzed C–X cross-coupling reactions involving oxidative addition–reductive elimination, SET, and HAT are found to be highly unfavourable for the Mn(II) and Zn(II)-catalyzed etherification reactions. We further investigated the plausibility of a σ-bond metathesis pathway. Though the classical mechanism for the C–O coupling reactions predicted the unfavourability of σ-bond metathesis, some recent studies have reported σ-bond metathesis as an appealing pathway for the O-arylation reaction.25 In a σ-bond metathesis reaction pathway, the oxidation state of the metal remains intact throughout the catalytic cycle.61 Thus it could be considered as a redox-neutral mechanistic pathway. Generally, the σ-bond metathesis mechanism features a typical cyclic four-membered transition state, resulting in simultaneous carbon–halogen bond cleavage and C–O bond formation. Here, the mechanism initiates via the reaction of aryl halide with the active catalysts for the investigated reactions, which results in the formation of a cyclic four-membered transition state. Furthermore, the products are formed directly from the transition states with the regeneration of the active catalyst. The plausible catalytic cycle for the σ-bond metathesis mechanism for the manganese and zinc-catalyzed O-arylation of phenol is shown in Schemes 4, 5 and 6, respectively.
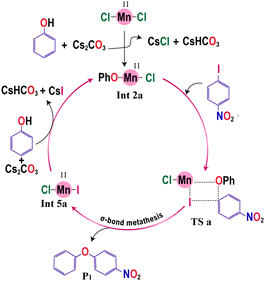 |
| | Scheme 4 Plausible catalytic cycle for ligand-free Mn catalysed C–O cross-coupling reactions. | |
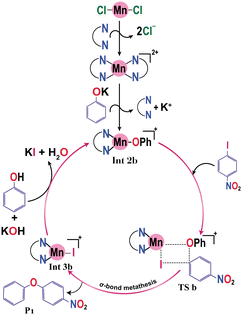 |
| | Scheme 5 Plausible catalytic cycle for ligand-assisted Mn catalyzed C–O cross-coupling reactions. | |
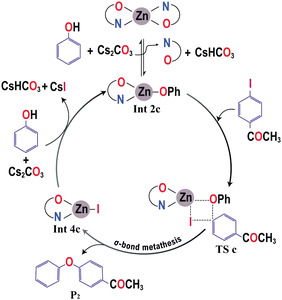 |
| | Scheme 6 Plausible catalytic cycle for ligand-assisted Zn catalysed C–O cross-coupling reactions. | |
In this σ-bond metathesis mechanism, the Csp2–I bond of the aryl iodides is broken simultaneously with the formation of the Csp2–O bond. The single imaginary frequencies (−395.11 cm−1, −404.76 cm−1 and −422.46 cm−1) confirmed the existence of four-membered transition states for the model reactions of ligand-free and ligand-assisted manganese and zinc-catalyzed O-arylation reactions, respectively. In the transition states, it appears that the metal–oxygen bond lengths in the active catalyst and the Csp2–I bond lengths of the aryl halides are increased. Additionally, there occurs a bond formation between the sp2 carbon of the 4-iodonitrobenzene/4-iodoacetophenone and the oxygen atom of the active catalyst. The variation of the bond lengths between the atoms during each reaction pathway is enlisted in Tables 1, 2 and 3, respectively. The simultaneous bonding interactions between metal and iodine atoms and Csp2–OPh atoms revealed that the mechanism proceeds in a concerted manner. The activation barrier obtained for the σ-bond metathesis pathway for the ligand-free and ligand-assisted manganese-catalyzed C–O coupling reactions is 33.8 kcal mol−1 and 32.4 kcal mol−1, respectively. At a temperature of 110 °C, the reactants can readily exceed this activation energy, which is consistent with the experimental results.62 Similarly, for the proline-ligated zinc-catalyzed O-arylation reaction, an activation barrier of 35.0 kcal mol−1 was obtained, which is concomitant with the high temperature (145 °C) required for the model system (Scheme 1c). Fig. 2a–c show the Gibbs free energy profile diagram for the σ bond metathesis mechanism for the ligand-free and ligand-assisted manganese(II) and zinc(II)-catalyzed O-arylation of phenol with the aryl halides, respectively. Thus, the theoretical investigation carried out on the model systems revealed that a concerted mechanism involving σ-bond metathesis turned out to be the most favourable pathway for Mn(II) and Zn(II) catalyzed O-arylation reactions irrespective of the metal ions and ligands used in the chemical transformation. The optimized geometries of the reactants and transition states of the σ-bond metathesis mechanism for the investigated reactions are depicted in Fig. 4 and 5.
