
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Manipulating vapochromic or solvatochromic properties of platinum(II) pincer complexes through ligand modifications
Clare L.
Stubbs
a,
Mathew J.
Bryant
a,
Lauren E.
Hatcher
 b and
Paul R.
Raithby
*a
b and
Paul R.
Raithby
*a
aDepartment of Chemistry, University of Bath, Bath, BA2 7AY, UK. E-mail: p.r.raithby@bath.ac.uk
bSchool of Chemistry, Cardiff University, Main Building, Park Place, Cardiff CF10 3AT, UK
First published on 27th October 2025
Abstract
Colour-changing chemical sensors have found important applications in the detection of low concentrations of volatile organic compounds (VOCs). Among the most promising molecular materials are platinum pincer complexes that display rapid colour changes when exposed to a variety of VOCs. In the solid state, these rapid responses have been linked to changes in non-covalent interactions between the platinum pincer molecules and the guest VOCs. To gain a better understanding of the interactions involved, we have studied the manipulation of vapochromic or solvatochromic properties in a series of square planar platinum(II) pincer complexes through structural modification of the monodentate ligand occupying the fourth coordination site. The platinum(II) complexes are based on the 1,3-di(pyridine)benzene tridentate linker (N^C^N) skeleton with the fourth site occupied by a monodentate, anionic ligand L. The formulae of the complexes synthesised are [Pt(N^C(C(O)OMe)^N)(L)] (L = (NCO) (6), (NCS) (7), (OC(O)Me) (8), (OC(O)CF3) (9), (OS(O)2CF3) (10), and (OS(O)2(C6H4Me)) (11)), and the crystalline solids and solutions of these materials have been tested for solvatochromic or vapochromic changes with VOCs including dichloromethane, acetonitrile, diethyl ether, methanol and water. The complexes 6 and 7 crystallised as yellow solids, with no solvent voids in the crystal lattice. Single-crystal X-ray analyses showed that the intermolecular Pt⋯Pt separations were too long for direct Pt⋯Pt interactions. Neither material displayed solvatochromic or vapochromic properties. However, when the fourth ligand was an acetate group, complex 8, the solid displayed vapochromism, changing colour from an orange water-containing crystalline form to an anhydrous yellow form, and a blue form when treated with methanol vapour. A crystal structure analysis showed that in the orange form adjacent pincer molecules were linked together through a hydrogen bonding network involving lattice water molecules, supported by π⋯π stacking interactions. Complexes 9 and 10 both showed solvatochromism, forming a bright yellow solid when crystallised from dichloromethane but forming an orange solid when recrystallised from acetonitrile. Complex 11 displayed both vapochromic and solvatochromic properties. This complex was isolated as a purple solid, turning yellow upon treatment with methanol droplets or vapour, but this colour change could be reversed upon addition of acetonitrile.
Introduction
The search for effective vapochromic or solvatochromic materials that can change colour, either reversibly or irreversibly, when exposed to gases or solvent vapours continues to grow because of relevance to the development of sensors to monitor changes in environmental conditions.1,2 A wide range of solid-state materials have been shown to have vapochromic properties.3 Solid–gas/vapour reactions are easier to control than liquid–gas/vapour reactions so the emphasis has been on the solid-state, and the current strategy for developing effective materials is based on the design of the crystalline solid with a view to improving the stability against degradation under vapochromic conditions, and broadening the environmental conditions under which they can operate. Generally, the crystalline materials should be soft, with high structural order, and have a flexible response to incoming guest molecules.4 Promising vapochromic materials include organic crystals,5–11 organometallic and coordination complexes,12–17 coordination polymers and metal organic frameworks (MOFs).18–24 Coordination polymers of Zn and Cd supported by naphthylvinylpyridine ligands undergo visible-light induced structural changes associated with colour change, to detect Pd(II) ions and explosive nitroaromatic compounds.25,26 Zr-based MOFs with pyrazolate ligands have proved to be selective and efficient in capturing formaldehyde vapour.27The two largest classes of crystalline materials that show vapochromic or solvatochromic properties are luminescent complexes of platinum(II)28–31 and gold(I)32–39 where the vapochromic behaviour is often linked to changes in the Pt⋯Pt or Au⋯Au intermolecular interactions during the absorption or loss of the guest vapour molecules, resulting in a colour change in the host material.
Platinum(II) complexes, with their four-coordinate, square planar coordination geometry and d8 electron configuration, are particularly suited to solid-state stacking involving intermolecular Pt⋯Pt interactions through dz2⋯dz2 orbital overlap and π⋯π stacking interactions through coordinated aromatic and heteroaromatic ligands. In the crystalline state, once the intermolecular Pt⋯Pt separation is below ca. 3.5 Å overlap of the occupied 5dz2 orbitals occurs, forming bonding dσ and antibonding dσ* orbitals. The destabilised dσ* orbital is the highest occupied molecular orbital (HOMO) within the assembly, whereas the π* orbital of the coordinated aromatic chelating ligand is the lowest unoccupied molecular orbital (LUMO).31 The assembly, with the Pt⋯Pt interactions, will display characteristic phosphorescence from the triplet metal–metal-to-ligand charge transfer (3MMLCT) that originates from the HOMO–LUMO transition. The energy of the dσ* is highly dependent on the Pt dz2⋯dz2 orbital overlap, which results in significant shifts in the position of the 3MMLCT transition and is responsible for the colour changes when the solid complex is exposed to vapours or solvents that alter the structure of the assembly.
The first report of the vapochromic behaviour of a one-dimensionally stacked platinum(II) complex appeared in 1995, when it was shown that Magnus' green salt, [Pt(p-CN(C6H4))(C10H21)4][Pd(CN)4], displayed a reversible colour change in the presence of a variety of volatile organic solvent (VOC) vapours.40 This was quickly followed up by the idea of using this class of crystalline materials as a chemical sensor for VOCs.41 Since then, a plethora of square planar platinum(II) complexes have shown vapochromic responses to a wide range of volatile solvent vapours and gases.42,43 Recent studies, have shown the versatility of soft Pt(II) salts, consisting of oppositely charged complexes, for showing multi-stimuli responses, through photoluminescence.44–46 Multi-stimuli responsive Pt(II) complexes have found applications in information storage and in anti-counterfeiting applications, where the change in luminescence is related to changes in the crystal packing arrangements and in changes in intermolecular Pt⋯Pt or π⋯π stacking interactions.47
Within the class of luminescent platinum(II) complexes, those supported by tridentate pincer ligands, such as 2,2′;6′,2′′-perpyridine (terpy) and its derivatives, have shown a range of vapochromic and solvatochromic responses.48–54 For terpy-based materials, due to the presence of the Pt(II) centre and the neutral tridentate ligand, even when an anionic ligand is bound in the fourth coordination site the complexes are cationic, requiring the presence of an anionic counterion to balance the charge. In the solid state, the presence of the counterion has an influence on the crystal packing and thus on the vapochromic behaviour of the compound. If the terpy-based pincer ligands are replaced by N^C^N dipyridylbenzene ligands that are capable of coordinating to the Pt(II) centre through cyclometallation, one of the positive charges on the Pt(II) centre is balanced, and the requirement for a counterion is removed.55 This provides a simplified design strategy for the development of Pt(II) vapochromic materials, eliminating the limitation of a counterion. These materials have shown intriguing photoluminescent and photoresponsive supramolecular properties56,57 including mechanochromism and piezochromism.58,59
It has been found that the properties of Pt(II) square planar pincer complexes are dependent on the structure of the pincer ligand and upon the nature of the ligand in the fourth coordination site.60,61 The complex [Pt(N^C(R)^N)(CN)] (1, R = C(O)OMe) exhibits rapidly reversible absorptive and emissive vapochromism when water or methanol is added to the solid (Fig. 1).62 In its anhydrous form, the complex is yellow and it converts reversibly on a subsecond timescale to a red hydrate form in the presence of water vapour. Exposure of the anhydrous form to methanol vapour results in a rapidly reversible colour change from yellow to blue, resulting from the formation of a methanol solvate. Spectroscopic and crystallographic studies showed that the colour changes are caused by the water and methanol molecules forming intermolecular hydrogen bonds with the cyanide ligand of the complex, altering the crystal packing and resulting in changes in the Pt⋯Pt stacking distance within the solid. The presence of methyl ester substituent on the pincer ligand reduces the packing density compared to that in similar unsubstituted cyclometallated complexes,63 which assists in the diffusion of solvent molecules through the solid. Replacement of the methyl ester group in complex 1 by different-sized substituents leads to the formation of the complexes [Pt(N^C(R)^N)(CN)] (2, R = C(O)Me; 3, R = C(O)OEt; 4, R = C(O)OPh) (Fig. 1).64
 | ||
| Fig. 1 Diagram of the structure of the [Pt(N^C(R)^N)(CN)] complexes showing the position of the substituents on the tridentate pincer ligand and the position of the cyanide ligand.64 | ||
The vapochromic or solvatochromic properties of the complexes 2–4 differ from those of 1. The presence of the smaller acyl group in 2 results in a hydrated, green form at ambient conditions, which changes to yellow upon heating above 140 °C. The colour change is irreversible and is associated with the loss of water from the hydrated green form. There is no evidence of other vapochromic or solvatochromic behaviour in this system. However, both the complexes with the larger ethyl (3) and phenyl (4) ester substituents exhibit complex, rapidly reversible vapochromic or solvatochromic behaviour with a variety of solvents and display some solvent selectivity. This suggests that a larger spacer group R attached at the para-position of the cyclometallated ring of the pincer ligand is required to facilitate vapochromic or solvatochromic behaviour in the solid state. For these complexes, the presence of the strong-field cyanide ligand acts as a hydrogen-bonding site for solvent binding upon uptake. The presence of the cyanide also alters the orbital structure of the complex, helping to stabilise HOMOs with which it shares π-orbital interactions, thus affording a prominent non-bonding dz2 orbital above the frontier orbitals to facilitate intermolecular Pt⋯Pt interactions.62
In order to further probe the properties of the molecular materials, we now report an investigation exploring the effect that changing the ligand at the fourth coordination site from cyanide to other monoanionic ligands, with varying ligand field strengths and hydrogen bonding acceptor properties, while retaining the platinum methyl-ester substituted pincer ligand as the core of the molecule, that has shown the greatest variety of rapidly reversible vapochromic and solvatochromic effects in the series of complexes studied.62,64
Experimental
The reactions were all carried out under an atmosphere of dry nitrogen using standard Schlenk line techniques. The solvents used were dried using an automated solvent purification system or through distillation over an appropriate drying agent and were stored over molecular sieves prior to use. The starting materials were purchased from commercial sources and used without further purification unless otherwise stated. Methyl-3,5-di(pyridin-2-yl)benzoate was prepared by a Negishi reaction,65 and methyl-3,5-di(pyridin-2-yl)benzolate platinum(II) chloride (5) was prepared in 98% yield using the published literature method.55 Their formulations were confirmed by 1H NMR spectroscopy (see SI).IR spectra were recorded on a Perkin Elmer Spectrum One spectrometer using neat solids. 1H and 19F NMR spectra were recorded on either a Bruker Advance 400 MHz or 500 MHz instrument. Chemical shifts (expressed in parts per million) were referenced to residual solvent peaks. UV-visible absorbance spectra were measured in solution using a Perkin Elmer Lambda 650 UV/vis spectrometer. Photoluminescence spectra were recorded on a Perkin Elmer LS55 luminescence spectrometer.
Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 solvent mixture of MeCN:CH2Cl2. The mixture was heated to reflux at 85 °C for 48 h under an inert atmosphere. The solution was cooled, and the solvent was removed. 50 ml of CH2Cl2 was added to the residue and then filtered to remove the AgCl. The CH2Cl2 was then removed, leaving behind a yellow solid. Crystals were grown from the slow evaporation of a mixture of solvents. Yield: 0.084 g/83% FTIR (cm−1) ν 2222 (s, br), 1698 (s), 1592 (s), 1458 (s), 1325 (s), 871 (m). 1H NMR (500 MHz, chloroform-d): δ 9.04–8.83 (2 H, m), 8.12–7.97 (4 H, m), 7.81 (2 H, ddd, 3JH–H = 8.1, 0.8 Hz), 7.37 (2 H, ddd, 3JH–H = 7.3, 1.4 Hz), 3.96 (3 H, s). Calculated CHN (%): C 43.35, H 2.49, N 7.98. Found CHN (%): C 43.19, H 2.76, N 6.89.
:1 solvent mixture of MeCN:CH2Cl2. The mixture was heated to reflux at 85 °C for 48 h under an inert atmosphere. The solution was cooled, and the solvent was removed. 50 ml of CH2Cl2 was added to the residue and then filtered to remove the AgCl. The CH2Cl2 was then removed, leaving behind a yellow solid. Crystals were grown from the slow diffusion of diethyl ether into a concentrated acetonitrile solution at −18 °C. Yield: 0.092 g/92%. FTIR ν(cm−1): 2921 (s), 2861 (s), 2080 (s), 1698 (s), 1476 (s), 1236 (s). 1H NMR (500 MHz, chloroform-d): δ 9.10 (s, 2H), 7.96–7.81 (m, 4H), 7.69–7.41 (m, 2H), 7.29–7.05 (m, 2H), 3.92–3.71 (m, 3H). Calculated CHN (%): C 42.07, C 2.42, N 7.75. Found CHN (%): C 41.96, H 2.18, N 7.49.
:1 solvent mixture of MeCN:CH2Cl2. The mixture was heated to reflux at 85 °C for 24 h. The solution was cooled, and the solvent was removed. 50 ml of CH2Cl2 was added to the residue and then filtered to remove the AgCl. The CH2Cl2 was then removed. Crystals were grown from the vapour diffusion of di-ethyl ether into a CH2Cl2 solution at −18 °C and from the slow evaporation of a water and acetone mix. Yield: 0.052 g/52%. FTIR ν(cm−1): 3448 (w, br), 2166 (m), 2033 (m), 1693 (s), 1605 (s), 1587 (s), 1239 (s). 1H NMR (500 MHz, chloroform-d): δ 9.41–9.26 (m, 2H), 8.09 (s, 2H), 7.99 (td, 3JH–H = 7.8, 1.6 Hz, 2H), 7.78 (ddd, 3JH–H = 7.9, 0.7 Hz, 2H), 7.34 (ddd, 3JH–H = 7.3, 1.4 Hz, 2H), 3.95 (s, 3H), 2.02 (d, 3JH–H = 1.0 Hz, 3H). Calculated for 8. 3.5 (H2O) CHN (%): C 39.61, H 3.82, N 4.62. Found CHN (%): C 39.24, H 3.11, N 4.68.
:1 solvent mixture of MeCN:CH2Cl2. The mixture was heated to reflux at 85 °C for 48 h under an inert atmosphere. The solution was cooled, and the solvent was removed. 50 ml of CH2Cl2 was added to the residue and then filtered to remove the AgCl. The CH2Cl2 was then removed, leaving behind an orange solid. Yield: 0.062 g/62%. FTIR ν(cm−1): 1714 (s), 1687 (s), 1416 (s), 1182 (s), 1115 (s), 839 (s). 1H NMR (500 MHz, chloroform-d): δ 8.71 (dt, 3JH–H = 5.7, 1.0 Hz, 2H), 8.04–7.89 (m, 4H), 7.72 (ddd, 3JH–H = 7.9, 1.4, 0.8 Hz, 2H), 7.30 (ddd, 3JH–H = 7.3, 1.5 Hz, 2H), 4.27 (s, 3H). 19F NMR (470 MHz, chloroform-d): δ −74.58, −74.74–−75.20 (m). Calculated CHN (%): C 40.21, H 2.19, N 4.69. Measured CHN (%): C 39.52, H 2.53, N 4.02.
:1 solvent mixture of MeCN:CH2Cl2. The mixture was heated to reflux at 85 °C for 48 h under an inert atmosphere. The solution was cooled, and the solvent was removed. 50 ml of CH2Cl2 was added to the residue and then filtered to remove the AgCl. The CH2Cl2 was then removed leaving behind an orange solid. Yield: 0.039 g/39% FTIR ν(cm−1): 3453 (m, br), 1720 (s), 1610 (s), 1239 (s), 1023 (s). 1H NMR (500 MHz, acetonitrile-d3): δ 8.15 (d, 3JH–H = 5.5 Hz, 2H), 7.94 (dd, 3JH–H = 8.6, 7.0 Hz, 2H), 7.62–7.54 (m, 4H), 7.25–7.19 (m, 2H), 3.89 (s, 3H). 19F NMR (470 MHz, acetonitrile-d3) δ −79.33. Calculated CHN (%): C 36.03, H 2.07, N 4.42. Measured CHN (%): C 36.12, H 2.21, N 4.62.
:1 solvent mixture of MeCN:CH2Cl2. The mixture was heated to reflux at 85 °C for 48 h under an inert atmosphere. The solution was cooled, and the solvent was removed. 50 ml of CH2Cl2 was added to the residue and then filtered to remove the AgCl. The DCM was then removed, leaving behind a purple solid. Yield: 0.032 g/32% FTIR ν(cm−1): 1705 (s), 1607 (s), 1346 (s), 1248 (s), 1114(s). 1H NMR (500 MHz, chloroform-d): δ 9.45–9.33 (m, 2H), 8.18–8.07 (m, 3H), 8.01 (td, 3JH–H = 7.8, 1.6 Hz, 2H), 7.81 (t, 3JH–H = 7.6 Hz, 3H), 7.44–7.33 (m, 3H), 7.12 (d, 3JH–H = 7.8 Hz, 1H), 3.98 (d, 3JH–H = 8.5 Hz, 3H), 3.64 (d, 3JH–H = 4.9 Hz, 3H). Calculated CHN (%): C 45.80, H 3.08, N 4.27. Measured CHN (%): C 45.01, H 3.73, N 4.42.
:1 solvent mixture of MeCN:CH2Cl2. The mixture was heated to reflux at 85 °C for 48 h under an inert atmosphere. The solution was cooled, and the solvent was removed. 50 ml of CH2Cl2 was added to the residue and then filtered to remove the AgCl. The CH2Cl2 was then removed, leaving behind a yellow solid. Crystals were grown from the slow evaporation of a mixture of solvents. Yield: 0.084 g/83% FTIR (cm−1) ν 2222 (s, br), 1698 (s), 1592 (s), 1458 (s), 1325 (s), 871 (m). 1H NMR (500 MHz, chloroform-d): δ 9.04–8.83 (2 H, m), 8.12–7.97 (4 H, m), 7.81 (2 H, ddd, 3JH–H = 8.1, 0.8 Hz), 7.37 (2 H, ddd, 3JH–H = 7.3, 1.4 Hz), 3.96 (3 H, s). Calculated CHN (%): C 43.35, H 2.49, N 7.98. Found CHN (%): C 43.19, H 2.76, N 6.89.
:1 solvent mixture of MeCN:CH2Cl2. The mixture was heated to reflux at 85 °C for 48 h under an inert atmosphere. The solution was cooled, and the solvent was removed. 50 ml of CH2Cl2 was added to the residue and then filtered to remove the AgCl. The CH2Cl2 was then removed, leaving behind a yellow solid. Crystals were grown from the slow diffusion of diethyl ether into a concentrated acetonitrile solution at −18 °C. Yield: 0.092 g/92%. FTIR ν(cm−1): 2921 (s), 2861 (s), 2080 (s), 1698 (s), 1476 (s), 1236 (s). 1H NMR (500 MHz, chloroform-d): δ 9.10 (s, 2H), 7.96–7.81 (m, 4H), 7.69–7.41 (m, 2H), 7.29–7.05 (m, 2H), 3.92–3.71 (m, 3H). Calculated CHN (%): C 42.07, C 2.42, N 7.75. Found CHN (%): C 41.96, H 2.18, N 7.49.
