
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBeyond hydrogen bonds: unveiling the structure-directing role of side-on X⋯π interactions in adamantane–thiourea crystals
Lamya H.
Al-Wahaibi
 a,
Olivier
Blacque
b,
Aamal A.
Al-Mutairi
c,
Ali A.
El-Emam
*d,
Rosa M.
Gomila
e,
Edward R. T.
Tiekink
*e and
Antonio
Frontera
*e
a,
Olivier
Blacque
b,
Aamal A.
Al-Mutairi
c,
Ali A.
El-Emam
*d,
Rosa M.
Gomila
e,
Edward R. T.
Tiekink
*e and
Antonio
Frontera
*e
aDepartment of Chemistry, College of Sciences, Princess Nourah bint Abdulrahman University, Riyadh 11671, Saudi Arabia
bDepartment of Chemistry, University of Zurich, Winterthurerstrasse 190, 8057 Zurich, Switzerland
cDepartment of Chemistry, College of Sciences, Imam Mohammad Ibn Saud Islamic University (IMSIU), Riyadh 11623, Saudi Arabia
dDepartment of Medicinal Chemistry, Faculty of Pharmacy, Mansoura University, Mansoura 35516, Egypt. E-mail: elemam@mans.edu.eg
eDepartment of Chemistry, Universitat de les Illes Balears, Crta de Valldemossa km 7.5, 07122 Palma de Mallorca, Spain. E-mail: edward.tiekink@uib.es; toni.frontera@uib.es
First published on 17th October 2025
Abstract
This study presents a comprehensive experimental and computational investigation into the role of variable side-on X⋯π (X = O, S, Cl) interactions complementing conventional hydrogen-bonding in the molecular packing of four differently substituted 3-(adamantan-1-yl)-1-[(E)-(arylmethylidene)amino]thiourea derivatives (1–4). Single-crystal X-ray diffraction analyses reveal distinct molecular packing modes across the series: conventional hydrogen-bonding dominates in 1, while non-conventional non-covalent interactions involving the aryl rings are prominent in 2–4. Molecular electrostatic potential (MEP) surface analysis precisely maps electrophilic (amino N–H) and nucleophilic (thione-S, nitro-O, F, Cl atoms) regions, consistent with observed hydrogen-bonding patterns and revealing positive potentials over the aromatic rings (0.7 to 6.9 kcal mol−1) that rationalize lone-pair (LP)⋯π interactions. Combined quantum theory of atoms in molecules (QTAIM) and non-covalent interaction (NCI) plot analyses confirm the presence of LP⋯π contacts in 2 and 3 (S⋯π) and Cl⋯π interactions in 4, characterised by specific bond critical points and extended RDG isosurfaces. The Energy Decomposition Analysis (EDA) for LP⋯π-mediated dimers of 2–4 reveals that these interactions are predominantly stabilised by dispersion and correlation effects, with electrostatic contributions being minor. This joint crystallographic and computational approach elucidates how side-on X⋯π interactions contribute to crystal architecture, confirming their structure-directing role and offering insights for rational crystal engineering.
1. Introduction
The crucial role of π-systems in assembling molecular species is well-established.1 Probably the most well-recognised among these are parallel, often off-set, stacking interactions between aromatic rings.2–8 However, it is now understood that such interactions are not restricted to aromatic hydrocarbons alone, but can involve rings incorporating heteroatoms,9,10 chelate rings, interactions with an arene ring11,12 or other chelate rings,12,13 and non-aromatic systems assembled with an arene ring.14–16 The second key motif is the T-shaped interaction, whereby a hydrogen atom of one aromatic ring is directed towards the centroid (Cg) of a second aromatic ring; the importance of such interactions has been highlighted, in particular, by Nishio et al.17,18 Analogous interactions have been noted where the aromatic ring is a chelate ring.19–21 T-shaped interactions where the hydrogen atom is substituted by some other entity are also known. Thus, metal centres, in charged22 and neutral23 forms may interact with an arene ring. In addition, lone-pair⋯π(arene) interactions with the donor atom perpendicular to the plane of the arene ring resemble T-shaped interactions.24–26 Halogens, too, are well-known to interact with arene rings.26–29When a halide (X) occupies a position above the C–X bond approximately perpendicular to the arene plane, the nature of bonding associated with the ensuing non-covalent interaction is rationalised in terms of the σ-hole concept.30–32 In essence, this theory indicates the presence of an electron-deficient region at the end of the C–X bond with compensating build-up of electron density in the equatorial region around the X atom. In this way, C–X⋯Cg, and related interactions, can be strong and directional, akin to conventional hydrogen-bonding.33–35 The electron-rich regions of X can also form significant non-covalent interactions by engaging in π-hole interactions.36–38 It turns out σ- and π-hole interactions can occur cooperatively between molecules39,40 and intriguingly, involve the same atom.41–43
Far from being restricted to halides, other species such as nitro44–46 and sulphur47–50 can participate in σ- and π-hole interactions with arene rings, with investigations often prompted by biological considerations. In the presence of side-on X⋯π(arene), X = O, S and Cl, are interactions that form the focus of the present joint crystallographic and computational chemistry investigation. The four investigated molecules, 1–4 as shown in Fig. 1, feature functionalised arene rings that engage in conventional (1) hydrogen-bonding and non-conventional (2–4) non-covalent interactions involving arene rings, and which are related to a recently studied series of molecules having the same central backbone.51
 | ||
| Fig. 1 Chemical diagrams for molecules 1–4 studied herein. | ||
Molecules 1–4 were originally synthesised in the context of investigating their biological activity. The chemotherapeutic efficiency of adamantane-based derivatives was discovered early after the development of amantadine and its related derivatives as effective antiviral drugs.52–55 The adamantane nucleus was recognized as the key pharmacophore in several biologically-active compounds.56–58 Among the major chemotherapeutic activities displayed by adamantane derivatives, the anti-HIV,59,60 anti-cancer,61,62 anti-microbial63,64 and anti-malarial65 derivatives are the most interesting ones. In addition, several thiosemicarbazide and thiosemicarbazone derivatives were reported to possess marked chemotherapeutic properties.66–68
In an earlier study, a series of adamantane–thiosemicarbazone hybrid derivatives were reported that possess marked in vitro anti-microbial and anti-proliferative activities.69 Building upon this, the present joint crystallographic and computational chemistry investigation primarily focuses on the structural insights and electronic properties of four adamantane–thiosemicarbazone derivatives, 1–4. As shown in Fig. 1, the investigated molecules feature functionalised arene rings that engage in conventional hydrogen-bonding (in 1) and various non-conventional non-covalent interactions involving arene rings (in 2–4). This study aims to discern whether these identified X⋯π interactions are merely coincidental artifacts of global molecular packing or if they exert a deliberate, structure-directing influence on the crystal architecture by employing a variety of computational chemistry methods.
2. Experimental
2.1 Synthesis
The investigated compounds 1–4 were prepared via the reaction of 1-adamantyl isothiocyanate A with hydrazine to yield 4-(adamantan-1-yl)-3-thiosemicarbazide B with subsequent reaction with the corresponding aromatic aldehyde to yield the target arylideneamino derivatives, 1–4 (Scheme 1), as previously described.69 Single crystals of compounds 1–4 were obtained by slow evaporation of their respective solutions in ethanol/chloroform (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) held at room temperature for 48 h.
:1, v/v) held at room temperature for 48 h.
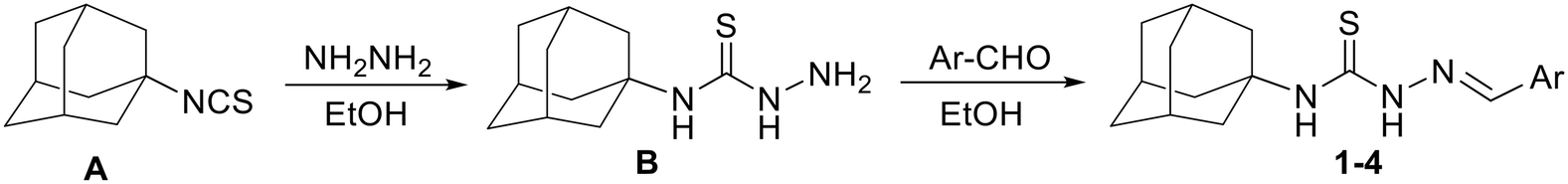 | ||
| Scheme 1 Outline of the synthesis of 1–4. | ||
3-(Adamantan-1-yl)-1-[(E)-(2-hydroxybenzylidene)amino]thiourea 1: colourless plates, M. pt: 194–196 °C; yield 70%.