Table 1 Variation of the bond lengths between Mn and coordinated atoms involved in the mechanistic pathway for the ligand-free O-arylation reaction. All the values of bond length are given in Å units
| Species |
Mn–Cl |
Mn–OPh |
Mn–I |
Csp2–I |
Csp2–OPh |
| ArI–NO2 |
— |
— |
— |
2.14 |
— |
| PhOMnCl |
2.34 |
1.92 |
— |
— |
— |
| TS1a |
2.35 |
2.05 |
3.06 |
2.42 |
1.87 |
| MnICl |
2.34 |
— |
2.74 |
— |
— |
| Ether |
— |
— |
— |
— |
1.36 |
Table 2 Variation of bond lengths between the Mn and coordinated atoms during the mechanistic pathway for the ligand-assisted pathway. All the values of bond length are given in Å units
| Species |
Mn–N1 |
Mn–N2 |
Mn–OPh |
Mn–I |
Csp2–I |
Csp2–OPh |
| ArI–NO2 |
— |
— |
— |
— |
2.14 |
— |
|
L-MnOPh |
2.25 |
2.22 |
1.94 |
— |
— |
— |
| TS1b |
2.25 |
2.24 |
2.07 |
3.68 |
2.40 |
1.81 |
|
L-MnI |
2.23 |
2.23 |
— |
2.75 |
— |
— |
| Ether |
— |
— |
— |
— |
— |
1.36 |
Table 3 Variation of bond lengths between Zn and coordinated atoms of the ligands in the mechanistic pathway. All the values of bond length are given in Å units
| Species |
Zn–OL |
Zn–NL |
Zn–OPh |
Zn–I |
Csp2–I |
Csp2–OPh |
| ArICOCH3 |
— |
— |
— |
— |
2.13 |
— |
| Int 2 |
2.02 |
2.17 |
1.94 |
— |
— |
— |
| TS |
2.02 |
2.15 |
2.02 |
3.30 |
2.50 |
1.88 |
| Product |
2.05 |
2.14 |
— |
2.66 |
— |
1.37 |
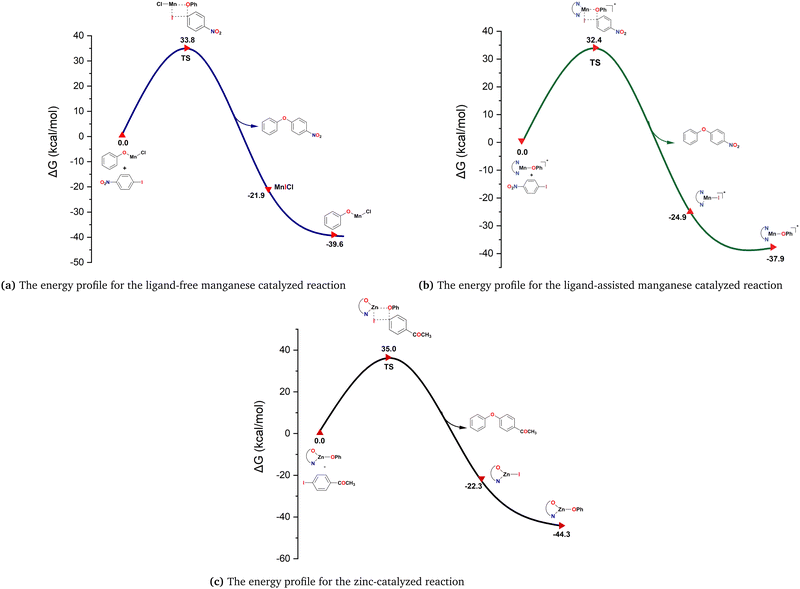 |
| | Fig. 2 Gibbs free energy profile for the σ-bond metathesis mechanism of (a) ligand-free Mn(II)-catalyzed O-arylation reaction (b) ligand-assisted Mn(II)-catalyzed O-arylation reaction and (c) ligand-assisted Zn(II) catalyzed O-arylation reaction. All the energy values are given with respect to the reactants in kcal mol−1. | |
 |
| | Fig. 3 Binuclear intermediate (Int 6a) formed in the ligand-free manganese-catalyzed O-arylation reaction. | |
 |
| | Fig. 4 The optimized geometries of the aryl halide (a), active catalyst (b), and transition state (c) involved in the proposed σ-bond metathesis mechanism for the zinc-catalyzed O-arylation reaction (bond lengths are given in Å). | |
 |
| | Fig. 5 The optimized geometries of the aryl halide (a), active catalysts (b) and (c), and transition states (d) and (e) involved in the proposed σ-bond metathesis mechanism for the ligand-free and ligand-assisted Mn-catalyzed O-arylation reaction (bond lengths are given in Å). | |
4. Electronic effects of various functional groups