:1 solvent mixture of MeCN:CH2Cl2. The mixture was heated to reflux at 85 °C for 24 h. The solution was cooled, and the solvent was removed. 50 ml of CH2Cl2 was added to the residue and then filtered to remove the AgCl. The CH2Cl2 was then removed. Crystals were grown from the vapour diffusion of di-ethyl ether into a CH2Cl2 solution at −18 °C and from the slow evaporation of a water and acetone mix. Yield: 0.052 g/52%. FTIR ν(cm−1): 3448 (w, br), 2166 (m), 2033 (m), 1693 (s), 1605 (s), 1587 (s), 1239 (s). 1H NMR (500 MHz, chloroform-d): δ 9.41–9.26 (m, 2H), 8.09 (s, 2H), 7.99 (td, 3JH–H = 7.8, 1.6 Hz, 2H), 7.78 (ddd, 3JH–H = 7.9, 0.7 Hz, 2H), 7.34 (ddd, 3JH–H = 7.3, 1.4 Hz, 2H), 3.95 (s, 3H), 2.02 (d, 3JH–H = 1.0 Hz, 3H). Calculated for 8. 3.5 (H2O) CHN (%): C 39.61, H 3.82, N 4.62. Found CHN (%): C 39.24, H 3.11, N 4.68.
:1 solvent mixture of MeCN:CH2Cl2. The mixture was heated to reflux at 85 °C for 48 h under an inert atmosphere. The solution was cooled, and the solvent was removed. 50 ml of CH2Cl2 was added to the residue and then filtered to remove the AgCl. The CH2Cl2 was then removed, leaving behind an orange solid. Yield: 0.062 g/62%. FTIR ν(cm−1): 1714 (s), 1687 (s), 1416 (s), 1182 (s), 1115 (s), 839 (s). 1H NMR (500 MHz, chloroform-d): δ 8.71 (dt, 3JH–H = 5.7, 1.0 Hz, 2H), 8.04–7.89 (m, 4H), 7.72 (ddd, 3JH–H = 7.9, 1.4, 0.8 Hz, 2H), 7.30 (ddd, 3JH–H = 7.3, 1.5 Hz, 2H), 4.27 (s, 3H). 19F NMR (470 MHz, chloroform-d): δ −74.58, −74.74–−75.20 (m). Calculated CHN (%): C 40.21, H 2.19, N 4.69. Measured CHN (%): C 39.52, H 2.53, N 4.02.
:1 solvent mixture of MeCN:CH2Cl2. The mixture was heated to reflux at 85 °C for 48 h under an inert atmosphere. The solution was cooled, and the solvent was removed. 50 ml of CH2Cl2 was added to the residue and then filtered to remove the AgCl. The CH2Cl2 was then removed leaving behind an orange solid. Yield: 0.039 g/39% FTIR ν(cm−1): 3453 (m, br), 1720 (s), 1610 (s), 1239 (s), 1023 (s). 1H NMR (500 MHz, acetonitrile-d3): δ 8.15 (d, 3JH–H = 5.5 Hz, 2H), 7.94 (dd, 3JH–H = 8.6, 7.0 Hz, 2H), 7.62–7.54 (m, 4H), 7.25–7.19 (m, 2H), 3.89 (s, 3H). 19F NMR (470 MHz, acetonitrile-d3) δ −79.33. Calculated CHN (%): C 36.03, H 2.07, N 4.42. Measured CHN (%): C 36.12, H 2.21, N 4.62.
:1 solvent mixture of MeCN:CH2Cl2. The mixture was heated to reflux at 85 °C for 48 h under an inert atmosphere. The solution was cooled, and the solvent was removed. 50 ml of CH2Cl2 was added to the residue and then filtered to remove the AgCl. The DCM was then removed, leaving behind a purple solid. Yield: 0.032 g/32% FTIR ν(cm−1): 1705 (s), 1607 (s), 1346 (s), 1248 (s), 1114(s). 1H NMR (500 MHz, chloroform-d): δ 9.45–9.33 (m, 2H), 8.18–8.07 (m, 3H), 8.01 (td, 3JH–H = 7.8, 1.6 Hz, 2H), 7.81 (t, 3JH–H = 7.6 Hz, 3H), 7.44–7.33 (m, 3H), 7.12 (d, 3JH–H = 7.8 Hz, 1H), 3.98 (d, 3JH–H = 8.5 Hz, 3H), 3.64 (d, 3JH–H = 4.9 Hz, 3H). Calculated CHN (%): C 45.80, H 3.08, N 4.27. Measured CHN (%): C 45.01, H 3.73, N 4.42.
X-ray crystallography
Results and discussion
Synthesis and characterisation of methyl-3,5-di(pyridin-2-yl)benzoate platinum cyanate (6) and methyl-3,5-di(pyridin-2-yl)benzoate platinum thiocyanate (7)
Previous studies have shown that the complex [Pt(N^C(C(O)OMe)^N)(Cl)] (5), with the π-donating chloride ligand in the fourth coordination site, does not display vapochromism or solvatochromism when the solid is exposed to a range of solvents and vapours.70 In contrast, the related complexes [Pt(N^C(R)^N)(CN)] (1, R = C(O)OMe) where the fourth coordination site is occupied by the strongly π-accepting cyanide ligand, do show extensive vapochromism or solvatochromism.62,64 Our recent work has indicated that a bulky R substituent in the para-position of the cyclometallated ring of the pincer ligand is required for vapochromism to occur.64 From our earlier work we hypothesise that the cyanide ligand must also play an important role in the vapochromic process due to the strong ligand field effect that it introduces, both by facilitating the Pt⋯Pt interactions in the solid state and also through its ability to hydrogen bond to solvents and vapours. In order to probe these factors we first prepared the complexes [Pt(N^C(C(O)OMe)^N)(NCO)] (6) and [Pt(N^C(C(O)OMe)^N)(NCS)] (7) where the cyanate and thiocyanate ligands are intermediate between the chloride and cyanide ligands in the spectrochemical series, that runs in the order Cl < NCO < NCS < CN of increasing ligand field strength.71 Both these ligands also have the capability to become involved in hydrogen bonding through the terminal oxygen and sulphur atoms, respectively.The complexes 6 and 7 were prepared from the chloride complex 5 by a simple ligand exchange reaction (Fig. 2). Complex 5 was treated with the silver salt [AgL] (L = NCO; NCS) in a 50:50 MeCN:CH2Cl2 mixture, and heated to 85 °C for 48 h. Both complexes were isolated as yellow solids and recrystallised from the solvent mixture. They were initially characterised by IR spectroscopy and 1H NMR spectroscopy. The IR spectrum of 6 displays strong bands at ν 2222 (s, br) and 1698 (s) cm−1 consistent with the stretching ν(N–C–O) and bending ν(N–C–O) modes of the coordinated cyanate ligand, and the IR spectrum of 7 shows band at 2080 (s) cm−1 consistent with the ν(N–C–S) stretching mode of the thiocyanate ligand.72
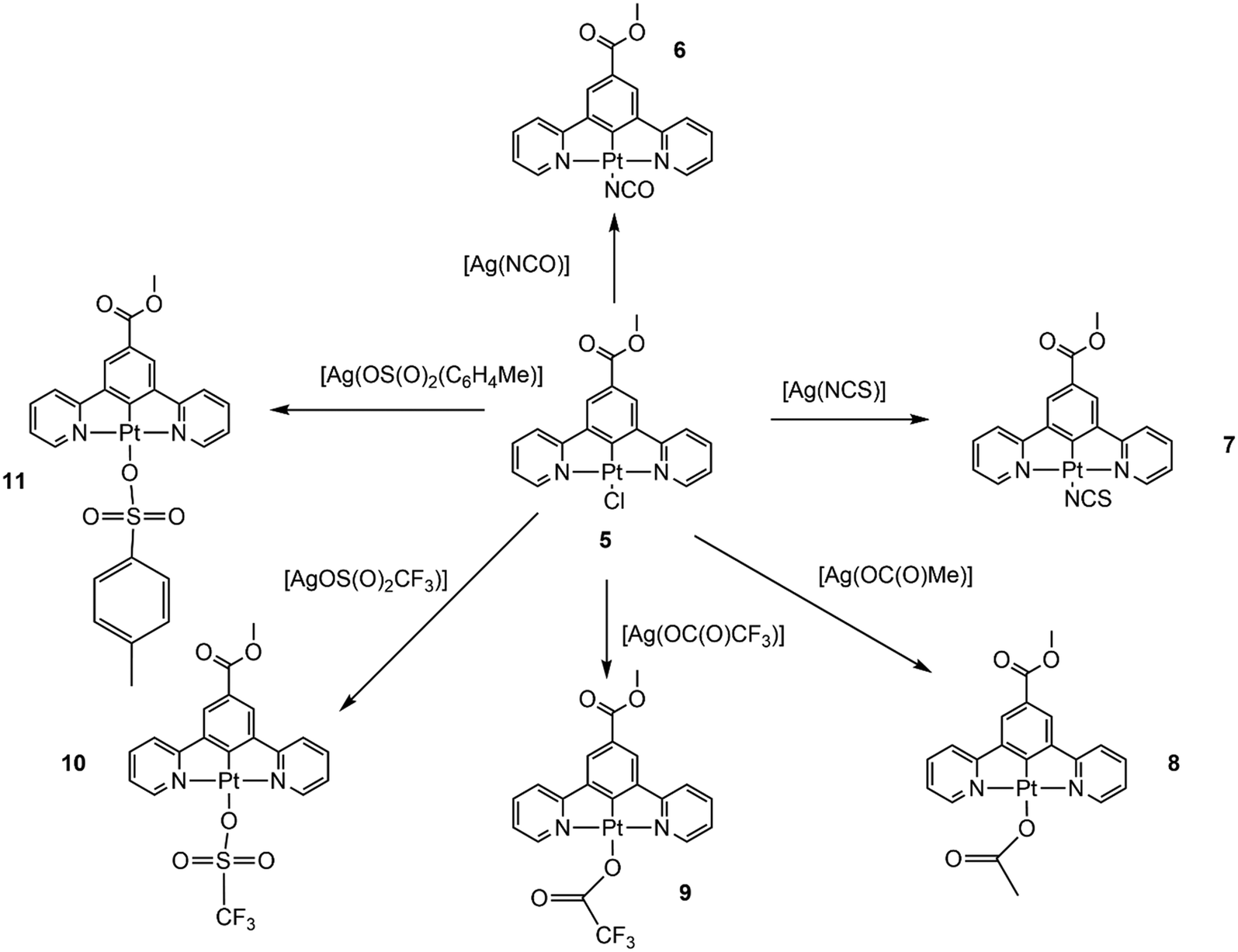 | ||
| Fig. 2 The formation of the pincer complexes with monodentate anionic ligands in the fourth coordination site. In each reaction complex 5 and the silver salt of the anion were heated to 85 °C for 48 h in a 1:1 solvent mixture of MeCN:CH2Cl2. | ||
The solution state UV/vis absorption and emission spectra of 6 and 7 measured at a concentration of 1 × 10−5 M in CH2Cl2 were very similar to those previously reported for 5 and are shown in Fig. S1 and S3 (SI). At higher concentrations, 1 × 10−3 M in CH2Cl2, λex 400 nm, the emission spectrum of 6 displays a broad peak between 600–750 nm, in addition to the pronounced vibrational structure of the 3π–π* transition (Fig. S2). This broadband is indicative of excimer formation, which can occur in high concentrations of Pt(II) complexes due to self-quenching or aggregation between the molecules.70 At higher concentrations, 7 also shows excimer formation (Fig. S4), but the effect is not as pronounced as with 6.
