
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solvent induced structural transformation of a cerium(III) 2,5-furandicarboxylate metal–organic framework
Satarupa
Das
a,
Jeremiah P.
Tidey
 b,
Jie
Liu
b,
Katie S.
Pickering
a,
James C.
Coe
a,
Marc
Walker
b and
Richard I.
Walton
*a
b,
Jie
Liu
b,
Katie S.
Pickering
a,
James C.
Coe
a,
Marc
Walker
b and
Richard I.
Walton
*a
aDepartment of Chemistry, University of Warwick, Coventry, CV4 7AL, UK. E-mail: r.i.walton@warwick.ac.uk
bDepartment of Physics, University of Warwick, Coventry, CV4 7AL, UK
First published on 22nd September 2025
Abstract
The synthesis and structural characterisation of a cerium(III) MOF, UOW-11, is reported. The material is constructed using the sustainably sourced 2,5-furandicarboxylate (FDC) linker under solvothermal conditions in N,N-dimethylformamide (DMF) solvent and scalable to gram-scale production. Single-crystal analysis using 3D electron diffraction reveals a unique framework architecture, P21/n space group, with composition [Ce4(FDC)7(DMF)2.36](DMA)2 that has a structure containing four cerium(III) sites that are connected via bridging FDC ligands to give a 3D extended anionic framework counterbalanced by occluded dimethylammonium (DMA) cations, formed via partial decomposition of DMF. Upon exposure to water, UOW-11 undergoes a structural transformation, forming a second, structurally distinct Ce(III) coordination polymer, [Ce2(FDC)2(H2O)10]FDC·6H2O, UOW-2(Ce), which crystallises in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . The asymmetric unit comprises two Ce3+ ions, coordinated by two FDC2− ligands, ten water molecules, with one additional uncoordinated FDC2− ligand, and six occluded water molecules. The transformation leads to significant changes in coordination geometry and composition, including the loss of DMA and a different Ce
. The asymmetric unit comprises two Ce3+ ions, coordinated by two FDC2− ligands, ten water molecules, with one additional uncoordinated FDC2− ligand, and six occluded water molecules. The transformation leads to significant changes in coordination geometry and composition, including the loss of DMA and a different Ce![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :FDC ratio, consistent with a dissolution–recrystallisation mechanism. Monitoring the conversion using in situ infra-red spectroscopy shows the transformation is complete after ∼3 hours at room temperature.
:FDC ratio, consistent with a dissolution–recrystallisation mechanism. Monitoring the conversion using in situ infra-red spectroscopy shows the transformation is complete after ∼3 hours at room temperature.
Introduction
Metal–organic frameworks of cerium are rapidly emerging due to their structural diversity and range of properties that may be suitable for practical applications.1 In particular, they are of special interest due to the combination of porosity, Ce(III)/(IV) redox properties and the presence of low lying 4f orbitals which facilitates fast electron transfer that may be relevant for applications in energy storage, gas separation, and sensing applications.2 A number of reports have been made on replacement of Zr4+ by Ce4+ in the well-known material UiO-66,3,4 and some novel structures containing Ce4+ have also been discovered with unique building units.5 Many more MOFs of Ce3+ are known, some of which have structures that are analogues of other large lanthanide cations such as La3+.6 The reason that most of the reported cerium MOFs contain Ce(III) is likely due to the strong oxidising nature of Ce(IV); the standard reduction potential for the Ce4+/Ce3+ couple is +1.61 V versus the standard hydrogen electrode, and hence it can be easily reduced during the typical solvothermal synthesis conditions used for their crystallisation.7Unlike Ce(IV)-MOFs, which exhibit a limited variety of SBUs and crystal structures, Ce(III)-MOFs show a wide range of SBUs and topologies.8 The reported Ce(III)-MOFs are often constructed from either 1D infinite building units, or discrete oligonuclear secondary building units (SBUs). In these structures, cerium is typically coordinated by carboxylate groups, along with oxo/hydroxo bridging oxygen atoms or water/solvent molecules. Among the various carboxylate linkers used for MOFs, 2,5-furan dicarboxylate (FDC) is important to consider as it can be sustainably synthesised from lignocellulosic biomass.9 To date, however, there is only one report of the synthesis of a distinct crystal structure of a cerium MOF with the 2,5-furan dicarboxylate (FDC) linker.10 This material, named CAU-28, contains cerium(IV) in the cluster [Ce6O4(OH)4]12+ as the secondary building unit (the same cluster as seen in UiO-66), which is bound to 8 carboxylate groups from 8 distinct FDC ligands, and is isostructural to a Zr4+ analogue.