It was gleaned from the experiment that irrespective of the metal catalysts involved, the reported etherification reactions proceed efficiently in the presence of electron-withdrawing substituents on aryl iodides. Consequently, a detailed computational study was conducted to probe the role of functional groups on aryl halide in controlling the feasibility of the reaction. The electronic structure calculations were conducted by varying the functional groups at the para position of the aryl halides. Even though the substrate scope of unsubstituted and p-methyl aryl iodides is not studied in the experiment, we have considered them in our study to account for the trends observed in the experiment. The activation barrier for each O-arylation reaction was calculated to probe the electronic effects of the substituents, by characterizing the transition states involved. It was objectified that the addition of electron-withdrawing groups at the para position of the aryl iodide considerably decreased the activation barrier for the σ-bond metathesis pathway despite the difference in the catalytic metal centers involved. These findings vindicate the high yields observed in the experiment when the reaction was performed using the para-substituted aryl iodides. (More details are provided in the ESI†).
5. Discussion
In the domain of the copper-catalyzed Ullmann-type O-arylation reactions, existing studies delineated mechanistic pathways encompassing oxidative addition–reductive elimination, halogen atom transfer, and single electron transfer mechanisms contingent upon the reaction conditions. Conversely, our own investigation revealed a redox-neutral mechanism involving a σ-bond metathesis mechanism for the cobalt-catalyzed O-arylation reaction. Accordingly, the realm of first-row transition metals manifests discernible mechanistic heterogeneity, contingent upon the intricate interplay of reaction conditions. Such pathways stand in stark contrast to the mechanisms for the second-row transition metals, which predominantly adhere to the conventional redox-active oxidative addition–reductive elimination pathway. Interestingly in the present study, a redox-neutral σ-bond metathesis mechanism emerged as the most promising mechanism for the Mn and Zn-catalyzed C–O cross-coupling reactions similar to that of Co(II) catalyzed O-arylation reaction.25 The mechanism is found to remain invariant irrespective of the metal, nature of the ligand, and the base involved in the reaction. Hence, for the high-valent M(II) (M = Mn, Co, and Zn) catalyzed O-arylation reactions, we have elucidated a mechanism distinct from that observed in Cu(I) catalyzed O-arylation reactions. This could be attributed to the following reasons:
•The high-valent state of the central metal prevents it from going into a higher oxidation state (IV). This could arise from the inability of the ligands to stabilize the central metal in its higher oxidation state.
•Moreover, the manganese and zinc exhibit an inherent stability owing to their d5 and d10 electronic configurations.
•The ease of bond formation between the cross-coupling partners (Csp2–O bond formation).