Crystal structure determinations of 6 and 7
The yellow complex 6 crystallises in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (No. 2), with one molecule of the complex in the asymmetric unit and no solvent of crystallisation present. The molecular structure is shown in Fig. 3 and selected bond parameters are presented in Table 1. The molecule displays the expected square planar geometry at the Pt(II) centre. There is a slight distortion with the N(1)–Pt(1)–N(2) angle of 161.15(17)° being attributed to the bite angle of the pincer ligand.62 The cyanate ligand is essentially linear with the N(3)–C(19)–O(3) angle of 178.9(6)°, and the multiple bond character in the N(3)–C(19) and C(19)–O(3) is indicated by the bond lengths of 1.154(6) and 1.207(7) Å, respectively. Overall, the molecule is approximately planar with torsion angles close to 0° and the methyl ester group lies parallel to the pincer ligand.
(No. 2), with one molecule of the complex in the asymmetric unit and no solvent of crystallisation present. The molecular structure is shown in Fig. 3 and selected bond parameters are presented in Table 1. The molecule displays the expected square planar geometry at the Pt(II) centre. There is a slight distortion with the N(1)–Pt(1)–N(2) angle of 161.15(17)° being attributed to the bite angle of the pincer ligand.62 The cyanate ligand is essentially linear with the N(3)–C(19)–O(3) angle of 178.9(6)°, and the multiple bond character in the N(3)–C(19) and C(19)–O(3) is indicated by the bond lengths of 1.154(6) and 1.207(7) Å, respectively. Overall, the molecule is approximately planar with torsion angles close to 0° and the methyl ester group lies parallel to the pincer ligand.
 | ||
| Fig. 3 The molecular structure of 6 showing the atom numbering scheme. The displacement ellipsoids are drawn at 50%. | ||
| a. Bond lengths | ||
|---|---|---|
| Atom | Atom | Length/Å |
| Pt1 | N1 | 2.033(4) |
| Pt1 | N2 | 2.033(4) |
| Pt1 | N3 | 2.091(4) |
| Pt1 | C6 | 1.908(5) |
| N3 | C19 | 1.154(6) |
| O3 | C19 | 1.207(7) |
| b. Bond angles | |||
|---|---|---|---|
| Atom | Atom | Atom | Angle/° |
| N1 | Pt1 | N2 | 161.15(17) |
| N1 | Pt1 | N3 | 98.76(17) |
| N2 | Pt1 | N3 | 100.10(16) |
| C6 | Pt1 | N1 | 80.57(18) |
| C19 | N3 | Pt1 | 177.1(4) |
| C6 | Pt1 | N3 | 178.78(15) |
| N3 | C19 | O3 | 178.9(6) |
| c. Torsion angles | ||||
|---|---|---|---|---|
| A | B | C | D | Angle/° |
| N3 | Pt1 | N2 | C16 | −3.6(5) |
| N3 | Pt1 | N1 | C1 | −0.7(5) |
| Pt1 | N1 | C5 | C7 | −0.2(5) |
| Pt1 | N2 | C12 | C11 | 1.2(6) |
| Pt1 | C6 | C7 | C5 | −0.8(6) |
| Pt1 | C6 | C11 | C12 | −2.2(6) |
| C6 | Pt1 | N3 | C19 | 9(16) |
| Pt1 | N3 | C19 | O3 | −111(34) |
In the crystal lattice the molecules of 6 pack in a head-to-tail fashion (Fig. S5) with a Pt⋯Pt intermolecular separation of 5.545(1) Å, showing that there is no Pt⋯Pt interaction present. This Pt⋯Pt separation is intermediate between the value of 3.663(1) Å found in the yellow form of [Pt(N^C(C(O)OMe))^N)(CN)] (1)62 and the 7.404(1) Å in the chloride complex [Pt(N^C(C(O)OMe))^N)(Cl)] (5)55 both of which crystallise in the monoclinic space group P21/c (or (P21/n). Interestingly, the Kitaigorodskii packing indices (KPIs) for the three molecules are similar, with values of 71.9% (for 6), 73.8% (for 1) and 71.3% (for 5).73 The crystal structure of 6 does display π⋯π stacking interactions between symmetry related pyridine rings on adjacent molecules with centroid⋯centroid distances of 3.677 Å (between ring N2C12C13C14C15C16 and the same ring related by the symmetry operator 1 − x, 1 − y, z) and 3.883 Å (between ring N2C12C13C14C15C16 and ring N1C1C2C3C4C5 related by the symmetry operator 2 − x, 1 − y, 1 − z). There are also relatively long and non-linear C–H⋯O hydrogen bond interactions between the cyanate oxygen atom and several of the pyridine hydrogen atoms (Table S8, SI).
Complex 7 crystallises in the monoclinic space group P21/c, with one molecule in the asymmetric unit and with no solvent molecules present. The molecular structure is shown in Fig. 4, and selected bond parameters are presented in Table 2. As with 6, complex 7 displays the expected square planar configuration with the N-bound thiocyanate ligand occupying the fourth coordination site. The thiocyanate ligand is linear with an N(3)–C(19)–S(1) angle of 179.5(7)°, and N(3)–C(19) and C(19)–S(1) bond lengths of 1.139(8) and 1.637(7) Å, respectively. The remainder of the bond parameters within 7 are similar to those found in 6.
 | ||
| Fig. 4 The molecular structure of complex 7 showing the atom numbering scheme. Displacement ellipsoids are drawn at the 50% probability level. | ||
| a. Bond lengths | ||
|---|---|---|
| Atom | Atom | Length/Å |
| Pt1 | C6 | 1.909(7) |
| Pt1 | N2 | 2.036(6) |
| Pt1 | N1 | 2.034(6) |
| Pt1 | N3 | 2.097(6) |
| S1 | C19 | 1.637(7) |
| C19 | N3 | 1.139(8) |
| b. Bond angles | |||
|---|---|---|---|
| Atom | Atom | Atom | Angle/° |
| N1 | Pt1 | N2 | 161.0(2) |
| N1 | Pt1 | N3 | 99.0(2) |
| C6 | Pt1 | N1 | 80.3(3) |
| C6 | Pt1 | N3 | 178.0(3) |
| C19 | N3 | Pt1 | 175.8(7) |
| N3 | C19 | S1 | 179.5(7) |
| c. Torsion angles | ||||
|---|---|---|---|---|
| A | B | C | D | Angle/° |
| N2 | Pt1 | N3 | C19 | −157(11) |
| N1 | Pt1 | N3 | C19 | 21(11) |
As with complex 6, complex 7 displays two significant π–π interactions between the pyridyl rings and the central phenyl ring of the pincer ligand in adjacent layers of the crystal lattice. The molecules stack in a head-to-tail manner, similar to 6, with centroid⋯centroid distances of 3.913(2) Å between the ring (C6C7C8C9C10C11) and the ring (N2C12C13C14C15C16) related by the symmetry operator 1 − x, 1 − y, 1 − z and 3.659(1) Å between the ring (N1C1C2C3C4C5C6) and the ring (N2C12C13C14C15C16) related by the symmetry operator 2 − x, 1 − y, 1 − z. There are three intermolecular hydrogen bonds involving the thiophene S(1) atom and the carboxylate oxygen atoms and aromatic C–H hydrogen atoms on one of the pyridyl rings (Table S16, SI). Within the crystal packing, the shortest intermolecular Pt⋯Pt distance is 4.948(1) Å, again beyond the distance expected for any Pt⋯Pt interactions and the KPI index for this molecule is 73.9%, similar to that found in complex 1.