3-(Adamantan-1-yl)-1-[(E)-(4-nitrobenzylidene)amino]thiourea 2: yellow plates, M. pt: 153–155 °C; yield 85%.
3-(Adamantan-1-yl)-1-[(E)-(2,4-difluorobenzylidene)amino]thiourea 3: colourless plates, M. pt: 216–218 °C; yield 72%.
3-(Adamantan-1-yl)-1-[(E)-(2,6-dichlorobenzylidene)amino]thiourea 4: colourless needles, M. pt: 238–240 °C; yield 75%.
2.2 X-ray crystallography
Intensity data for 1–4 were measured at 160 K on a Rigaku OD Synergy/Hypix diffractometer using CuKα radiation (λ = 1.54184 Å) from a dual wavelength X-ray source and an Oxford Instruments Cryojet XL cooler. The selected suitable single crystal was mounted using polybutene oil on a flexible loop fixed on a goniometer head and immediately transferred to the diffractometer. Analytical absorption corrections were applied and data processing was with CrysAlis Pro.70 The structures were solved by dual space direct methods using ShelXT71 and the refinements were by full-matrix least-squares (on F2) with anisotropic displacement parameters for all non-hydrogen atoms;72 the C-bound hydrogen atoms were included in the models in their calculated positions while O- and N-bound hydrogen atoms were refined freely. A weighting scheme of the form w = 1/[σ2(Fo2) + (aP)2 + bP], where P = (Fo2 + 2Fc2)/3, was introduced in each case. The crystal structure of 1 was refined as a non-merohedral 2-component twin. The twin law was identified and indexed using the Ewald Explorer tool of CrysAlisPro.70 The minor component is rotated by −179.93° around vector (0.71 −0.00 −0.71) in the reciprocal space (hkl) and around vector (0.89 0.00 −0.46) in the direct space (uvw). The scale factor of the major component is refined to 0.5791(9). The crystallographic analysis also included the use of the programs WinGX,73 ORTEP-3 for Windows,73 DIAMOND74 and PLATON.75 Selected crystal data and refinement details for 1–4 are collated in Table 1.| Compound | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| Formula | C18H23N3OS | C18H22N4O2S | C18H21F2N3S | C18H21Cl2N3S |
| Formula weight | 329.45 | 358.45 | 349.44 | 382.34 |
| Colour | Colourless | Yellow | Colourless | Colourless |
| Crystal size/mm3 | 0.04 × 0.12 × 0.33 | 0.03 × 0.05 × 0.22 | 0.05 × 0.07 × 0.29 | 0.06 × 0.09 × 0.28 |
| Space group | P21/n |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P |
| a/Å | 14.2535(3) | 6.3418(1) | 6.3669(1) | 7.2877(1) |
| b/Å | 6.5278(2) | 11.5247(3) | 11.8140(1) | 11.4798(2) |
| c/Å | 18.3623(4) | 12.2090(2) | 11.8768(1) | 11.5478(1) |
| α/° | 90 | 96.592(2) | 78.145(1) | 71.734(1) |
| β/° | 102.641(2) | 97.243(2) | 84.270(1) | 79.215(1) |
| γ/° | 90 | 104.197(2) | 79.081(1) | 83.190(1) |
| V/Å3 | 1667.09(7) | 848.26(3) | 856.746(17) | 899.36(2) |
| Z | 4 | 2 | 2 | 2 |
| D c/g cm−3 | 1.313 | 1.403 | 1.355 | 1.412 |
| λ(CuKα)/mm−1 | 1.782 | 1.863 | 1.895 | 4.359 |
| Measured data | 40669 |
18883 |
20080 |
24977 |
| θ range/° | 3.6–74.6 | 3.7–77.3 | 3.8–77.4 | 4.1–77.3 |
| Unique data | 40669 |
3566 | 3604 | 3802 |
| Observed data (I ≥ 2.0σ(I)) | 37646 |
3243 | 3507 | 3672 |
| No. parameters | 221 | 234 | 226 | 226 |
| R, obs. data; all data | 0.035; 0.095 | 0.036; 0.098 | 0.030; 0.082 | 0.027; 0.066 |
| a; b in weighting scheme | 0.058; 0.272 | 0.056; 0.298 | 0.043; 0.244 | 0.029; 0.429 |
| R w, obs. data; all data | 0.038; 0.097 | 0.039; 0.101 | 0.030; 0.082 | 0.030; 0.068 |
| Range of residual electron density peaks/e Å−3 | −0.23–+0.28 | −0.43–+0.29 | −0.17–+0.33 | −0.25–+0.35 |
2.3 Computational methods
All quantum chemical calculations were performed using the TURBOMOLE 7.7 program package76 on the experimentally determined X-ray fractional atomic coordinates. This approach was chosen because the focus of the study is to analyse the interactions as they stand in the solid-state, rather than determining the most stable gas-phase dimer. The electronic structure analyses were carried out at the PBE0-D4 level of theory. The PBE0 hybrid functional77 was employed for exchange–correlation, augmented by Grimme's D4 empirical dispersion78,79 correction to accurately account for non-covalent interactions. The def2-TZVP basis set was used for all atoms.80 The interaction energies were not corrected for the basis set superposition error (BSSE) due to the use of a large basis set.The PBE0-D4/def2-TZVP level was selected as it has been consistently demonstrated in the literature to provide an excellent balance of accuracy and computational cost for modelling non-covalent interactions, including dispersion-bound supramolecular complexes, yielding reliable interaction energies and electron density properties.31,77
To provide a detailed energetic breakdown of the intermolecular interactions, energy decomposition analysis (EDA) was performed using the Kitaura–Morokuma method.81 This also includes a dispersion term computed using the D4 approximation. Further characterisation of the non-covalent interactions, including quantum theory of atoms in molecules (QTAIM) and non-covalent interaction (NCI) plot analyses,82,83 was conducted at the same PBE0-D4/def2-TZVP level of theory using the AIMAll program.84 Molecular electrostatic potential (MEP) surfaces were also generated from the wavefunctions.