Again, to rationalize the effect of different substituents on the feasibility of manganese and zinc-catalyzed C–O coupling reactions, the frontier molecular orbital theory (FMO) analysis of the species involved in the turn-over limiting steps was conducted to calculate the HOMO–LUMO gap. A reaction having a small HOMO–LUMO gap could lead to increased bonding interactions amongst the species involved. The FMOs involved in the O-arylation reaction could be the HOMO of the metal–phenoxide complex and the LUMO of aryl halides. The observed results show that the inclusion of the electron-withdrawing group at the para position of the aryl halide lowers the energy of the LUMO (see ESI,† for more details). Accordingly, the HOMO–LUMO energy gap of the concerned reactants gets reduced, thereby reducing the activation barrier for the reaction.
6. Conclusions
In our study, we have done a detailed computational investigation on the mechanisms of manganese and zinc-catalyzed etherification reactions between aryl halides with phenols using the density functional theory method. Among the commonly proposed mechanisms for the Ullmann-type cross-coupling reactions, the σ-bond metathesis is found to be the most promising mechanism for the investigated O-arylation reactions. The mechanism remains invariant regardless of the nature of the metal, ligands, etc. Despite the significant attempts, a transition state for the oxidative addition of organic halide to the active catalysts is not achieved. The radical mechanisms for the O-arylation reactions such as SET and HAT, have been ruled out theoretically due to the high activation barrier obtained for the reaction under these mechanisms. The favourability of non-redox mechanisms could be ascribed due to the stable electronic configurations and high valence states of the transition metal center. The effect of substituents on the aryl halide reveals that the electron-withdrawing group (EWG) on the aryl halide promotes the reactivity by lowering the LUMO energy, thereby reducing the HOMO–LUMO gap, and consequently lowering the activation barrier of the reaction. The results gained from this study will aid in designing better reaction protocols for manganese and zinc-catalyzed O-arylation.
Data availability
The data supporting this article have been included as part of the ESI.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The work was supported by the University Grants Commission, Govt. of India via the research grant no: 2368-MRP/15-16/KLMG002/UGC-SWRO. C. Rajalakshmi thanks the University Grants Commission (UGC, India) for the Senior Research Fellowship (SRF). The authors gratefully acknowledge DST-FIST for providing the financial support. The authors also thank the Mahatma Gandhi University Innovation Foundation (MGUIF) for providing the High Performance Computing facilities (MGUIF Paramastra).
Notes and references
- E.-i Negishi, Angew. Chem., Int. Ed., 2011, 50, 6738–6764 CrossRef CAS PubMed.
- S. Zhang, Z. Zhu and Y. Ding, Dalton Trans., 2012, 41, 13832–13840 RSC.
- F. Ullmann and J. Bielecki, Ber. Dtsch. Chem. Ges., 1901, 34, 2174–2185 CrossRef CAS.
- F. Ullmann, Ber. Dtsch. Chem. Ges., 1903, 36, 2382–2384 CrossRef.
- F. Ullmann and P. Sponagel, Ber. Dtsch. Chem. Ges., 1905, 38, 2211–2212 CrossRef CAS.
- I. Goldberg, Ber. Dtsch. Chem. Ges., 1906, 39, 1691–1692 CrossRef.
- S. Yamamura and S. Nishiyama, J. Synth. Org. Chem., Jpn., 1997, 55, 1029–1039 CrossRef CAS.
- I. S. Chung and S. Y. Kim, J. Am. Chem. Soc., 2001, 123, 11071–11072 CrossRef CAS PubMed.
- S. Mondal, ChemTexts, 2016, 2, 1–11 CrossRef CAS.
- A. Biffis, M. Zecca and M. Basato, J. Mol. Catal. A: Chem., 2001, 173, 249–274 CrossRef CAS.
- P. Gautam and B. M. Bhanage, J. Org. Chem., 2015, 80, 7810–7815 CrossRef CAS PubMed.
- I. Kondolff, H. Doucet and M. Santelli, Organometallics, 2006, 25, 5219–5222 CrossRef CAS.