Vapochromic/solvatochromic tests on complexes 6 and 7
The two complexes formed yellow crystals and crystallised without solvent present in the crystal lattice. Examination of the crystal structures showed no solvent accessible void space. By analogy with the studies on complex 5,55 the colour and the crystal packing suggested that it was unlikely that the two complexes would exhibit vapochromic or solvatochromic properties. However, the complexes were tested in the solution state by dissolving in a variety of solvents and in the solid state by exposure to a series of vapours; however, no change was observed in the UV-visible spectra, and it was concluded that the two complexes did not exhibit vapochromism/solvatochromism under the conditions used. These results are consistent with the hypothesis that the ligand field strength of the cyanate and thiocyanate ligands is not sufficient to induce Pt⋯Pt intermolecular interactions, as is the case in the cyanide complex 1.The synthesis and characterisation of (methyl-3,5-dipyridyn-2-yl)benzoate platinum(II) acetate (8), (methyl-3,5-dipyridyn-2-yl)benzoate platinum(II) trifluoroacetate (9), (methyl-3,5-dipyridyn-2-yl)benzoate platinum(II) triflate (10) and (methyl-3,5-dipyridyn-2-yl)benzoate platinum(II) tosylate (11)
In the next step of the investigation, the linear ligand in the fourth coordination site was replaced by a series of bulkier groups, all of which could act as hydrogen bond acceptors. The four molecules prepared were the acetate (8), trifluoroacetate (9), triflate (10) and tosylate (11) derivatives (Fig. 2). As with 6 and 7, the new neutral Pt(II) complexes were obtained from the chloride complex 5 by a simple ligand exchange reaction. The complexes were initially characterised in solution by IR and multinuclear NMR spectroscopy.Complex 8_water was isolated as an orange/beige solid in good yield by recrystallisation from a water/acetone mixture. The IR spectrum confirmed the presence of organic carbonyl groups with ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) stretching frequencies at 1693(s) and 1605(s) cm−1. The solution state UV-visible absorption and emission spectra are similar to those observed for 5 (Fig. S6) since the majority of the transitions observed in solution originate from the pincer ligand and not from the ligand in the fourth coordination site.
O) stretching frequencies at 1693(s) and 1605(s) cm−1. The solution state UV-visible absorption and emission spectra are similar to those observed for 5 (Fig. S6) since the majority of the transitions observed in solution originate from the pincer ligand and not from the ligand in the fourth coordination site.
Recrystallisation of the orange form of the crystals with dry dichloromethane resulted in a colour change to pale yellow. Employing vapochromic techniques, the same colour change was observed when a thin film of the orange complex was exposed to dichloromethane vapour or a stream of dry nitrogen gas. Similarly, exposure of the orange thin film to methanol vapour turned the film blue. The colours of the thin films are shown in Fig. 5, while Fig. 6 shows the solid-state UV-vis absorption spectra with three distinct profiles for the forms of the complex.
 | ||
| Fig. 5 Thin films of the acetate derivative complex 8 in the presence of water (orange), when dried (yellow) and when exposed to methanol vapour (blue). | ||
 | ||
| Fig. 6 The solid-state UV-vis absorption spectra for the water (orange), the dry (yellow) and the methanol (blue) forms of complex 8. | ||
Crystal structure determinations of the orange form of complex 8_water
Single crystal X-ray structure determinations have previously been helpful in understanding the nature of the processes that occur during solid-state vapochromic transformations.62 A single crystal of the orange form was obtained by slow evaporation of a solution of 8 in water/acetone mixture. The asymmetric unit of the triclinic space group, P (No. 2), contains two independent molecules of the square planar Pt(II) complex linked together by a network of five hydrogen-bonded water molecules. These water molecules sit in channels between stacks of Pt(II) complexes in the solid state. The network and the orientation of the two Pt(II) molecules are shown in Fig. 7.
 | ||
| Fig. 7 The crystal structure of orange, hydrated form of complex 8, showing the two independent molecules of the complex and the five water molecules. Displacement ellipsoids are drawn at the 50% probability level. | ||
As with the previous complexes in the series, the two Pt(II) centres adopt the expected square planar coordination geometry. The molecules, as a whole, are essentially planar except for the acetyl groups that occupy the fourth coordination site on the two Pt(II) centres. The dihedral angle between the central Pt(1)N(1)N(2)C(6)O(3) plane and the plane of the acetyl group O(3)O(4)C(19)C(20) is 75.3(3)° while that for the plane Pt(2)N(3)N(4)C(26)O(7) and the acetyl plane O(7)O(8)C(39)C(40) is 122.5(3)°. The bond parameters within the molecules (Table 3) are similar to those reported for complexes 6 and 7, and related platinum pincer complexes.64
| a. Bond lengths | ||
|---|---|---|
| Atom | Atom | Length/Å |
| C6 | Pt1 | 1.894(6) |
| C26 | Pt2 | 1.909(6) |
| N1 | Pt1 | 2.033(5) |
| N3 | Pt2 | 2.021(6) |
| O3 | Pt1 | 2.139(5) |
| O7 | Pt2 | 2.154(4) |
| C19 | O4 | 1.254(8) |
| C2 | O1 | 1.215(8) |
| C22 | O5 | 1.212(8) |
| C39 | O8 | 1.233(8) |
| b. Bond angles | |||
|---|---|---|---|
| Atom | Atom | Atom | Angle/° |
| N2 | Pt1 | N1 | 162.3(2) |
| C6 | Pt1 | N1 | 81.6(2) |
| C6 | Pt1 | O3 | 174.0(2) |
| C19 | O3 | Pt1 | 123.0(5) |
| N3 | Pt2 | N4 | 162.0(2) |
| C26 | Pt2 | N3 | 81.5(2) |
| C26 | Pt2 | O7 | 174.8(2) |
| C39 | O7 | Pt2 | 123.2(4) |
| c. Torsion angles | ||||
|---|---|---|---|---|
| A | B | C | D | Angle/° |
| Pt2 | O7 | C39 | O8 | −22(1) |
| Pt1 | O3 | C19 | O4 | −3(1) |
An analysis of the intermolecular interactions shows that the Pt⋯Pt distance between the two Pt centres in the independent adjacent molecules is 5.588(1) Å which is significantly beyond the limit expected for any direct Pt⋯Pt interactions, and substantially longer than the Pt⋯Pt distances of 3.319(1) and 3.341(1) Å found in the red form of [Pt(N^C(R)^N)(CN)] (1, R = C(O)OMe) where water molecules link adjacent Pt(II) complexes together.62 However, there are significant π⋯π stacking interactions between the central arene rings of the two independent molecules and pyridyl rings on the adjacent molecules (Table 4). With the water molecules present in the crystal lattice, there are no solvent voids, and the KPI value is 71.7%. However, if the water molecules are removed from the structural model, channels along the a-axis are apparent in the lattice with a void space of 14.7% (286.93 Å3) of the unit cell volume (Fig. 8a),74 and the KPI value drops to 63.5%.
| Plane 1a | Plane 2a | Symmetry relation | Interplanar angle (°) | Centroid⋯centroid distance (Å) | Shift distance (Å) |
|---|---|---|---|---|---|
| a #1 C3 C4 C5 C6 C7 C8; #2 N1 C9 C10 C11 C12 C13; #3 N2 C14 C15 C16 C17 C18; #4 C24 C25 C26 C27 C28; #5 N3 C23 C24 C25 C26 C27 C28 C29; #6 N4 C34 C35 C36 C37 C38. | |||||
| #1 | #4 | −1 + x, y, z | 1.965 | 3.774 | 1.475 |
| #1 | #4 | x, y, z | 1.965 | 3.675 | 1.494 |
| #3 | #6 | −1 + x, y, z | 2.068 | 3.585 | 1.344 |
| #3 | #6 | x, y, z | 2.068 | 3.522 | 0.773 |
 | ||
| Fig. 8 (a) Packing diagram of complex 8 viewed down the a-axis (with the water molecules removed) showing the void space. (b) Stacks of the molecules of complex 8 showing the channels filled with water molecules and the hydrogen bonding network (red and light blue dotted lines) linking alternate molecules together. | ||
What is of significance is the hydrogen bonding network in the crystal that links the stacks of molecules together and supports the π⋯π stacking interactions as shown in Fig. 8(b). Water molecules are hydrogen bonded to each other and to the oxygen atoms of the acetyl groups: O(3), O(4), O(7) and O(8). These hydrogen bonds are listed in Table 5 and the water–acetate interaction network is illustrated in Fig. 7.
| Donor | H | Acceptor | Symm opa | D–H (Å) | H⋯A (Å) | D⋯A (Å) | Angle |
|---|---|---|---|---|---|---|---|
| a Symmetry operator codes: [1555] = x, y, z; [1655] = −1 + x, y, z; [2665] = 1 − x, 1 − y, −z; [2765] = 2 − x, 1 − y, −z. | |||||||
| O9 | –H9A | ⋯O4 | [1555] | 0.75(7) | 2.14(8) | 2.866(8) | 166(8) |
| O9 | –H9B | ⋯O3 | [1655] | 1.24(11) | 1.66(11) | 2.892(7) | 174(10) |
| O10 | –H10A | ⋯O9 | [1555] | 0.79(7) | 1.96(7) | 2.736(8) | 172(8) |
| O10 | –H10B | ⋯O13 | [1655] | 0.93(8) | 1.85(7) | 2.787(8) | 179(10) |
| O11 | –H11A | ⋯O10 | [1555] | 0.78(7) | 1.99(7) | 2.742(8) | 162(7) |
| O11 | –H11B | ⋯O12 | [1555] | 0.82(7) | 1.94(7) | 2.725(9) | 161(8) |
| O12 | –H12A | ⋯O7 | [2665] | 0.90(6) | 1.95(7) | 2.836(7) | 169(7) |
| O12 | –H12B | ⋯O8 | [2765] | 0.84(7) | 2.06(7) | 2.828(8) | 152(6) |
| O13 | –H13A | ⋯O11 | [1555] | 0.64(7) | 2.11(7) | 2.734(8) | 165(9) |
It is likely that the hydrogen bonding interactions between the water molecules in the channels and acetyl groups influence the intermolecular distance between the Pt(II) molecules, and that the separation is responsible for the colour of the complex. The loss of water will then result in the observed colour change from orange to yellow.