3. Results and discussion
3.1 Molecular structures
The molecular structures of 1–4 are shown in Fig. 2 and selected geometric parameters are listed in Tables 2 and S1. The molecule in 1 comprises a strictly planar S2CN core: r.m.s. deviation = 0.0028 Å with maximum deviation of 0.0049(12) Å for atom C1. The C1–S1(thione) bond length is 1.6958(16) Å with the C1–N1 [1.330(2) Å] and C1–N2 [1.3627(18) Å] bonds being disparate despite each secondary amine atom carrying a hydrogen bond. This reflects the substitution pattern in the molecule whereby the N1 atom is bound to an adamantan-1-yl group whereas the N2 atom is connected to the tertiary-N3 atom [N2–N3 = 1.3751(19) Å] which engages in an imine bond; C12–N3 = 1.2828(18) Å. These results are consistent with significant delocalisation of electron density in the central part of the molecule. Systematic variations in the angle subtended at the C1 atom also substantiate this conclusion with the widest angles involving the thione-S1 atom, i.e. S1–C1–N1 = 126.95(11)° and S1–C1–N2 = 118.08(11)°, with the widest of these involving the more tightly bound N1 atom; N1–C1–N2 = 114.96(14)°. As anticipated, the key torsion angles in this region of the molecule are close to planarity, i.e. N1–C1–N2–N3, C1–N2–N3–C12 and N2–N3–C12–C13, see Table 2. Globally, the amine-N1–H and amine-N2–H atoms are anti to each other with the latter lying to the same side of the molecule as the thione-S1 atom. This orientation allows for the formation of intramolecular amine-N1–H⋯N3(imine) hydrogen bonds within non-symmetric, five-membered {⋯N2CNH} synthons; geometric parameters are included in Table 3. | ||
| Fig. 2 (a)–(d) The molecular structures of 1–4, respectively, showing atom-labelling schemes and displacement ellipsoids at the 50% probability level, and (e) overlay diagram with 1 represented by the red image, inverted-2 (blue), inverted-3 (green) and 4 (pink). The molecules have been overlapped so the central N(S)N atoms are coincident. | ||
| Parameter | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| N1–C1–N2–N3 | −5.4(2) | 9.35(19) | 8.06(15) | −7.14(17) |
| N2–C1–N1–C2 | 165.91(14) | −167.86(13) | −165.14(10) | 173.77(12) |
| C1–N2–N3–C12 | −173.56(13) | 177.91(13) | 173.53(10) | −177.93(12) |
| N2–N3–C12–C13 | 178.28(12) | −177.58(12) | −178.70(9) | 177.85(11) |
| N3–C12–C13–C14 | −11.1(2) | 2.6(2) | −5.51(16) | −35.14(19) |
| N3–C12–C13–C18 | 170.45(13) | −177.69(14) | 174.65(11) | 147.10(12) |
| C1,N1,N2,S1/C13–C18 | 10.86(8) | 12.38(10) | 10.23(8) | 37.66(6) |
| A | Y | B | Y⋯B | A⋯B | A–Y⋯B | Symmetry operation |
|---|---|---|---|---|---|---|
| 1 | ||||||
| N1 | H1n | N3 | 2.17(2) | 2.6316(17) | 115.0(19) | x, y, z |
| N2 | H2n | O1 | 2.20(2) | 2.9959(17) | 153.9(17) | 2 − x, −y, 1 − z |
| O1 | H1o | S1 | 2.36(3) | 3.1034(11) | 151(2) | 2 − x, −y, 1 − z |
| C11 | H11a | Cg(C13–C18) | 2.83 | 3.6314(16) | 138 | −1/2 + x, 1/2 − y, −1/2 + z |
| C10 | H10a | H11b | 2.37 | 4.0313(19) | 144 | 1/2 − x, 1/2 + y, 1/2 − z |
| 2 | ||||||
|---|---|---|---|---|---|---|
| N1 | H1n | N3 | 2.139(19) | 2.6237(17) | 114.0(16) | x, y, z |
| N2 | H2n | S1 | 2.55(2) | 3.4030(13) | 161.6(17) | 3 − x, 1 − y, 2 − z |
| C12 | H12 | S1 | 2.86 | 3.7042(14) | 149 | 3 − x, 1 − y, 2 − z |
| C1 | S1 | Cg(C13–C18) | 3.7822(7) | 4.0981(15) | 88.42(5) | 2 − x, 1 − y, 2 − z |
| N4 | O1 | Cg(C13–C18) | 3.7257(15) | 4.3180(15) | 110.85(10) | 1 + x, y, z |
| C7 | H7a | H3a | 2.34 | 4.1538(17) | 148 | 1 − x, 1 − y, 1 − z |
| 3 | ||||||
|---|---|---|---|---|---|---|
| N1 | H1n | N3 | 2.155(15) | 2.6075(13) | 111.7(11) | x, y, z |
| N2 | H2n | S1 | 2.551(17) | 3.3993(10) | 165.9(16) | −x, 1 – y, 2 − z |
| C17 | H17 | F2 | 2.51 | 3.3671(16) | 149 | 1 − x, 2 − y, 2 − z |
| C1 | S1 | Cg(C13–C18) | 3.7325(6) | 4.1196(12) | 90.84(4) | 1 − x, 1 − y, 2 − z |
| C11 | H11a | H10a | 2.33 | 4.087(2) | 143 | 1 − x, −y, 1 − z |
| 4 | ||||||
|---|---|---|---|---|---|---|
| N1 | H1n | N3 | 2.113(16) | 2.5736(16) | 114.2(14) | x, y, z |
| N2 | H2n | S1 | 2.551(17) | 3.3919(12) | 169.3(16) | −x, 1 − y, −z |
| C18 | Cl2 | Cg(C13–C18) | 3.4802(7) | 4.0292(14) | 95.25(5) | −1 − x, 1 − y, 1 − z |
| C10 | H10b | Cl2 | 2.83 | 3.3746(13) | 115 | 1 + x, −1 + y, z |
The bond lengths and angles in 1–3 bear a close resemblance to those in 1, as indicated in SI Table S1. As indicated in Fig. 2(e), there is also a close similarity between the conformations of central cores and adamantan-1-yl groups across 1–4. However, significantly greater differences are apparent between the relative orientations of the terminal aryl groups. This is indicated by the range of N3–C12–C13–C14 torsion angles, i.e. 2.6(2)° for 2 to −35.14(19)° for 4, with the latter being the outlier. For 2, the terminal nitro group is slightly twisted out of the plane of the aryl ring with the greatest twist reflected in the C15–C16–N4–O2 torsion angle of 172.15(15)°.
3.2 Molecular packing
It will be demonstrated in the section that four distinct packing modes are observed due to i) different hydrogen-bonding motifs, i.e.1 compared to 2–4 and ii) different interactions involving the aryl rings, i.e. in 2–4. Geometric parameters characterising the identified intermolecular contacts are listed in Table 3.In the crystal of 1, complementary amine-N2–H⋯O(hydroxyl) and hydroxyl-O–H⋯S(thione) hydrogen bonds occur across a centre of inversion to generate a non-symmetric, six-membered {⋯HNCS⋯HO} synthon, as evident from both images comprising Fig. 3; this synthon is encompassed within a larger, centrosymmetric and 18-membered {⋯HOC3N2CS}2 synthon. The dimeric aggregates assemble in a supramolecular layer parallel (1 0 −1) with methylene-C11–H⋯π(aryl) interactions among the interactions between the dimers, as shown in Fig. 3(a). Close H⋯H contacts occur between adamantan-1-yl groups, as shown in Fig. 3(b).
 | ||
| Fig. 3 Molecular packing in the crystal of 1: (a) a slightly tilted side-on view of the supramolecular layer featuring amine-N–H⋯O(hydroxyl) and hydroxyl-O–H⋯S(thione) hydrogen bonds, and methylene-C–H⋯π(aryl) interactions shown as brown, orange and violet dashed lines, respectively, and (b) a view of the unit-cell contents in projection down the b-axis with H⋯H contacts shown as bright-green dashed lines. | ||
In the crystal of 2, in which the amine-N2–H atom is the only conventional hydrogen bond donor, amine-N2–H⋯S(thione) hydrogen-bonding occurs between centrosymmetrically related molecules within a centrosymmetric, eight-membered {⋯HNCS}2 synthon; peripheral to the synthons are imine-C–H⋯S(thione) interactions, as listed in Table 3. The aryl ring forms two side-on interactions, one with the thione-S1 atom and the other with the nitro-O1 atom. The S1⋯Cg(aryl) and O1⋯Cg(aryl) separations of 3.7822(7) and 3.7257(15) Å, respectively, are longer than the respective sum of the van der Waals radii of 3.70 and 3.42 Å;75 where the van der Waals radius of an aryl ring is assumed to be 1.90 Å.85 However, a closer inspection of the separations between the thione-S1 atom and the ring indicates a close S1⋯C18 contact of 3.5850(18) Å. With the next closest contact being 3.7303(16) Å, the S1⋯Cg(aryl) interaction may be classified as a localised contact, where the interacting atom is close to one carbon of the ring in contrast to a delocalised interaction where the interacting atom is close to all constituent atoms of the aryl ring.86 A similar conclusion pertains to the O1⋯Cg(aryl) interaction where the closest O1⋯C13 separation is 3.263(2) Å and the next shortest contact being 3.575(2) Å. The aforementioned interactions occur within a linear, supramolecular chain aligned along [1 0 0], Fig. 4(a). The chains assemble into layers parallel to (1 0 1) with the closest contacts between them being of the type H⋯H. The layers stack along the b-axis with the shortest interaction between a nitro-O1⋯H9(adamantan-1-yl) contact of 2.75 Å, just 0.03 Å beyond the sum of the van der Waals radii; a view of the unit-cell contents is shown in Fig. 4(b).