In this paper, we report the synthesis of a cerium(III) 2,5-furandicarboxylate MOF, designated as UOW-11. Upon soaking in water, UOW-11 undergoes a structural transformation, leading to the formation of a distinct coordination polymer structure, UOW-2(Ce). This involves a structural rearrangement triggered solely by water exposure, which has not been previously reported before for a cerium MOF to our knowledge.
Results and discussion
The material UOW-11 has a chemical formula of [Ce4(FDC)7(DMF)2.36](DMA)2 and crystallises in the monoclinic crystal system with space group P21/n (Table S1). The 3D framework is anionic with occluded dimethylammonium (DMA) cations balancing the charge. The presence of DMA can be explained by the decomposition of the DMF solvent during the crystallisation, due to the presence of small amounts of water, as has been seen before in the synthesis of MOFs.11 In the asymmetric unit, there are 4 unique cerium atoms as depicted in Fig. 1.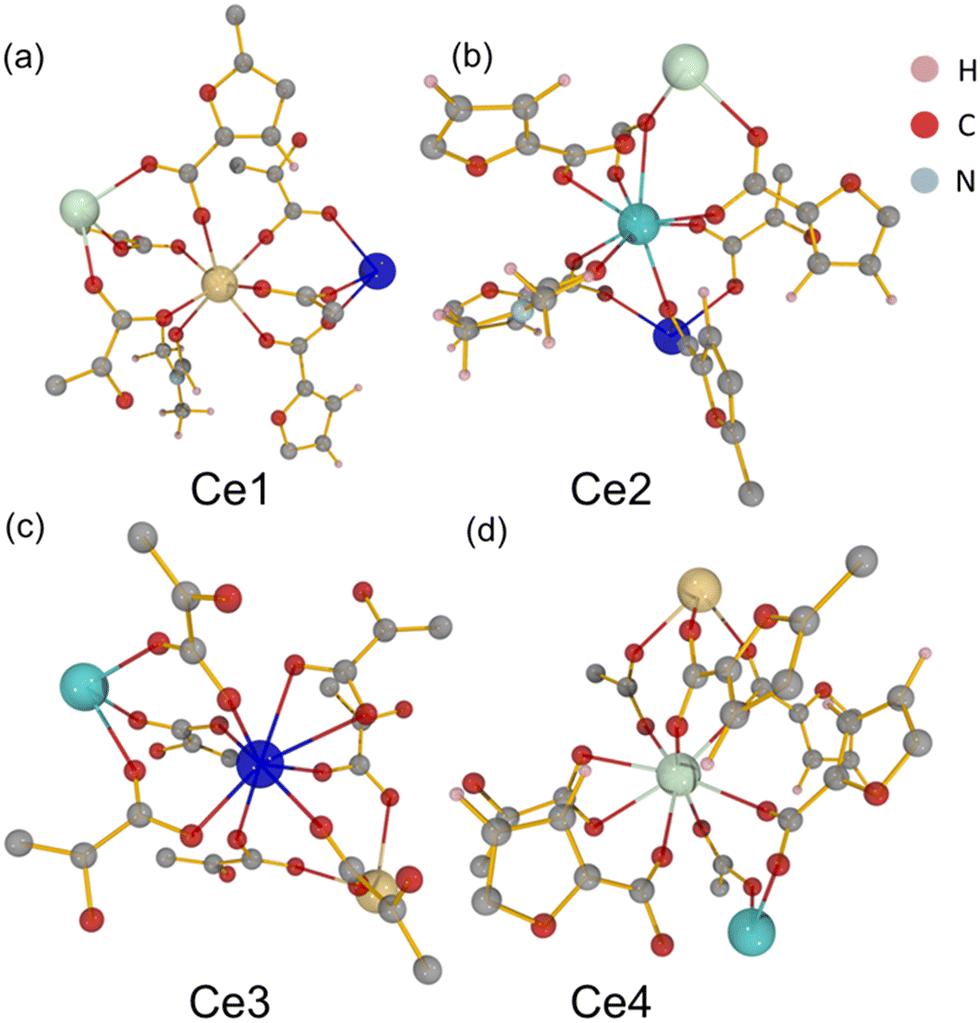 | ||
| Fig. 1 Local environments of the four distinct cerium centres (Ce1–Ce4) in UOW-11, (a–d), highlighting their local coordination environments. Hydrogen (pink), carbon (grey), nitrogen (blue), oxygen (red), and cerium (large spheres, Ce1 yellow, Ce2 sky blue, Ce3 royal blue, Ce4 sea green) atoms are shown. | ||