X-ray powder diffraction studies on the vapochromic orange, yellow and blue forms of complex 8
It was not possible to obtain suitable quality single crystals of the yellow and blue forms of complex 8 but these could be obtained as microcrystalline powders. It is hypothesised that in the blue form methanol molecules would be involved in hydrogen bonding to the acetyl groups in a similar way to that observed for the water molecules in the orange form of 8_water. The blue colour of the complex is similar to that observed in the methanol form of [Pt(N^C(C(O)OMe)^N)(CN)] (1).62 The yellow powder was postulated to be an anhydrous form.X-ray powder diffraction patterns were obtained of the three vapochromic forms of complex 8 and the patterns were compared. The yellow anhydrous form was isolated by loading a capillary with the complex and placing it under vacuum before flame-sealing the glass, which has the same effect as using dry nitrogen gas. The blue methanolic form was isolated by placing a small piece of methanol-soaked cotton wool at the bottom of the capillary and then filling it with the complex and flame-sealing. As the methanol vapour diffused through the capillary, the methanolic form of the complex was isolated. The comparison of the powder diffraction patterns is shown in Fig. 9. The patterns for the three structures are similar, with essentially the same peak pattern between 2θ values of 25–30°, but some differences between the peak positions and/or splitting in the 2θ range of 5–10°. This suggests that the crystal packing is similar in the three cases, and presumably similar to the single crystal structure of the orange form, containing channels through which solvents can pass.
 | ||
| Fig. 9 The X-ray powder diffraction patterns for the hydrated orange form (orange line), the anhydrous yellow form (yellow line) and the methanolic blue form (blue line). Grey boxes highlight the major differences between the patterns. | ||
Solvatochromic and vapochromic studies on (methyl-3,5-dipyridyn-2-yl)benzoate platinum(II) trifluoroacetate (9)
In the next step of the investigation, the ligand in the fourth coordination site of the square planar platinum(II) complex was modified by replacing the methyl of the acetyl group with a CF3 group. This modification is likely to enhance the solubility of the complex in polar solvents through enhanced molecule/solvent interactions. The complex was isolated in good yield as a bright yellow solid and characterised by IR, 1H and 19F NMR spectroscopies, and by X-ray powder diffraction analysis. The 19F NMR signal at δ −74.58 confirms the presence of the CF3 group.Complex 9 shows enhanced solubility in dichloromethane and acetonitrile compared to 8. The profile of the UV-vis spectra are similar to those recorded for 5. The peaks below 300 nm can be assigned to the 1π–π* transition, and the broad peak between 350–340 nm is the result of the dominant 3π–π* transition. However, in acetonitrile, the 3π–π* transition is shifted compared to the spectrum recorded in dichloromethane. The emission profiles are concentration dependent, and at higher concentrations, a broad band is observed (Fig. S7 and S8). As seen in previous complexes in the series, this peak is caused by excimer formation because of the higher concentrations in solution. The UV-vis spectra for complex 9 in dichloromethane and acetonitrile are compared in Fig. 10. The significant shift in the 3π–π* transition maxima suggests that the complex exhibits solvatochromism, being yellow in dichloromethane and orange in acetonitrile.
 | ||
| Fig. 10 Comparison of the different solvatochromic forms of complex 9, measured from their UV-vis spectra with a solution concentration 1 × 10−4 M. | ||
The solvatochromic and vapochromic properties of 9 were further explored in the solid-state. When initially recrystallised from dichloromethane the yellow colour of the complex suggests, by analogy with other complexes in the series, that Pt⋯Pt stacking in the crystalline structure is absent, and that the complex is unlikely to display vapochromic behaviour. However, when the complex is recrystallised from an acetonitrile/diethyl ether mixture, the colour of the crystalline solid changes from yellow to orange. The yellow form of 9 also changes from yellow to orange when the yellow crystals are exposed to acetonitrile vapour or solvent in the presence of diethyl ether. However, because of the high solubility of the complex the crystals dissolve rapidly in the acetonitrile solvent to give a bright orange solution. The removal of the solvent does not recover the yellow form, but instead, a deep red crystalline solid is obtained. The yellow-coloured solid is only recovered by recrystallisation with dichloromethane. The series of colour changes is illustrated in Fig. 11.
 | ||
| Fig. 11 The colour changes observed as the orange crystals of 9 are formed by treatment with acetonitrile, following the material being dissolved in acetonitrile, finally leaving a deep red residue. a) Orange crystals grown from acetonitrile/diethyl ether vapour diffusion; b) following evaporation of the diethyl ether, the remaining acetonitrile starts to dissolve the crystals while they retain their orange colour; c) upon evaporation of the remaining acetonitrile a deep red crystalline solid remains. | ||
Because of the tendency of crystals of 9 to desolvate, it was not possible to grow crystals of a suitable quality for a single-crystal X-ray diffraction study, but bulk powder samples could be studied by X-ray powder diffraction. The yellow form was recrystallised from dichloromethane, but the orange form was obtained by placing a small sample of the yellow powder in a capillary with a small piece of acetonitrile-soaked cotton wool and flame-sealing the tube. Acetonitrile was allowed to diffuse through until the solid was converted to the orange form. The X-ray powder patterns for the two forms are shown in Fig. S9. The two patterns show similarities at higher 2θ values but significant differences at lower 2θ values, suggesting that while the two forms are structurally similar, the solvent exchange processes do result in substantial differences in the long-range crystal-packing arrangements in the powder, and inferring that solvent molecules are being incorporated into the crystalline lattice.
Solvatochromic and vapochromic studies on (methyl-3,5-dipyridyn-2-yl)benzoate platinum(II) triflate (10)
In the next complex to be investigated a triflate group was substituted in the fourth coordination site on the platinum(II) complex. The complex 10 was isolated in good yield as a yellow solid, and was characterised by IR, 1H and 19F NMR spectroscopies and by X-ray powder diffraction. The presence of the triflate group was confirmed by the 19F NMR spectroscopic signal at δ −79.33.The UV-vis absorption and emission spectra are similar to those measured in dichloromethane for the previous complexes in the series, showing a similar concentration dependence (Fig. S10). Attempts were made to recrystallise 10 from dichloromethane and acetonitrile, and as with 9, the dichloromethane solution appeared yellow while the acetonitrile solution was orange, exhibiting solvatochromism. A comparison of the UV-vis spectra for the complex in the two solvents shows a small shift in the peak maxima (Fig. 12), which is not as large as in the case of 9, but the blue shift of 10 nm is consistent with the observed colour change.
 | ||
| Fig. 12 A comparison of the UV-vis spectra of the different solvatochromic forms of complex 10, solution concentration 1 × 10−4 M; dichloromethane solution (yellow trace), acetonitrile solution (orange trace). | ||
An orange film can be formed with the acetonitrile solvent when the solution is dropped onto a slide. When the excess acetonitrile on the slide evaporates, it leaves an orange powder instead of the deep red residue obtained for complex 9. The two forms could only be accessed by recrystallisation from the appropriate solvent and not by using vapochromic techniques.
While it was not possible to obtain suitable single crystals of 9 for an X-ray study, the yellow and orange powders were investigated using X-ray powder diffraction. The two powders show very similar traces (Fig. S11), which suggests that any structural changes associated with the colour change do not have significant long-range order, unlike those observed for 8 and 9. It is interesting that the solvatochromic transition occurs more slowly than that in the acetate 8 and suggests that the bulkier triflate group slows the diffusion process through the crystalline material in the solid state.
Solvatochromic and vapochromic studies on (methyl-3,5-dipyridyn-2-yl)benzoate platinum(II) tosylate (11)
In the final member of the series, the fourth coordination site on the platinum(II) centre is substituted by the bulkier tosylate substituent. The complex 11 was synthesised in good yield by the ligand exchange method used in the formation of the previous members of the series, and isolated as a purple solid from the acetonitrile-containing reaction mixture. The complex 11 was characterised by IR and 1H spectroscopies and by X-ray powder diffraction.Upon dissolving the purple solid in dichloromethane, a pale-yellow solution was obtained. The UV–vis spectrum exhibits the same profile as the other complexes in the series and displays a similar concentration dependence (Fig. S12).
The vapochromic and solvatochromic properties of 11 were investigated using dichloromethane, acetonitrile and methanol as the solvents or vapours. The solvatochromic behaviour was investigated using UV-vis spectroscopy. Since the complex turns yellow in dichloromethane and methanol, comparative solution-state spectroscopy has been carried out in acetonitrile and dichloromethane. A comparison of the spectra for the different solvated forms is shown in Fig. 13. As previously assigned, the peaks between 300–350 nm can be assigned to the 1π–π* transitions and the dominant peak between 340–420 nm is assigned to the 3π–π* transition, with some 1π–π* character. The yellow line for the dichloromethane spectrum follows the typical profile observed for all the other complexes in the series. However, the acetonitrile signal (purple line) is blue-shifted with respect to the dichloromethane signal, and the solution turns orange.
 | ||
| Fig. 13 A comparison of the UV-vis spectra of the different solvatochromic forms of complex 11, solution concentration 1 × 10−4 M. 3π–π* transition highlighted. The purple trace represents the acetonitrile solvent spectrum, the yellow trace represents the dichloromethane solvent spectrum. | ||
In addition to running the pure solvent systems, solution state spectroscopy was performed on solvent mixtures. Interestingly, when the solution strength is 10% of acetonitrile, the profile of the spectrum changes from the profile of the dichloromethane spectrum to that of the complex when it is measured in pure acetonitrile (Fig. 14). This change has not been observed in the other solvatochromic complexes in the series and may suggest that the complex is more stable in acetonitrile because of complex–solvent interactions.
 | ||
| Fig. 14 The monitoring of mixed dichloromethane/acetonitrile solvent systems of complex 11 using UV-vis spectroscopy. Concentration of 1 × 10−4 M solution in dichloromethane, with acetonitrile added stepwise with the added volumes of acetonitrile given in the legend. The black arrow highlights the shift of the maximum to lower wavenumber and lower intensity as the concentration of acetonitrile increases. | ||
The emission spectra for the two solvent systems for complex 11 have also been measured. A comparison of the spectra for the two solvents shows that there is no change in the low wavelength emission, between 450–550 nm, which is characteristic of the 3π–π* transition. However, the excimer emission is blue-shifted when the solvent is changed from dichloromethane to acetonitrile (Fig. S13), which may have implications for the luminescent properties of the complex. The emission spectra for the mixed solvent system displayed in Fig. S14, show that in dichloromethane with 5% of acetonitrile added, the excimer emission starts to shift, but at above 30% of acetonitrile, there is no further change.