 | ||
Fig. 4 Molecular packing in the crystal of 2: (a) a side-on view of the supramolecular chain along [1 0 0] featuring amine-N–H ⋯S(thione) hydrogen bonds, and localised C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S⋯π(aryl) and N–O⋯π(aryl) interactions shown as orange, pink and red dashed lines, respectively, and (b) a view of the unit-cell contents in projection down the a-axis highlighting the stacking of layers along the b-axis with H⋯H contacts shown as bright-green dashed lines. S⋯π(aryl) and N–O⋯π(aryl) interactions shown as orange, pink and red dashed lines, respectively, and (b) a view of the unit-cell contents in projection down the a-axis highlighting the stacking of layers along the b-axis with H⋯H contacts shown as bright-green dashed lines. | ||
In the crystal of 3, as for 2, the amine-N–H⋯S(thione) hydrogen-bonding persists as does the centrosymmetric {⋯HNCS}2 synthon. These are complemented by aryl-C–H⋯F interactions that occur within centrosymmetric, eight-membered {⋯HC2F}2 synthons. The aryl ring forms a side-on interaction with the thione-S1 atom. The S1⋯Cg(aryl) separation of 3.7325(6) Å is about 0.03 Å longer than the respective sum of the van der Waals radii of 3.70 Å.85 The range of S1⋯C(aryl) separations is 3.8081(11) to 4.1519(13) Å, suggesting this is best classified as a weak, localised S⋯π(aryl) interaction.86 The mentioned intermolecular interactions occur within supramolecular layers parallel to (1 1 0), as illustrated in Fig. 5(a). The closest interactions between the layers, which stack along the c-axis, are of the type H⋯H, as highlighted in the view of the unit-cell contents shown in Fig. 5(b).
 | ||
| Fig. 5 Molecular packing in the crystal of 3: (a) a plane view of the supramolecular layer parallel to (1 1 0) featuring amine-N–H ⋯S(thione) hydrogen bonds, and aryl-C–H⋯F and weak delocalised CS⋯π(aryl) interactions shown as orange, blue and pink dashed lines, respectively, and (b) a view of the unit-cell contents in projection down the a-axis highlighting the stacking of layers along the c-axis with H⋯H contacts shown as bright-green dashed lines. | ||
Finally, in the crystal of 4, as for 2 and 3, the amino-N–H⋯S(thione) hydrogen-bonding is formed with a centrosymmetric {⋯HNCS}2 synthon. Linear chains of these dimeric aggregates feature aryl-C–Cl⋯π(aryl) interactions between centrosymmetrically molecules; the chain is approximately aligned along [−1 1 3], as shown in Fig. 6(a). The Cl⋯Cg(aryl) separation of 3.4802(7) Å is considerably shorter than the sum of the van der Waals radii of chlorine (1.75 Å (ref. 75)) and an aryl ring (1.90 Å (ref. 85)), i.e. 3.65 Å; the closest C⋯C contact between the rings is 3.4916(19) Å compared to the sum of the van der Waals of 3.40 Å. Given the closest individual Cl⋯C separation is considerably longer, i.e. 3.5783(14) Å, this interaction is best classified as a localised contact.86 The angle subtended at the chlorine atom in this interaction is 95.25(5)°, indicating this is a side-on interaction. The chains are assembled into a three-dimensional architecture with methylene-C–H⋯Cl interactions being the shortest contacts between them, as shown in Fig. 6(b).
 | ||
| Fig. 6 Molecular packing in the crystal of 4: (a) a view of the supramolecular chain approximately along [−1 1 3] featuring amine-N–H ⋯S(thione) hydrogen bonds, and aryl-C–Cl⋯π(aryl) interactions shown as orange and violet dashed lines, respectively, and (b) a view of the unit-cell contents in projection down the a-axis highlighting methylene-C–H⋯Cl interactions shown as turquoise dashed lines. | ||
3.3 Hirshfeld surface analysis
To assess the impact of the X⋯π (X = H, Cl, O and S) interactions upon the calculated Hirshfeld surfaces, relevant calculations were conducted with Crystal Explorer 17.5 (ref. 87) using standard protocols.88 The results for the salient surface contacts are listed in Table 4 with the full listing given in SI Table S2.| Compound | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| Y = O | Y = O | Y = F | Y = Cl | |
| Contact | ||||
| H⋯H | 61.0 | 48.8 | 49.2 | 49.7 |
| H⋯S/S⋯H | 13.4 | 17.3 | 9.8 | 11.6 |
| H⋯Y/Y⋯H | 4.3 | 9.4 | 16.8 | 17.2 |
| C⋯S/S⋯C | 0.0 | 1.5 | 1.8 | 0.0 |
| C⋯Y/Y⋯C | 0.4 | 2.4 | 0.7 | 4.9 |
As expected for molecules containing bulky adamantan-1-yl groups,89 most surface contacts in the crystals of 1–4 involve hydrogen, i.e. 97.1, 92.9, 92.7 and 90.0%, respectively. The most prominent surface contacts are of the type H⋯H with the maximum of 61.0% noted in the crystal of 1. The falloff in percentage contributions by H⋯H contacts in 2–4 is correlated with increasing significance of other contacts. For 2, there is an increase in both H⋯S/S⋯H contacts and of H⋯Y/Y⋯H contacts, involving the nitro-O atoms. More dramatic are the increases in H⋯Y/Y⋯H contacts in 3 and 4, Y = F or Cl, respectively, even though the Y⋯H separations are generally greater than the respective sums of the van der Waals radii, a short C–H⋯Cl interaction was noted in 4 (see above).
With respect to the calculated surface contacts not involving H-atoms, these are generally small or non-existent, as shown in SI Table S2. Of interest in consideration of the side-on Y⋯π(aryl) interactions described above are C⋯S/S⋯C and C⋯Y/Y⋯C surface contacts; two-dimensional fingerprint plots for these interactions are available in SI Fig. S1. For 2, these amount to 1.5 and 2.4%, respectively, consistent with the localised S, O⋯π(aryl) interactions. For 3, the contribution from C⋯S/S⋯C surface contacts [1.8%] corresponds to the long, delocalised S⋯π(aryl) interactions. The most significant contribution [4.9%] from side-on interactions of this type is found in 4, where relatively close, delocalised Cl⋯π(aryl) interactions were identified.
The perennial question in analyses such as these is whether the identified side-on Y⋯π(aryl) interactions in the crystals of 2–4 arise due to global molecular packing or are structure-directing. Hence, a comprehensive density functional theory (DFT) analysis was undertaken.
3.4 DFT calculations
To complement the experimental structural insights gleaned from X-ray diffraction analyses of compounds 1–4, a density functional theory (DFT) study was undertaken to further dissect the nature and energetic contributions of the non-covalent interactions identified in their molecular packing. While conventional hydrogen bonding is predominant in many supramolecular assemblies, the crystallographic findings highlight the presence of intriguing side-on X⋯π (X = O, S, Cl) interactions in 2–4, which warrant deeper theoretical investigation. Specifically, the aim was to characterise the lone-pair (LP)⋯π interactions involving the thione-S1 atom (in 2 and 3), as well as the significant C–Cl⋯π interaction in 4. Through detailed molecular electrostatic potential (MEP) surface analysis, regions of electrophilicity and nucleophilicity are mapped to rationalise the propensity for these interactions. Furthermore, a combined quantum theory of atoms in molecules (QTAIM) and non-covalent interaction (NCI) plot analysis is employed to provide a topological characterisation of the electron density in these contacts, confirming their non-covalent nature. Finally, energy decomposition analysis (EDA) is utilised to quantify the energetic contributions of the various components (electrostatic, exchange-repulsion, orbital, correlation and dispersion) within the self-assembled LP⋯π dimers, offering a quantitative understanding of their stability and directionality in the solid-state. This computational approach will ascertain whether these X⋯π interactions are merely a consequence of global molecular packing considerations or if they play a structure-directing role, thereby contributing to the rational design of new materials.Fig. 7 presents the calculated molecular electrostatic potential (MEP) surfaces for compounds 2 (a), 3 (b), and 4 (c). These maps visualise the charge distribution and potential sites for intermolecular interactions. In all three compounds, the most positive MEP value, indicating the most electrophilic region, is consistently located at the hydrogen atom of the exposed amino group (the one that does not participate in the intramolecular N–H⋯N hydrogen bond). These maxima range from 42.0 kcal mol−1 in 4 to 46.4 kcal mol−12, clearly highlighting these sites as preferential hydrogen bond donors. Conversely, the most negative MEP regions, representing nucleophilic sites, are observed at the thione-S atom in 3 (−30.1 kcal mol−1) and 4 (−31.8 kcal mol−1), and at the nitro-O atoms in 2 (−28.8 kcal mol−1) followed by the thione-S atom (−26.3 kcal mol−1). These negative minima are consistent with the observed hydrogen-bonding interactions in the solid state, where these atoms act as hydrogen bond acceptors. Furthermore, the fluorine atoms in 3 and the chlorine atoms in 4 also exhibit small negative MEP values (−6.7 kcal mol−1 and −5.0 kcal mol−1, respectively), indicating their potential to act as weak hydrogen bond acceptors or participate in other minor attractive interactions. Crucially, the MEP analysis of the aromatic rings reveals a slightly positive electrostatic potential in all compounds, ranging from a minimum of 0.7 kcal mol−1 in 4 to a maximum of 6.9 kcal mol−1 in 2. This positive potential over the π-system directly rationalises the propensity for the observed LP⋯π interactions, where electron-rich lone-pairs from neighbouring atoms can engage with the electrophilic face of the aromatic ring.