Ce1 is seven-coordinate and is connected to Ce3 and Ce4 by three distinct FDC ligands via μ2-O bridging with the rest of the coordination completed by binding to one DMF oxygen. Ce2 displays a mixed coordination environment: in the absence of DMF (64% of the unit cells), Ce2 is seven-coordinate, while in the presence of a partially occupied O-bound DMF molecule (occupancy 0.364(17)), Ce2 becomes eight-coordinate. This DMF is disordered about an inversion centre located approximately at the nitrogen atom of the molecule. In both cases, Ce2 is linked to Ce3 by three different oxygen atoms from three separate FDC ligands via syn–syn bidentate carboxylate bridges (Fig. 2a) and connected to Ce4 by a μ2- and μ3-O from two FDC ligands, as well as to another FDC by a μ2-η1:η1 bridge. Ce3 and Ce4 are also both eight-coordinate. Cerium chains connected by oxygen linkages to another layer of cerium by tetradentate bridging FDC resulting in a 3D network. (Fig. 2b and c).
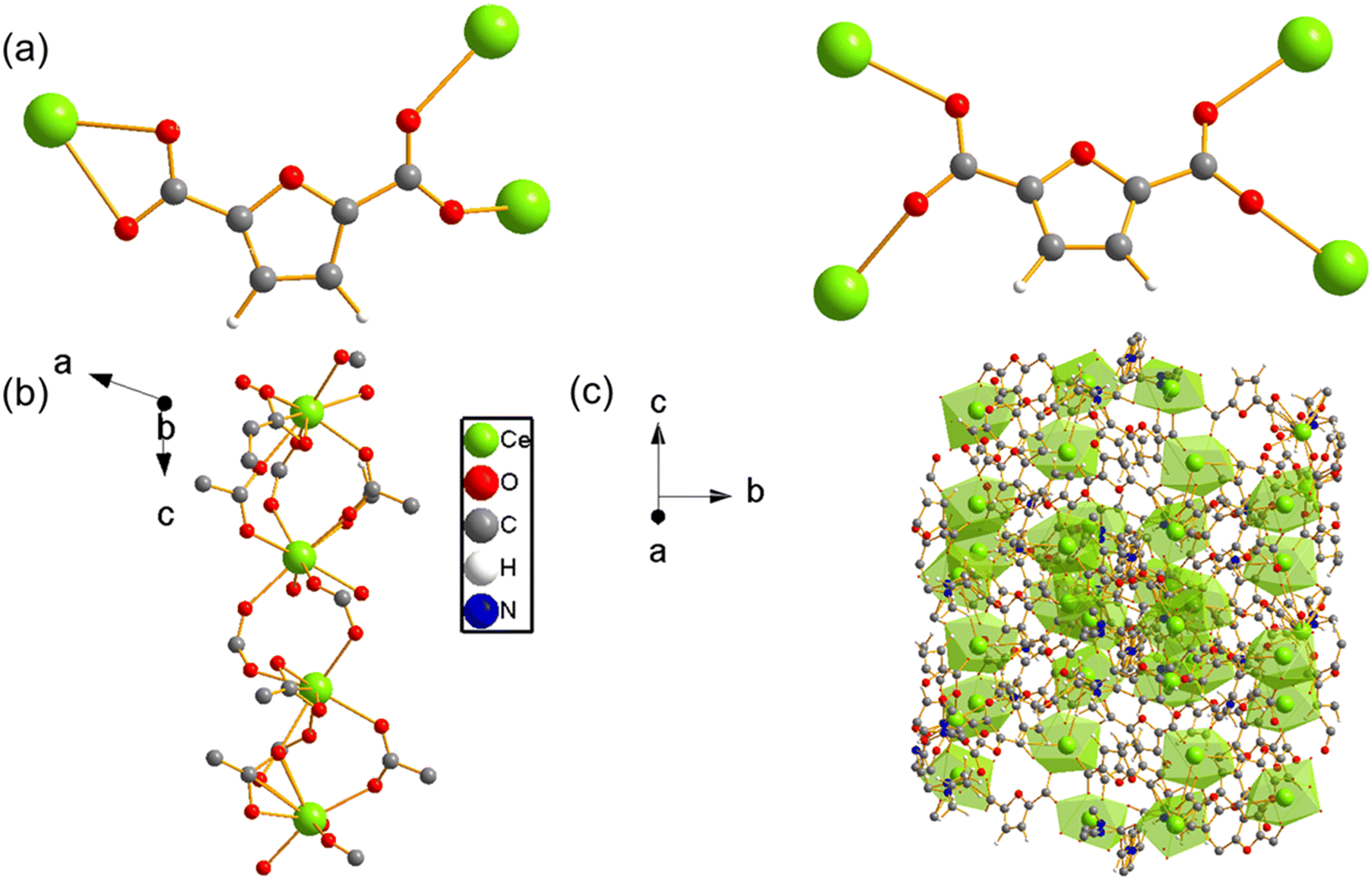 | ||
| Fig. 2 (a) Binding modes of the 2,5-furandicarboxylate (FDC) ligand in UOW-11, showing single and bridging coordination to Ce centres. (b) The resulting Ce–FDC chain-like secondary building unit, viewed along the crystallographic b-axis showing the projection onto the a–c plane. (c) The extended 3-dimensional framework structure of UOW-11, viewed along the [100] direction. Ce atoms are shown in green, O in red, C in grey, H in white, and N in blue. | ||
To assess the phase purity of the bulk sample and to confirm consistency with the 3D-ED unit cell parameters, Pawley fitting was performed to the high-resolution powder X-ray diffraction data collected from the bulk sample. As observed in Fig. 3 and Table S2, the structure parameters of the powder sample are in good agreement with the determined structure. We have further plotted the experimentally obtained UOW-11 powder XRD pattern along with simulated patterns from the single crystal structure, 2,5-furan dicarboxylic acid and CeO2, a likely side product, Fig. S1, which shows the absence of these impurities The unit cell parameters determined from the room temperature powder diffraction are larger than the single crystal structure determination at 175 K, as expected, but otherwise no indication of any phase transition was observed and the 3D-ED-derived structure matched the powder diffraction data well.