- D. Kundu, P. Maity and B. C. Ranu, Org. Lett., 2014, 16, 1040–1043 CrossRef CAS PubMed.
- K. S. Sindhu, S. M. Ujwaldev, K. K. Krishnan and G. Anilkumar, J. Catal., 2017, 348, 146–150 CrossRef CAS.
- D. Kundu, M. Tripathy, P. Maity and B. C. Ranu, Chem. – Eur. J., 2015, 21, 8727–8732 CrossRef CAS PubMed.
- E. N. Jacobsen, W. Zhang, A. R. Muci, J. R. Ecker and L. Deng, J. Am. Chem. Soc., 1991, 113, 7063–7064 CrossRef CAS.
- K. K. Krishnan, N. A. Harry, S. M. Ujwaldev and G. Anilkumar, ChemistrySelect, 2018, 3, 3984–3988 CrossRef CAS.
-
F. A. Cotton, G. Wilkinson, C. A. Murillo and M. Bochmann, Advanced inorganic chemistry, John Wiley and Sons, Inc., 1999 Search PubMed.
- S. K. Bose and T. B. Marder, Org. Lett., 2014, 16, 4562–4565 CrossRef CAS PubMed.
- R. Bedford, N. Gower, M. Haddow, J. Harvey, J. Nunn, R. Okopie and R. Sankey, Angew. Chem., Int. Ed., 2012, 51, 5435–5438 CrossRef CAS PubMed.
- S. Tang, L. Zeng, Y. Liu and A. Lei, Angew. Chem., Int. Ed., 2015, 54, 15850–15853 CrossRef CAS PubMed.
- K. Keerthi Krishnan, S. Saranya, K. Rohit and G. Anilkumar, J. Catal., 2019, 372, 266–271 CrossRef CAS.
- K. Rohit, S. Saranya, N. A. Harry and G. Anilkumar, ChemistrySelect, 2019, 4, 5150–5154 CrossRef CAS.
- C. Rajalakshmi, G. Krishnaveni, B. Varghese, A. Gopan and V. I. Thomas, J. Phys. Org. Chem., 2025, 38, e4671 CrossRef CAS.
- C. Rajalakshmi, A. Radhakrishnan, S. M. Ujwaldev, G. Anilkumar and V. I. Thomas, J. Organomet. Chem., 2022, 972, 122385 CrossRef CAS.
- C. Rajalakshmi, S. Jibin, R. Sulay, S. Asha, V. Ipe Thomas and G. Anilkumar, Polyhedron, 2021, 193, 114869 CrossRef CAS.
- C. Rajalakshmi, A. Krishnan, S. Saranya, G. Anilkumar and V. I. Thomas, Org. Biomol. Chem., 2022, 20, 4539–4552 RSC.
-
R. A. Gaussian09, Gaussian. Inc., Wallingford CT, 2009, 121, 150–166.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098 CrossRef CAS PubMed.
- M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. DeFrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654–3665 CrossRef CAS.
- V. A. Rassolov, J. A. Pople, M. A. Ratner and T. L. Windus, J. Chem. Phys., 1998, 109, 1223–1229 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS.
- W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284–298 CrossRef CAS.
- G. A. Halil Gökce and C. AlaÅŸalvar, J. Coord. Chem., 2019, 72, 1075–1096 CrossRef.
- M. Dolg, U. Wedig, H. Stoll and H. Preuss, J. Chem. Phys., 1987, 86, 866–872 CrossRef CAS.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- K. Fukui, Acc. Chem. Res., 1981, 14, 363–368 CrossRef CAS.
- C. Gonzalez and H. B. Schlegel, J. Chem. Phys., 1989, 90, 2154–2161 CrossRef CAS.
- H. Dumont, A. Marbeuf, J.-E. Bourée and O. Gorochov, J. Mater. Chem., 1993, 3, 1075–1079 RSC.