 | ||
| Fig. 7 The molecular electrostatic potential (MEP) surfaces of (a) 2, (b) 3, and (c) 4. The colour scale, ranging from −32 kcal mol−1 (red) to 48 kcal mol−1 (blue), indicates regions of negative and positive electrostatic potential, respectively. Key MEP values are shown on the surfaces. The isovalue = 0.001 a.u. | ||
Fig. 8 presents a combined quantum theory of atoms in molecules (QTAIM) and non-covalent interaction (NCI) plot analysis for the self-assembled LP⋯π dimers of (a) 2 and (b) 3, along with (c) showing an energy decomposition analysis (EDA) of their respective interaction energies. The QTAIM analysis vividly confirms the LP⋯π nature of the primary interaction: a bond critical point (BCP, depicted as a red sphere) and a corresponding bond path connect the thione-S atom of one molecule to a carbon atom of the aromatic π-system of a neighbouring molecule. The extended green RDG (reduced density gradient) isosurface from the NCIplot analysis, which broadly embraces the interacting π-systems, further substantiates the delocalised, non-covalent nature of this interaction. Importantly, the analysis reveals that the exocyclic and conjugated iminothiourea system also actively participates in the binding mechanism. This is evidenced by two symmetrically equivalent BCPs interconnecting carbon and nitrogen atoms within this exocyclic π-system, accompanied by a green RDG isosurface that occupies the space between these interacting regions, indicating their involvement in stabilising interactions.
 | ||
| Fig. 8 A combined QTAIM/NCIplot analysis and EDA for the self-assembled LP⋯π dimers: (a) dimer of 2, highlighting the S(LP)⋯π interaction, (b) dimer of 3, showing LP⋯π and C−H⋯F interactions. Red spheres denote bond critical points (BCPs) with corresponding bond paths as dashed bonds. Only intermolecular interactions are represented. Green isosurfaces represent attractive non-covalent interactions (RDG < 0). (c) A bar plot showing the energy components (in kcal mol−1) from EDA for the dimers of 2 and 3. | ||
In the case of 3, as shown in Fig. 8(b), two additional C–H⋯F contacts are observed. These interactions are characterised by specific BCPs, bond paths and small, green RDG isosurfaces, indicating a weak, stabilising non-covalent contribution. The presence of these extra C–H⋯F contacts likely account for the slightly stronger overall interaction energy in the dimer of 3 (−12.6 kcal mol−1) compared to that of 2 (−11.5 kcal mol−1), as shown in the EDA summary. The significant dimerization energies obtained for both dimers underscore the important role of these self-assembled units in dictating the overall molecular packing.
The EDA presented in Fig. 8(c) provides quantitative insights into the nature of these interactions. Consistent with the MEP analysis showing low positive values over the π-systems, as shown in Fig. 7, the electrostatic term (Eel, light-blue bars) is relatively small, suggesting it contributes minimally to the overall stability. Instead, the interactions are primarily dominated by dispersion (Edisp, green bars), which is typical for π-stacking and LP⋯π interactions where long-range attractive forces are key. The correlation term (Ecor, purple bars) is also notably relevant, further emphasising the importance of electron correlation effects in these types of interactions. The exchange-repulsion term (Eex-rep, red bars) predictably shows repulsive contributions, balancing the attractive forces. Overall, the EDA confirms that these self-assembled dimers are primarily stabilised by dispersion and correlation effects, with electrostatic contributions playing a minor role.
Fig. 9 presents the combined QTAIM/NCIplot analysis and EDA for the self-assembled Cl⋯π dimer of compound 4. The QTAIM analysis confirms the Cl⋯π contacts through the presence of a BCP and a bond path connecting the chlorine atom to a carbon atom of the aromatic ring. While the QTAIM highlights the local interaction, the overall π-interaction is better visualised by the NCIplot analysis, which shows an extended green RDG isosurface typical of π-interactions. The dimerization energy for 4 is significantly smaller at −6.1 kcal mol−1, particularly when compared to the dimerization energies observed for compounds 2 and 3. This reduction in stabilisation energy can be attributed to the absence of the π-stacking interactions involving the iminothiourea groups, which were noted in the dimers of 2 and 3.
 | ||
| Fig. 9 (a) A combined QTAIM/NCIplot analysis and EDA for the self-assembled LP⋯π dimer of 4, showing the Cl(LP)⋯π interactions. The red spheres denote bond critical points (BCPs) with corresponding bond paths as dashed bonds. Only intermolecular interactions are represented. The green isosurfaces represent attractive non-covalent interactions (RDG < 0). (b) A bar plot showing the energy components (in kcal mol−1) from EDA for the dimer of 4. | ||
Consistent with previous analyses, the EDA shown in Fig. 9(b) further confirms the dominant role of dispersion (Edisp = −7.0 kcal mol−1) in stabilising this dimer, followed by a notable contribution from correlation (Ecor = −3.8 kcal mol−1). The electrostatic term (Eel = −1.6 kcal mol−1) is almost negligible, which is in good agreement with the small positive MEP value (0.7 kcal mol−1) observed over the aromatic ring in 4, Fig. 7(c). This analysis collectively highlights that the Cl⋯π interactions in 4 are primarily driven by dispersion forces, with electrostatic contributions playing a very minor role.
4. Conclusions
This study successfully synthesised and fully characterised four thiourea derivatives (1–4) through single-crystal X-ray diffraction, revealing their distinct molecular structures and diverse molecular packing motifs. The crystallographic analysis identified conventional hydrogen-bonding as a key organisational principle in 1, while compounds 2–4 exhibited fascinating non-conventional side-on X⋯π (X = O, S, Cl) interactions that play a significant role in their solid-state assembly.The integrated computational approach, employing molecular electrostatic potential (MEP) surfaces, combined quantum theory of atoms in molecules (QTAIM)/non-covalent interaction (NCI) plot analyses and energy decomposition analysis (EDA), provided crucial insights into the nature and energetics of these interactions. The MEP analysis effectively mapped the electrophilic and nucleophilic regions of the molecules, explaining the propensity for both classical hydrogen bonds and the more subtle LP⋯π interactions at the aromatic rings. The QTAIM and NCI analyses unequivocally confirmed the presence and delocalised nature of LP⋯π contacts involving sulphur and oxygen atoms (in 2 and 3) and Cl⋯π contacts (in 4), identifying specific bond critical points and extended attractive isosurfaces.
Quantitatively, the EDA revealed significant dimerization energies for the self-assembled units: −11.5 kcal mol−1 for 2, −12.6 kcal mol−1 for 3 and −6.1 kcal mol−1 for 4. These substantial energies confirm the importance of these assemblies in dictating the overall molecular packing. Critically, the EDA consistently demonstrated that dispersion forces constitute the dominant attractive component, followed by correlation energy, while the electrostatic term was found to be almost negligible. The comparatively lower dimerization energy of 4 was attributed to the absence of additional π-stacking contributions from the iminothiourea groups, which were present in the crystals of 2 and 3.