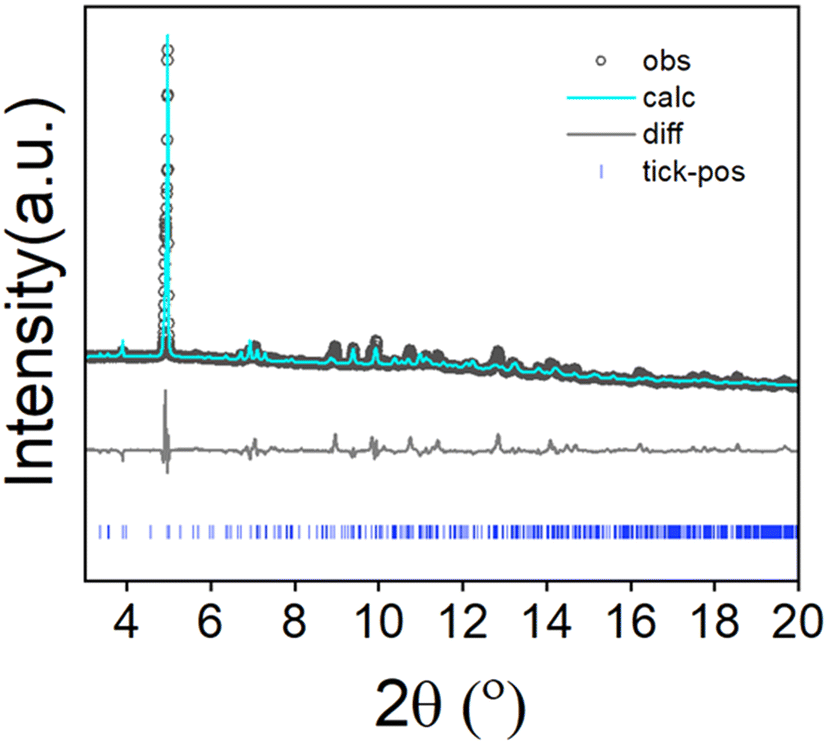 | ||
| Fig. 3 Pawley fit of high resolution powder X-ray diffraction data from UOW-11 (monoclinic space group) (λ = 0.82311 Å) at 293 K (a = 14.77(7) Å, b = 26.58(3) Å, c = 18.32(11) Å, V = 6505.63(12) Å3, Rw = 6%), cf. single-crystal measurements at 175 K (a = 14.85 (14) Å, b = 26.65 (18) Å, c = 17.97 (11) Å, V = 6557.0(10) Å3). | ||
To evaluate the thermal stability of UOW-11, thermogravimetric analysis (TGA) was performed (Fig. 4a). The TGA curve shows a significant mass loss starting at an onset temperature of 203 °C, which proceeds in three distinct steps and plateaus before 395 °C. This mass loss is attributed to the decomposition and combustion of the organic components, with DMF and DMA likely decomposing sequentially before the formation of CeO2. The theoretical residual mass is calculated to be approximately 34.6%, while the experimental value is around 31%, likely due to residual carbon trapped within the oxide matrix.
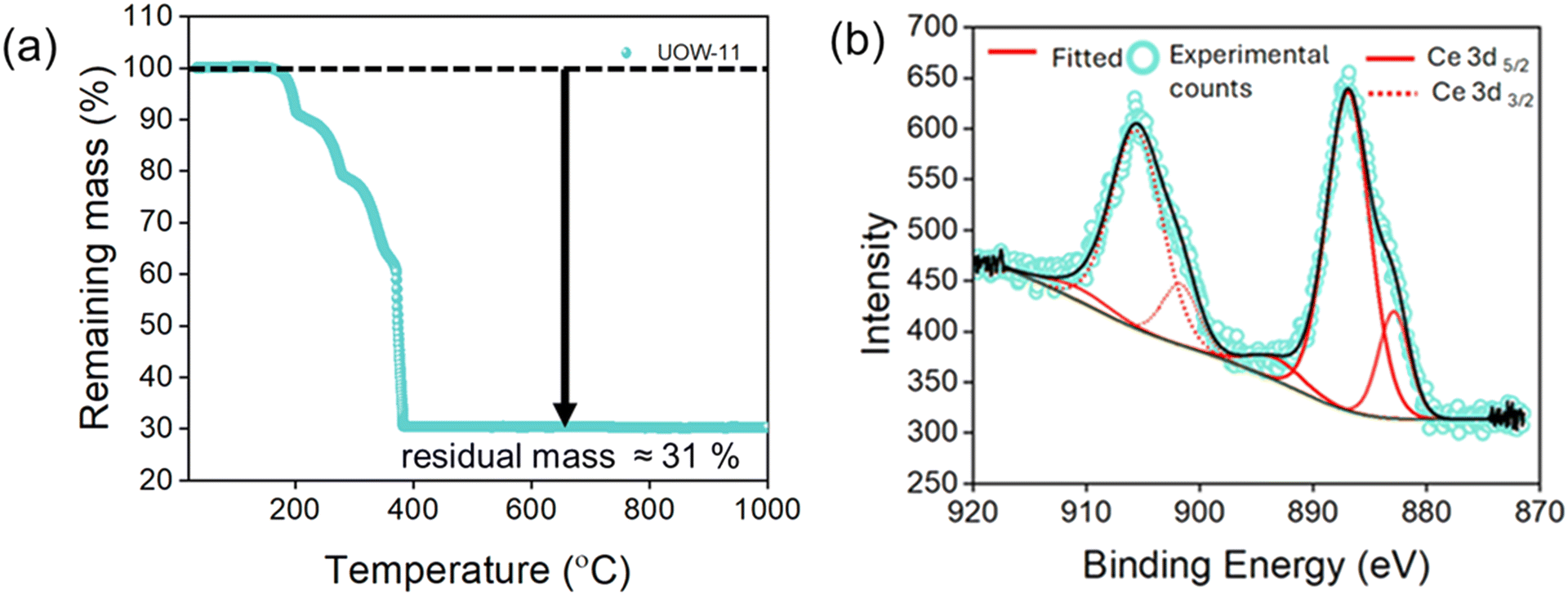 | ||
| Fig. 4 (a) TGA of as synthesised UOW-11 in air, (b) Ce 3d region of the XPS spectrum of UOW-11. | ||
X-ray photoelectron spectroscopy (XPS) was used analyse the oxidation state of Ce in UOW-11 (Fig. 4b). The Ce 3d3/2 signals are observed at approximately 901.6 and 905 eV, and the Ce 3d5/2 signals at around 882 and 886 eV, which are consistent with previously reported values for Ce3+.12 This confirms that cerium exists predominantly in the +3-oxidation state, supporting the structural analysis and the chemical formulation of the material.