- K. S. Ferraz, N. F. Silva, J. G. Da Silva, N. L. Speziali, I. C. Mendes and H. Beraldo, J. Mol. Struct., 2012, 1008, 102–107 CrossRef CAS.
- A. Nano, L. Brelot, G. Rogez, A. Maisse-François and S. Bellemin-Laponnaz, Inorg. Chim. Acta, 2011, 376, 285–289 CrossRef CAS.
- T. Darbre and M. Machuqueiro, Chem. Commun., 2003, 1090–1091 RSC.
- K. R. Reddy, C. V. Rajasekhar and G. G. Krishna, Synth. Commun., 2007, 37, 1971–1976 CrossRef CAS.
- C. Di Iulio, M. Middleton, G. Kociok-Köhn, M. D. Jones and A. L. Johnson, Eur. J. Inorg. Chem., 2013, 1541–1554 CrossRef CAS.
- M. H. Chisholm, J. C. Gallucci, H. Zhen and J. C. Huffman, Inorg. Chem., 2001, 40, 5051–5054 CrossRef CAS PubMed.
- E. R. Strieter, B. Bhayana and S. L. Buchwald, J. Am. Chem. Soc., 2009, 131, 78–88 CrossRef CAS PubMed.
- J. W. Tye, Z. Weng, A. M. Johns, C. D. Incarvito and J. F. Hartwig, J. Am. Chem. Soc., 2008, 130, 9971–9983 CrossRef CAS PubMed.
- R. Giri and J. F. Hartwig, J. Am. Chem. Soc., 2010, 132, 15860–15863 CrossRef CAS PubMed.
- S.-L. Zhang, L. Liu, Y. Fu and Q.-X. Guo, Organometallics, 2007, 26, 4546–4554 CrossRef CAS.
- S. Zhang and Y. Ding, Organometallics, 2011, 30, 633–641 CrossRef CAS.
- H. L. Aalten, G. van Koten, D. M. Grove, T. Kuilman, O. G. Piekstra, L. A. Hulshof and R. A. Sheldon, Tetrahedron, 1989, 45, 5565–5578 CrossRef CAS.
- C. Couture and A. J. Paine, Can. J. Chem., 1985, 63, 111–120 CrossRef CAS.
- S. Arai, M. Hida and T. Yamagishi, Bull. Chem. Soc. Jpn., 1978, 51, 277–282 CrossRef CAS.
- G. O. Jones, P. Liu, K. Houk and S. L. Buchwald, J. Am. Chem. Soc., 2010, 132, 6205–6213 CrossRef CAS PubMed.
- K. Ahmad, B. A. Khan, B. H. Akram, J. Khan, R. Mahmood and S. K. Roy, Comput. Theoret. Chem., 2018, 1134, 1–7 CrossRef CAS.
- S.-L. Zhang, L. Liu, Y. Fu and Q.-X. Guo, Organometallics, 2007, 26, 4546–4554 CrossRef CAS.
- S.-L. Zhang and H.-J. Fan, Organometallics, 2013, 32, 4944–4951 CrossRef CAS.
- G. Lefèvre, G. Franc, A. Tlili, C. Adamo, M. Taillefer, I. Ciofini and A. Jutand, Organometallics, 2012, 31, 7694–7707 CrossRef.
- Z. Lin, Coord. Chem. Rev., 2007, 251, 2280–2291 CrossRef CAS.
- H. Ryu, J. Park, H. K. Kim, J. Y. Park, S.-T. Kim and M.-H. Baik, Organometallics, 2018, 37, 3228–3239 CrossRef CAS.
|
| This journal is © the Owner Societies 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ab,
Parvathi
Santhoshkumar
a,
Lydia Elizabeth
Mathews
a,
Ann Miriam
Abraham
a,
K. R.
Rohit
c,
Gopinathan
Anilkumar
ab,
Parvathi
Santhoshkumar
a,
Lydia Elizabeth
Mathews
a,
Ann Miriam
Abraham
a,
K. R.
Rohit
c,
Gopinathan
Anilkumar


