In conclusion, this joint experimental and theoretical study unequivocally demonstrates that the side-on X⋯π interactions, alongside conventional hydrogen bonds, are not mere incidental contacts but are indeed structure-directing forces that profoundly influence the supramolecular architecture in these thiourea derivatives.
Author contributions
Lamya H. Al-Wahaibi: methodology, formal analysis and funding acquisition. Olivier Blacque: data acquisition. Aamal A. Al-Mutairi: methodology and investigation. Ali A. El-Emam: conceptualisation, writing and project administration. Rosa M. Gomila: DFT calculations and writing. Antonio Frontera: DFT calculations and writing. Edward R. T. Tiekink: crystallography, writing and project administration.Conflicts of interest
The authors declare no competing financial interest.Data availability
Spectroscopic data, crystallographic data, in the form of CIFs, and computational chemistry details are available from the corresponding authors.Supplementary information: table of geometric parameters and of percentage contributions to the calculated Hirshfeld surfaces, and two-dimensional fingerprint plots. See https://doi.org/10.1039/d5ce00817d.
CCDC 2481860–2481863 contain the supplementary crystallographic data for this paper.90a–d
Acknowledgements
This research was funded by the Princess Nourah bint Abdulrahman University Researchers Supporting Project No. PNURSP2025R3, Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia. The “Ministerio de Ciencia, Investigacion y Universidades/Agencia Estatal de Investigación” (MICIU/AEI/https://doi.org/10.13039/501100011033) of Spain (projects PID2020-115637GB-I00 and PID2023-148453NB-I00, ERDF a way of making Europe) is also thanked for support of this work.References
- E. R. T. Tiekink and J. Zukerman-Schpector, The importance of pi-interactions in crystal engineering: Frontiers in Crystal Engineering, John Wiley & Sons, Ltd, 2012 Search PubMed.
- G. R. Desiraju and A. Gavezzotti, From molecular to crystal structure; polynuclear aromatic hydrocarbons, J. Chem. Soc., Chem. Commun., 1989, 621–623, 10.1039/C39890000621.
- C. A. Hunter and J. K. M. Sanders, The nature of π-π interactions, J. Am. Chem. Soc., 1990, 112, 5525–5534, DOI:10.1021/ja00170a016.
- J. K. Klosterman, Y. Yamauchia and M. Fujita, Engineering discrete stacks of aromatic molecules, Chem. Soc. Rev., 2009, 38, 1714–1725, 10.1039/b901261n.
- D. B. Ninković, G. V. Janjić, D. Ž. Veljković, D. N. Sredojević and S. D. Zarić, What are the preferred horizontal displacements in parallel aromatic-aromatic interactions? Significant interactions at large displacements, ChemPhysChem, 2011, 12, 3511–3514, DOI:10.1002/cphc.201100777.
- S. E. Wheeler, Understanding substituent effects in noncovalent interactions involving aromatic rings, Acc. Chem. Res., 2013, 46, 1029–1038, DOI:10.1021/ar300109n.
- R. Thakuria, N. K. Nath and B. K. Saha, The nature and applications of π–π interactions: a perspective, Cryst. Growth Des., 2019, 19(2), 523–528, DOI:10.1021/acs.cgd.8b01630.
- S. E. Wheeler, Revisiting the Hunter-Sanders model for π−π interactions, J. Am. Chem. Soc., 2025, 147, 19738–19750, DOI:10.1021/jacs.5c03169.
- K. Takimiya, S. Shinamura, I. Osaka and E. Miyazaki, Thienoacene-based organic semiconductors, Adv. Mater., 2011, 23, 4347–4370, DOI:10.1002/adma.201102007.
- T. Mori, Classification of crystal structures of thiophene-containing organic semiconductors, CrystEngComm, 2023, 25, 6266–6278, 10.1039/D3CE00893B.
- Z. D. Tomić, D. N. Sredojević and S. D. Zarić, Stacking interactions between chelate and phenyl rings in square-planar transition metal complexes, Cryst. Growth Des., 2006, 6, 29–31, DOI:10.1021/cg050392r.
- D. P. Malenov, G. V. Janjiae, V. B. Medakoviae, M. B. Hall and S. D. Zarić, Noncovalent bonding: stacking interactions of chelate rings of transition metal complexes, Coord. Chem. Rev., 2017, 345, 318–341, DOI:10.1016/j.ccr.2016.12.020.
- D. N. Sredojević, Z. D. Tomić and S. D. Zarić, Evidence of chelate−chelate stacking interactions in crystal structures of transition-metal complexes, Cryst. Growth Des., 2010, 10, 3901–3908, DOI:10.1021/cg100312r.
- W. G. Jacob and S. E. Wheeler, Taking the aromaticity out of aromatic interactions, Angew. Chem., Int. Ed., 2011, 50, 7847–7849, DOI:10.1002/anie.201102982.
- A. J. Neel, M. J. Hilton, M. S. Sigman and F. D. Toste, Exploiting non-covalent π interactions for catalyst design, Nature, 2017, 543, 637–646, DOI:10.1038/nature21701.
- D. B. Ninković, J. P. Blagojević Filipović, M. B. Hall, E. N. Brothers and S. D. Zarić, What is special about aromatic-aromatic interactions? significant attraction at large horizontal displacement, ACS Cent. Sci., 2020, 6, 420–425, DOI:10.1021/acscentsci.0c00005.
- M. Nishio, CH/π hydrogen bonds in crystals, CrystEngComm, 2004, 6, 130–158, 10.1039/b313104a.
- M. Nishio, Y. Umezawa, J. Fantini, M. S. Weissd and P. Chakrabartie, CH–π hydrogen bonds in biological macromolecules, Phys. Chem. Chem. Phys., 2014, 16, 12648–12683, 10.1039/C4CP00099D.
- D. Chen, C. S. Lai and E. R. T. Tiekink, Crystal structures of 2,2′-bipyridine adducts of two cadmium O-alkyl dithiocarbonates: rationalisation of disparate coordination geometries based on different crystal packing environments, Z. Kristallogr. - Cryst. Mater., 2002, 218, 747–752, DOI:10.1524/zkri.218.11.747.20300.
- D. Sredojević, G. A. Bogdanović, Z. D. Tomić and S. D. Zarić, Stacking vs. CH–π interactions between chelate and aryl rings in crystal structures of square-planar transition metal complexes, CrystEngComm, 2007, 9, 793–798, 10.1039/B704302C.
- E. R. T. Tiekink and J. Zukerman-Schpector, Emerging supramolecular synthons: C–H⋯(chelate) interactions in metal bis(1,1-dithiolates), Chem. Commun., 2011, 47, 6623–6625, 10.1039/C1CC11173F.
- J. C. Ma and D. A. Dougherty, The cation−π interaction, Chem. Rev., 1997, 97, 1303–1324, DOI:10.1021/cr9603744.
- I. Caracelli, J. Zukerman-Schpector and E. R. T. Tiekink, Supramolecular synthons based on gold⋯π(arene) interactions, Gold Bull., 2013, 46, 81–89, DOI:10.1007/s13404-013-0088-7.
- M. Egli and R. V. Gessner, Stereoelectronic effects of deoxyribose O4' on DNA conformation, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 180–184, DOI:10.1073/pnas.92.1.180.
- T. J. Mooibroek, Patrick Gamez and J. Reedijk, Lone pair–π interactions: a new supramolecular bond?, CrystEngComm, 2008, 10, 1501–1515, 10.1039/B812026A.
- I. Caracelli, J. Zukerman-Schpector, I. Haiducc and E. R. T. Tiekink, Main group metal lone-pair⋯π(arene) interactions: a new bonding mode for supramolecular associations, CrystEngComm, 2016, 18, 6960–6978, 10.1039/c6ce01460g.
- S. Demeshko, S. Dechert and F. Meyer, Anion−π interactions in a carousel copper(II)−triazine complex, J. Am. Chem. Soc., 2004, 126, 4508–4509, DOI:10.1021/ja049458h.