In the solid state a powder sample of the purple form of complex 11 can be converted to the yellow form by blowing methanol vapour over the solid, and this can be reversed by adding acetonitrile droplets or vapour, as illustrated in Fig. 15. With the appropriate solvent, both forms are stable at room temperature and pressure, which contrasts the previous members of the series where only one form predominates under ambient conditions.
 | ||
| Fig. 15 The vapochromic properties of complex 11 when the purple solid is (a) exposed to methanol vapour, changing to a yellow colour, and (b) when the colour change is reversed by using acetonitrile vapour or droplets. | ||
Unfortunately, it was not possible to grow crystals suitable for a single-crystal X-ray diffraction study for either the purple or yellow forms of complex 11. However, X-ray powder diffraction patterns could be obtained. The yellow powder was the methanol form and was obtained by trapping a small amount of methanol at the end of a glass capillary with cotton wool and flame-sealing the glass, then adding the purple solid, and allowing the vapour to diffuse through the capillary, turning the purple solid to yellow. The X-ray diffraction patterns showed that the two forms had substantially different patterns (Fig. 16), consistent with the forms having different crystalline structures. This is not surprising because of the marked difference in colour between the two forms. The dark purple colour would suggest that there were Pt⋯Pt interactions in the structure, as found in the other highly coloured platinum(II) complexes in the series.62,64
 | ||
| Fig. 16 The X-ray powder diffraction patterns for the purple (purple trace) acetonitrile form and the yellow (yellow trace) for the methanolic form of complex 11. Grey boxes highlight the major differences between the patterns. | ||
Conclusion
In this investigation six platinum(II) pincer complexes with the formula [Pt(N^C(C(O)OMe)^N)(L)] (L = (NCO) (6), (NCS) (7), (OC(O)Me) (8), (OC(O)CF3) (9), (OS(O)2CF3) (10), and (OS(O)2(C6H4Me)) (11)) have been synthesised by exchange of the chloride ion in [Pt(N^C(C(O)OMe)^N)(Cl)] (5) and characterised spectroscopically and by X-ray diffraction techniques. Their solution and solid-state UV-vis absorption and emission spectra have been measured, and their solvatochromic and vapochromic properties have been investigated. These properties have been compared and contrasted with those of the previously reported series [Pt(N^C(R)^N)(CN)] ((1, R = C(O)OMe)); 2, R = C(O)Me); 3, R = C(O)OEt; 4, R = C(O)OPh)),62,64 with the aim of identifying the structural and electronic factors that influence the solvatochromic and vapochromic properties observed. There is a particular focus on the role of the monodentate ligand in the fourth coordination site of the platinum(II) square planar complexes and the contrast with the effect of the strong ligand field cyanide ion present in complexes 1–4.The two complexes 6 and 7, with the ligands NCO and NCS near the centre of the spectrochemical series, did not display vapochromic or solvatochromic properties in the solid state under any of the experimental conditions employed, with a range of VOCs. The crystals isolated were yellow, crystallised without solvent molecules in the crystal lattice, and from the crystal structure determinations, the intermolecular Pt⋯Pt distances were approximately 5 Å, substantially too long for any direct Pt⋯Pt interactions, although interligand π⋯π stacking interactions were present. The absence of Pt dz2⋯dz2 orbital overlap precludes 3MMLCT and resultant colour changes, and the yellow colour in crystalline platinum(II) samples often indicates that Pt⋯Pt interactions are absent. It may be concluded that the absence of a strong field ligand in the fourth coordination site would not favour solvatochromism or vapochromism because the strong field cyanide ligand would be required to raise the energy of the Pt(II) dz2 orbital, so it lies closer to the frontier orbitals, favouring the formation of dσ and dσ* interactions along a Pt⋯Pt chain. The cyanide group can also form strong hydrogen bonds with protic solvents that can manipulate the crystal packing and further enhance Pt⋯Pt interactions.
In the second part of the study, the fourth ligand position was substituted with acetate, trifluoroacetate, triflate, and tosylate ligands. While these were weak field, oxygen donor ligands, they do have the ability to form hydrogen bonds and interact with guest VOCs, as was the case with [Pt(N^C(C(O)OMe)^N)(CN)] (1).62 In these systems, it is the hydrogen-bonding network and the hydrogen bonding to the oxygen donor ligands in the complexes that influence the vapochromic behaviour so that the colour changes correlate to the crystal packing. The presence of cavities or channels within the crystalline lattice also facilitates the vapochromic or solvatochromic behaviour. All four of the complexes 8–11 do show vapochromic or solvatochromic behaviour, and in the case of the orange, hydrated form of the acetate complex, 8, the crystal structure shows that water molecules form a hydrogen bonding network with the acetate oxygen, and link adjacent pincer molecules together, being supported by π⋯π stacking interactions. In 8 the Pt⋯Pt separation, at approximately 5 Å, is too long for there to be a direct Pt⋯Pt interaction, as might be expected when a weak field ligand is in the fourth coordination site on the Pt centre. However, the blue methanol form and the anhydrous yellow form of 8, obtained by vapochromic techniques, confirm that it is possible to obtain vapochromic platinum(II) complexes with weak field ligands capable of hydrogen bonding to VOC guest molecules. It is unfortunate that crystal structure data is not obtainable for complexes 9, 10 and 11. Complexes 9 and 10, which contain a CF3 group as a substituent on the ligand in the fourth coordination site, are significantly more soluble than the previous members in the series. While it was not possible to isolate single crystals of these two complexes, in solution, they displayed solvatochromism, forming yellow solids when crystallised from dichloromethane and changing to orange solids when recrystallised from acetonitrile. Complex 11, with the larger tosylate ligand, is the most versatile in the series. The complex shows a reversible colour change from purple to yellow in the solid state when the methanol solvated form is exposed to acetonitrile. The change occurred when vapours of solvents were used, and the change could be observed with mixed solvent mixtures, with the presence of 10% of acetonitrile in the dichloromethane solution being sufficient to convert the solution to purple. This might suggest a more significant interaction between the acetonitrile solvent and the pincer molecule. While it was not possible to obtain a crystal structure for the purple form, the colour might be consistent with direct Pt⋯Pt interactions in the solid state. It appears that with the larger tosylate ligand, the transformation between the different coloured forms occurs more slowly, and this can perhaps be attributed to the bulkier substituent hindering solvent diffusion through the crystalline solid.
In summary, while the presence of a strong field ligand in the fourth coordination site of this series of platinum pincer complexes favours vapochromic or solvatochromic behaviour, the presence of weaker field ligands capable of hydrogen bonding with guest VOCs also permits solvatochromic and vapochromic behaviour to be observed. Through the colour changes, the series of complexes can differentiate between the presence of low concentrations of water, methanol, dichloromethane and acetonitrile.
Conflicts of interest
The authors have no conflicts of interest to declare.Data availability
The manuscript is accompanied by detailed supporting information available in PDF format, including synthetic protocols, crystal structure determination parameters, UV/vis absorption and emission data, and X-ray powder data. Further experimental data and information are available from the corresponding author via the University of Bath Research Portal on reasonable request. Supplementary information (SI) is available. See DOI: https://doi.org/10.1039/d5ce00747j.CCDC 2455364 (7) Experimental crystal structure determination 2025, 2455366 (8) Experimental crystal structure determination, 2025 and 2455458 (6) Experimental crystal structure determination 2025, contain the supplementary crystallographic data for this paper.75a–c
Acknowledgements
We are grateful to the Engineering and Physical Sciences Research Council (EPSRC) for a Programme Grant (grant no. EP/K004956/1). The project was also supported by EPSRC grant EP/101974X. LEH is grateful to the Royal Society for a University Research Fellowship (URF\R1\191104).References
- E. Li, K. Jie, M. Liu, X. Sheng, W. Zhu and F. Huang, Chem. Soc. Rev., 2020, 49, 1517–1544 RSC.
- O. S. Wenger, Chem. Rev., 2013, 113, 3686–3733 CrossRef.
- M. Kato, M. Yoshida, Y. Sun and A. Kobayashi, J. Photochem. Photobiol., C, 2022, 51, 100477 CrossRef.
- M. Kato, H. Ito, M. Hasegawa and K. Ishii, Chem. – Eur. J., 2019, 25, 5105–5112 CrossRef PubMed.
- B. Li, L. Cui and C. J. Li, Angew. Chem., Int. Ed., 2020, 59, 22012–22016 CrossRef PubMed.
- Q. Li, H. T. Z. Zhu and F. H. Huang, J. Am. Chem. Soc., 2019, 141, 13290–13294 CrossRef PubMed.
- H. Xia, D. Q. Liu, K. Song and Q. Miao, Chem. Sci., 2011, 2, 2402–2406 RSC.
- E. Takahashi, H. Takaya and T. Naota, Chem. – Eur. J., 2010, 16, 4793–4802 CrossRef.
- E. Takahashi, H. Takaya and T. Naota, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, C640 Search PubMed.
- L. You, D. Zha and E. V. Anslyn, Chem. Rev., 2015, 115, 7840–7892 CrossRef PubMed.
- R. Paolesse, S. Nardis, D. Monti, M. Stefanelli and C. Di Natale, Chem. Rev., 2017, 117, 2517–2583 CrossRef PubMed.
- M. Kato, Bull. Chem. Soc. Jpn., 2007, 80, 287–294 CrossRef.
- X. Zhang, B. Li, Z. H. Chen and Z. N. Chen, J. Mater. Chem., 2012, 22, 11427–11441 RSC.
- T. J. Wadas, Q. M. Wang, Y. J. Kim, C. Flaschenreim, T. N. Blanton and R. Eisenberg, J. Am. Chem. Soc., 2004, 126, 16841–16849 CrossRef.
- T. Seki, K. Sakurada and H. Ito, Angew. Chem., Int. Ed., 2013, 52, 12828–12832 CrossRef PubMed.
- M. Jin, T. Sumitani, H. Sato, T. Seki and H. Ito, J. Am. Chem. Soc., 2018, 140, 2875–2879 CrossRef PubMed.
- V. W.-W. Yam, V. K.-M. Au and S. Y.-L. Leung, Chem. Rev., 2015, 115, 7589–7728 CrossRef PubMed.