- E. Kanao, T. Morinaga, T. Kubo, T. Naito, T. Matsumoto, T. Sano, H. Maki, M. Yan and K. Otsuka, Separation of halogenated benzenes enabled by investigation of halogen–π interactions with carbon materials, Chem. Sci., 2020, 11, 409–418, 10.1039/C9SC04906A.
- E. R. T. Tiekink, Supramolecular architectures sustained by delocalised C–I⋯π(arene) interactions in molecular crystals and the propensity of their formation, CrystEngComm, 2021, 23, 904–928, 10.1039/d0ce01677b.
- P. Politzer and J. S. Murray, Halogen bonding: an interim discussion, ChemPhysChem, 2013, 14, 278–294, DOI:10.1002/cphc.201200799.
- M. H. Kolář and P. Hobza, Computer modeling of halogen bonds and other σ-hole interactions, Chem. Rev., 2016, 116, 5155–5187, DOI:10.1021/acs.chemrev.5b00560.
- S. Scheiner, Transition between the noncovalency and covalency of σ–hole bonds, J. Phys. Chem. A, 2023, 127, 9760–9770, DOI:10.1021/acs.jpca.3c06093.
- A. Mukherjee, S. Tothadi and G. R. Desiraju, Halogen bonds in crystal engineering: Like hydrogen bonds yet different, Acc. Chem. Res., 2014, 47, 2514–2524, DOI:10.1021/ar5001555.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, The halogen bond, Chem. Rev., 2016, 116, 2478–2601, DOI:10.1021/acs.chemrev.5b00484.
- R. H. Crabtree, Hypervalency, secondary bonding and hydrogen bonding: siblings under the skin, Chem. Soc. Rev., 2017, 46, 1720–1729, 10.1039/C6CS00688D.
- P. Politzer and J. S. Murray, Electrostatics and polarization in σ- and π-hole noncovalent interactions: an overview, ChemPhysChem, 2020, 21, 579–588, DOI:10.1002/cphc.201900968.
- S. Scheiner, Dissection of the origin of π-holes and the noncovalent bonds in which they engage, J. Phys. Chem. A, 2021, 125, 6514–6528, DOI:10.1021/acs.jpca.1c05431.
- N. Tarannam, R. Shukla and S. Kozuch, Yet another perspective on hole interactions, Phys. Chem. Chem. Phys., 2021, 23, 19948–19963, 10.1039/D1CP03533A.
- T. Lang, X. Li, L. Meng, S. Zheng and Y. Zeng, The cooperativity between the σ-hole and π-hole interactions in the ClO⋯XONO2/XONO⋯NH3 (X = Cl, Br, I) complexes, Struct. Chem., 2015, 26, 213–221, DOI:10.1007/s11224-014-0486-3.
- W. Zierkiewicz, M. Michalczyk and S. Scheiner, Noncovalent bonds through sigma and pi-hole located on the same molecule. guiding principles and comparisons, Molecules, 2021, 26, 1740, DOI:10.3390/molecules26061740.
- R. Shukla, N. Claiser, M. Souhassou, C. Lecomte, S. J. Balkrishna, S. Kumara and D. Chopra, Exploring the simultaneous σ-hole/π-hole bonding characteristics of a Br⋯π interaction in an ebselen derivative via experimental and theoretical electron-density analysis, IUCrJ, 2018, 5, 647–653, DOI:10.1107/S2052252518011041.
- E. R. T. Tiekink, Characterising supramolecular architectures in crystals featuring I⋯Br halogen bonding: Persistence of X⋯X' secondary-bonding in their congeners, Crystals, 2021, 11, 433, DOI:10.3390/cryst11040433.
- S. Scheiner, Competition between sigma and pi holes on the same atom, CrystEngComm, 2025, 27, 921–930, 10.1039/d4ce01194e.
- D. A. Parrish, J. R. Deschamps, R. D. Gilardi and R. J. Butcher, Polymorphs of picryl bromide, Cryst. Growth Des., 2008, 8, 57–62, DOI:10.1021/cg700727n.
- L. Huang, L. Massa and J. Karle, Calculated interactions of a nitro group with aromatic rings of crystalline picryl bromide, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 13720–13723, DOI:10.1073/pnas.0807218105.
- A. Bauzá, A. Frontera and T. J. Mooibroek, π-Hole interactions involving nitro aromatic ligands in protein structures, Chem. – Eur. J., 2019, 25, 13436–13443, DOI:10.1002/chem.201903404.
- K. S. C. Reid, P. F. Lindley and J. M. Thornton, Sulphur-aromatic interactions in proteins, FEBS Lett., 1985, 190, 209–213, DOI:10.1016/0014-5793(85)81285-0.
- K. N.-M. Daeffler, H. A. Lester and D. A. Dougherty, Functionally important aromatic–aromatic and sulfur−π interactions in the D2 dopamine receptor, J. Am. Chem. Soc., 2012, 134, 14890–14896, DOI:10.1021/ja304560x.
- R. F. N. Silva, A. C. S. Sacco, I. Caracelli, J. Zukerman-Schpector and E. R. T. Tiekink, Sulfur(lone-pair)⋯π interactions with FAD in flavoenzymes, Z. Kristallogr. - Cryst. Mater., 2018, 233, 531–537, DOI:10.1515/zkri-2018-2064.
- Y. Jin, W. Li, R. T. Saragi, M. Juanes, C. Pérez, A. Lesarri and G. Feng, Sulfur–arene interactions: the S⋯π and S–H⋯π interactions in the dimers of benzofuran⋯sulfur dioxide and benzofuran⋯hydrogen sulfide, Phys. Chem. Chem. Phys., 2023, 25, 12174–12181, 10.1039/D3CP01146A.
- J. Nath and J. B. Baruah, Positional isomers of (E)-2-(anthracen-9-ylmethylene)-N-(aryl)hydrazinecarbothioamide, zinc complexes and polymorphic solvates, CrystEngComm, 2025, 27, 46–54, 10.1039/D4CE00992D.
- W. L. Davies, R. R. Grunnert, R. F. Haff, J. W. McGahen, E. M. Neumeyer, M. Paulshock, J. C. Watts, T. R. Wood, E. C. Hermann and C. E. Hoffmann, Antiviral activity of 1-adamantanamine (amantadine), Science, 1964, 144, 862–863, DOI:10.1126/science.144.3620.862.
- H. A. Wendel, M. T. Snyder and S. Pell, Trial of amantadine in epidemic influenza, Clin. Pharmacol. Ther., 1966, 7, 38–43, DOI:10.1002/cpt19667138.
- F. G. Hayden and A. S. Monto, Oral rimantadine hydrochloride therapy of influenza A virus H3N2 subtype infection in adults, Antimicrob. Agents Chemother., 1986, 29, 339–341, DOI:10.1128/AAC.29.2.339.
- K. S. Rosenthal, M. S. Sokol, R. L. Ingram, R. Subramanian and R. C. Fort, Tromantadine: inhibitor of early and late events in herpes simplex virus replication, Antimicrob. Agents Chemother., 1982, 22, 1031–1036, DOI:10.1128/AAC.22.6.1031.
- L. Wanka, K. Iqbal and P. R. Schreiner, The lipophilic bullet hits the targets: medicinal chemistry of adamantane derivatives, Chem. Rev., 2013, 113, 3516–3604, DOI:10.1021/cr100264t.
- J. Liu, D. Obando, V. Liao, T. Lifa and R. Codd, The many faces of the adamantyl group in drug design, Eur. J. Med. Chem., 2011, 46, 1949–1963, DOI:10.1016/j.ejmech.2011.01.047.
- G. Lamoureux and G. Artavia, Use of the adamantane structure in medicinal chemistry, Curr. Med. Chem., 2010, 17, 2967–2978, DOI:10.2174/092986710792065027.
- M. E. Burstein, A. V. Serbin, T. V. Khakhulina, I. V. Alymova, L. L. Stotskaya, O. P. Bogdan, E. E. Manukchina, V. V. Jdanov, N. K. Sharova and A. G. Bukrinskaya, Inhibition of HIV-1 replication by newly developed adamantane-containing polyanionic agents, Antiviral Res., 1999, 41, 135–144, DOI:10.1016/s0166-3542(99)00006-6.