- H. N. Abdelhamid, Appl. Organomet. Chem., 2023, 37, e7078 CrossRef.
- G. S. Zhang, C. Fu, H. Y. Zhang and H. Zhang, CrystEngComm, 2021, 23, 4513–4521 RSC.
- L. Li, Y. Hua, Y. Guo, H. Y. Wang, X. N. Li and H. Zhang, New J. Chem., 2019, 43, 3428–3431 RSC.
- C. Chen, H. Z. Rao, S. Lin and J. Zhang, Dalton Trans., 2018, 47, 8204–8208 RSC.
- F. Drache, V. Bon, I. Senkovska, M. Adam, A. Eychmuller and S. Kaskel, Eur. J. Inorg. Chem., 2016, 4483–4489 CrossRef.
- P. Muller, F. M. Wisser, V. Bon, R. Grunker, I. Senkovska and S. Kaskel, Chem. Mater., 2015, 27, 2460–2467 CrossRef.
- Y. Cui, B. Chen and G. Qian, Coord. Chem. Rev., 2014, 273–274, 76–86 CrossRef.
- B. Dutta, A. Hazra, S. Datta, C. Sinha, P. Banerjee and M. H. Mir, ACS Appl. Polym. Mater., 2022, 4, 2841–2850 CrossRef.
- M. H. Mir, S. Bera, S. Khan, S. Maity, C. Sinha and B. Dutta, Chem. Commun., 2021, 57, 6197–6200 RSC.
- N. Sadovnik, P. Lyu, F. Nouar, M. Muschi, M. Qin, G. Maurin, C. Serre and M. Daturi, Nat. Commun., 2024, 15, 9456 CrossRef.
- A. Kobayashi and M. Kato, Eur. J. Inorg. Chem., 2014, 2014, 4469–4483 CrossRef.
- M. Albrecht, M. Lutz, A. L. Spek and G. van Koten, Nature, 2000, 406, 970–974 CrossRef.
- J. Fornies, S. Fuertes, J. A. Lopez, A. Martin and V. Sicilia, Inorg. Chem., 2008, 47, 7166–7176 CrossRef.
- M. Yoshida and M. Kato, Anal. Sci., 2025, 41, 1233–1249 CrossRef.
- R. L. White-Morris, M. M. Olmstead, F. L. Jiang, D. S. Tinti and A. L. Balch, J. Am. Chem. Soc., 2002, 124, 2327–2336 CrossRef.
- Y. A. Lee and R. Eisenberg, J. Am. Chem. Soc., 2003, 125, 7778–7779 CrossRef PubMed.
- T. Lasanta, M. E. Olmos, A. Laguna, J. M. Lopez-de-Luzuriaga and P. Naumov, J. Am. Chem. Soc., 2011, 133, 16358–16361 CrossRef.
- S. H. Lim, M. M. Olmstead and A. L. Balch, Chem. Sci., 2013, 4, 311–318 RSC.
- J. A. Pells, D. Guan and D. B. Leznoff, Eur. J. Inorg. Chem., 2022, 2022, e202200049 CrossRef.
- N. Mirzadeh, S. H. Priver, A. J. Blake, H. Schmidbaur and S. K. Bhargava, Chem. Rev., 2020, 120, 7551–7591 CrossRef.
- L. M. C. Luong, M. A. Malwitz, V. Moshayedi, M. M. Olmstead and A. L. Balch, J. Am. Chem. Soc., 2020, 142, 5689–5701 CrossRef PubMed.
- L. M. C. Luong, C. D. Lowe, A. V. Adams, V. Moshayedi, M. M. Olmstead and A. L. Balch, Chem. Sci., 2020, 11, 11705–11713 RSC.
- C. L. Exstrom, J. R. Sowa Jr., C. A. Daws, D. Janzen, K. R. Mann, G. A. Moore and F. F. Stewart, Chem. Mater., 1995, 7, 15–17 CrossRef.
- C. A. Daws, C. L. Exstrom, J. R. Sowa and K. R. Mann, Chem. Mater., 1997, 9, 363–368 CrossRef CAS.
- L. J. Grove, J. M. Rennekamp, H. Jude and W. B. Connick, J. Am. Chem. Soc., 2004, 126, 1594–1595 CrossRef.
- M. A. Soto, R. Kandel and M. J. MacLachlan, Eur. J. Inorg. Chem., 2021, 2021, 894–906 CrossRef.
- A. Rico, P. Le Poul, J. Rodríguez-López, S. Achelle and S. Gauthier, Dalton Trans., 2024, 53, 11417–11425 RSC.
- A. Rico, P. Le Poul, A. Lyčka, J. Rodríguez-López, S. Achelle and S. Gauthier, New J. Chem., 2025, 49, 8494–8501 RSC.
- A. Rico, P. Le Poul, J. Rodríguez-López, D. Jacquemin, S. Achelle and S. Gauthier, ChemPhotoChem, 2025, e202500164 CrossRef.
- Y. Zhang, J. Ni, Y. Zhu, Q. Zeng, Y. Ai and Y. Li, Chem. Eng. J., 2024, 498, 155049 CrossRef.
- L. J. Grove, A. G. Oliver, J. A. Krause and W. B. Connick, Inorg. Chem., 2008, 47, 1408–1410 CrossRef PubMed.
- S. D. Taylor, W. Howard, N. Kaval, R. Hart, J. A. Krause and W. B. Connick, Chem. Commun., 2010, 46, 1070–1072 RSC.
- Y. Shigeta, A. Kobayashi, T. Ohba, M. Yoshida, T. Matsumoto, H.-C. Chang and M. Kato, Chem. – Eur. J., 2016, 22, 2682–2690 CrossRef PubMed.
- S. Chatterjee, A. E. Norton, M. K. Edwards, J. M. Peterson, S. D. Taylor, S. A. Bryan, A. Andersen, N. Govind, T. E. Albrecht-Schmitt, W. B. Connick and T. G. Levitskaia, Inorg. Chem., 2015, 54, 9914–9923 CrossRef PubMed.
- V. M. Shingade, L. J. Grove and W. B. Connick, Dalton Trans., 2020, 49, 9651–9661 RSC.
- A. E. Norton, D. L. Zhao, S. D. Taylor, S. R. Kennedy, T. D. Ball, M. O. Bovee, W. B. Connick, S. Chatterjee and M. K. Abdolmaleki, Sens. Actuators, B, 2022, 359, 131502 CrossRef.
- A. E. Norton, S. D. Taylor, C. Williams, A. Zoller, W. Dietrich, W. B. Connick and S. Chatterjee, Dalton Trans., 2025, 54, 2357–2365 RSC.
- J. A. G. Williams, A. Beeby, E. S. Davies, J. A. Weinstein and C. Wilson, Inorg. Chem., 2003, 42, 8609–8611 CrossRef PubMed.
- Y. Ai, M. H.-Y. Chan, A. K.-W. Chan, M. Ng, Y. Li and V. W.-W. Yam, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 13856–13861 CrossRef CAS PubMed.
- B. Li, Y. Li, M. H.-Y. Chan and V. W.-W. Yam, J. Am. Chem. Soc., 2021, 143, 21676–21684 CrossRef CAS.
- C.-J. Lin, Y.-H. Liu, S.-M. Peng, T. Shinmyozu and J.-S. Yang, Inorg. Chem., 2017, 56, 4978–4989 CrossRef CAS.
- Y. Ai, Y. Li, M. H.-Y. Chan, G. Xiao, B. Zou and V. W.-W. Yam, J. Am. Chem. Soc., 2021, 143, 10659–10667 CrossRef CAS PubMed.
- C.-Y. Lien, Y.-F. Hsu, Y.-H. Liu, S.-M. Peng, T. Shinmyozu and J.-S. Yang, Inorg. Chem., 2020, 59, 11584–11594 CrossRef CAS PubMed.
- R. J. Salthouse, A. Sil, L. F. Gildea, D. S. Yufit and J. A. G. Williams, Inorg. Chem., 2023, 62, 12356–12371 CrossRef CAS.
- M. J. Bryant, J. M. Skelton, L. E. Hatcher, C. Stubbs, E. Madrid, A. R. Pallipurath, L. H. Thomas, C. H. Woodall, J. Christensen, S. Fuertes, T. P. Robinson, C. M. Beavers, S. J. Teat, M. R. Warren, F. Pradaux-Caggiano, A. Walsh, F. Marken, D. R. Carbery, S. C. Parker, N. B. McKeown, R. Malpass-Evans, M. Carta and P. R. Raithby, Nat. Commun., 2017, 8, 1800 CrossRef CAS PubMed.
- D. J. Cárdenas, A. M. Echavarren and M. C. Ramírez de Arellano, Organometallics, 1999, 18, 3337–3341 CrossRef.
- M. J. Bryant, S. Fuertes, L. E. Hatcher, L. H. Thomas and P. R. Raithby, Faraday Discuss., 2023, 244, 411–433 RSC.
- T. Bunlaksananusorn, K. Polborn and P. Knochel, Angew. Chem., Int. Ed., 2003, 42, 3941–3943 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. Nolze and W. Kraus, Powdercell 2.3, Federal Institute for Materials Research and Testing, Berlin, 2000 Search PubMed.
- S. J. Farley, D. L. Rochester, A. L. Thompson, J. A. K. Howard and J. A. G. Williams, Inorg. Chem., 2005, 44, 9690–9703 CrossRef CAS PubMed.
- C. E. Housecroft and A. G. Sharpe, Inorganic Chemistry, Pearson, Harlow, England, 5th edn, 2018 Search PubMed.
- R. A. Bailey, S. L. Kozak, T. W. Michelsen and W. N. Mills, Coord. Chem. Rev., 1971, 6, 407–445 CrossRef CAS.
- A. Kitaigorodskii, Acta Crystallogr., 1965, 18, 585–590 CrossRef CAS.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef CAS.
- (a) CCDC 2455364: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2nf09f; (b) CCDC 2455366: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2nf0ch; (c) CCDC 2455458: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2nf3bk.
| This journal is © The Royal Society of Chemistry 2025 |