- A. A. El-Emam, O. A. Al-Deeb, M. Al-Omar and J. Lehmann, Synthesis, antimicrobial, and anti-HIV-1 activity of certain 5-(1-adamantyl)-2-substituted thio-1,3,4-oxadiazoles and 5-(1-adamantyl)-3-substituted aminomethyl-1,3,4-oxadiazoline-2-thiones, Bioorg. Med. Chem., 2004, 12, 5107–5113, DOI:10.1016/j.bmc.2004.07.033.
- P. Lorenzo, R. Alvarez, M. A. Ortiz, S. Alvarez, F. J. Piedrafita and A. R. de Lera, Inhibition of IκB kinase-β and anticancer activities of novel chalcone adamantyl arotinoids, J. Med. Chem., 2008, 51, 5431–5440, DOI:10.1021/jm800285f.
- L. Farhana, M. I. Dawson, M. Leid, L. Wang, D. D. Moore, G. Liu, Z. Xia and J. A. Fontana, Adamantyl-substituted retinoid-related molecules bind small heterodimer partner and modulate the Sin3A repressor, Cancer Res., 2007, 67, 318–325, DOI:10.1158/0008-5472.CAN-06-2164.
- A. A. El-Emam, A. M. Al-Tamimi, M. A. Al-Omar, K. A. Alrashood and E. E. Habib, Synthesis and antimicrobial activity of novel 5-(1-adamantyl)-2-aminomethyl-4-substituted-1,2,4-triazoline-3-thiones, Eur. J. Med. Chem., 2013, 68, 96–102, DOI:10.1016/j.ejmech.2013.07.024.
- E. J. North, M. S. Scherman, D. F. Bruhn, J. S. Scarborough, M. M. Maddox, V. Jones, A. Grzegorzewicz, L. Yang, T. Hess, C. Morisseau, M. Jackson, M. R. McNeil and R. E. Lee, Design, synthesis and anti-tuberculosis activity of 1-adamantyl-3-heteroaryl ureas with improved in vitro pharmacokinetic properties, Bioorg. Med. Chem., 2013, 21, 2587–2599, DOI:10.1016/j.bmc.2013.02.028.
- X. Wang, Y. Dong, S. Wittlin, S. A. Charman, F. C. Chiu, J. Chollet, K. Katneni, J. Mannila, J. Morizzi, E. Ryan, C. Scheurer, J. Steuten, J. Santo Tomas, C. Snyder and J. L. Vennerstrom, Comparative antimalarial activities and ADME profiles of ozonides (1,2,4-trioxolanes) OZ277, OZ439, and their 1,2-dioxolane, 1,2,4-trioxane, and 1,2,4,5-tetraoxane isosteres, J. Med. Chem., 2013, 56, 2547–2555, DOI:10.1021/jm400004u.
- B. Shakya and P. N. Yadav, Thiosemicarbazones as potent anticancer agents and their modes of action, Mini-Rev. Med. Chem., 2020, 20, 638–661, DOI:10.2174/1389557519666191029130310.
- I. D'Agostino, G. E. Mathew, P. Angelini, R. Venanzoni, G. Angeles Flores, A. Angeli, S. Carradori, B. Marinacci, L. Menghini, M. A. Abdelgawad, M. M. Ghoneim, B. Mathew and C. T. Supuran, Biological investigation of N-methyl thiosemicarbazones as antimicrobial agents and bacterial carbonic anhydrases inhibitors, J. Enzyme Inhib. Med. Chem., 2022, 37, 986–993, DOI:10.1080/14756366.2022.2055009.
- L. R. P. de Siqueira, P. A. T. de Moraes Gomes, L. P. de Lima Ferreira, M. J. B. de Melo Rêgo and A. C. L. Leite, Multi-target compounds acting in cancer progression: focus on thiosemicarbazone, thiazole and thiazolidinone analogues, Eur. J. Med. Chem., 2019, 170, 237–260, DOI:10.1016/j.ejmech.2019.03.024.
- A. A. Al-Mutairi, M. A. Al-Alshaikh, F. A. M. Al-Omary, H. M. Hassan, A. M. El-Mahdy and A. A. El-Emam, Synthesis, antimicrobial, and anti-proliferative activities of novel 4-(adamantan-1-yl)-1-arylidene-3-thiosemicarbazides, 4-arylmethyl N'-(adamantan-1-yl)piperidine-1-carbothioimidates, and related derivatives, Molecules, 2019, 24, 4308, DOI:10.3390/molecules24234308.
- CrysAlis PRO, Rigaku Oxford Diffraction, Yarnton, Oxfordshire, England, 2024 Search PubMed.
- G. M. Sheldrick, SHELXT – Integrated space-group and crystal-structure determination, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8, DOI:10.1107/S2053273314026370.
- G. M. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8, DOI:10.1107/S2053229614024218.
- L. J. Farrugia, WinGX and ORTEP for windows: an update, J. Appl. Crystallogr., 2012, 45, 849–854, DOI:10.1107/S0021889812029111.
- K. Brandenburg, DIAMOND, Crystal Impact GbR, Bonn, Germany, 2006 Search PubMed.
- A. Spek, checkCIF validation ALERTS: what they mean and how to respond, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2020, 76, 1–11, DOI:10.1107/S2056989019016244.
- TURBOMOLE V7.7 2022, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, since 2007; available from http://www.turbomole.com.
- C. Adamo and V. Barone, Toward reliable Density Functional Methods without adjustable parameters: the PBE0 model, J. Chem. Phys., 1999, 110, 6158–6170, DOI:10.1063/1.478522.
- S. Grimme, S. Ehrlich and F. Goerigk, Effect of the damping Function in dispersion corrected Density Functional Theory, J. Comput. Chem., 2011, 32, 1456–1465, DOI:10.1002/jcc.21759.
- S. Grimme, A. Hansen, J. G. Brandenburg and F. Bannwarth, Dispersion-corrected Density Functional Theory: an overview of the D3 and D4 models, Chem. Rev., 2016, 116, 5105–5154, DOI:10.1021/acs.chemrev.5b00533.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305, 10.1039/b508541a.
- K. Kitaura and K. Morokuma, A new energy decomposition scheme for molecular interactions within the Hartree-Fock approximation, Int. J. Quantum Chem., 1976, 10, 325–340, DOI:10.1002/qua.560100211.
- R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, Oxford, 1990 Search PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, Revealing noncovalent interactions, J. Am. Chem. Soc., 2010, 132, 6498–6506, DOI:10.1021/ja100936w.
- T. A. Keith, AIMAll (Version 19.10.12), TK Gristmill Software, Overland Park KS, USA, 2019, http://aim.tkgristmill.com Search PubMed.
- C. Janiak, A critical account on π–π stacking in metal complexes with aromatic nitrogen-containing ligands, J. Chem. Soc., Dalton Trans., 2000, 3885–3896, 10.1039/B003010O.
- D. Schollmeyer, O. V. Shishkin, T. Rühl and M. O. Vysotsky, OH–π and halogen–π interactions as driving forces in the crystal organisations of tri-bromo and tri-iodo trityl alcohols, CrystEngComm, 2008, 10, 715–723, 10.1039/B716442D.
- P. R. Spackman, M. J. Turner, J. J. Mckinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, CrystalExplorer: a program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals, J. Appl. Crystallogr., 2021, 54, 1006–1011, DOI:10.1107/S1600576721002910.
- S. L. Tan, M. M. Jotani and E. R. T. Tiekink, Utilizing Hirshfeld surface calculations, non-covalent interaction (NCI) plots and the calculation of interaction energies in the analysis of molecular packing, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2019, 75, 308–318, DOI:10.1107/S2056989019001129.
- T. S. Basu Baul, R. Manne, A. Duthie, L. Y. Liew, J. Chew, S. M. Lee and E. R. T. Tiekink, Synthesis, structural and in vitro biological evaluation of diamondoid-decorated lipophilic organotin(IV) derivatives, J. Organomet. Chem., 2021, 941, 121802, DOI:10.1016/j.jorganchem.2021.121802.
- (a) CCDC 2481860: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2p9l0m; (b) CCDC 2481861: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2p9l1n; (c) CCDC 2481862: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2p9l2p; (d) CCDC 2481863: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2p9l3q.
| This journal is © The Royal Society of Chemistry 2025 |