UOW-11 undergoes structural evolution upon soaking in water at room temperature with growth of crystals suitable for structure determination by single crystal X-ray diffraction. Fig. 5a shows the resultant structure of the new material, which has the chemical formula of [Ce2(FDC)2(H2O)10]FDC·6H2O. This contains two cerium atoms in the asymmetric unit having each the same coordination and connected to each other by a FDC anion binding in μ2:η2 fashion (Fig. 5b) The Ce+3 ion is nine-coordinated in a slightly distorted monocapped square antiprism geometry, bound by four oxygen atoms from two FDC anions, five coordinated water oxygens and the bidentate carboxyl group of the tetradentate FDC anion which connects to the neighbouring cerium centres. The cerium atoms in UOW-2 are not directly connected by bridging carboxylate oxygen atom, unlike in UOW-11 (Fig. 5c). Fig. 5d shows the structure along [1−11] to show the rod-like motif. Interestingly, this material is isostructural to UOW-2, a Y3+-MOF which we have reported previously,13 and to Dy, Eu, and Gd analogues reported by others.14 Unlike for UOW-2(Ce), the yttrium analogue UOW-2(Y) was prepared directly under solvothermal conditions from a mixed solvent H2O/CH3CN/DMSO. The thermogravimetric analysis (TGA) of UOW-2 (Ce), Fig. S2, shows a first weight loss of ∼30% observed immediately on heating to ∼400 °C, can be attributed to the release of occluded and coordinated water (expected 27.9%). This is followed by a major decomposition step up to 600 °C, which can be ascribed to the breakdown of the FDC linkers within the framework. The final residue above 600 °C is slightly lower than the theoretically expected 33.4% for CeO2 which may be due to the presence of the excess organics released during its formation from UOW-11 (even though no crystalline impurities were detected), or may be evidence of some reduction of CeO2 to CeO2−x.
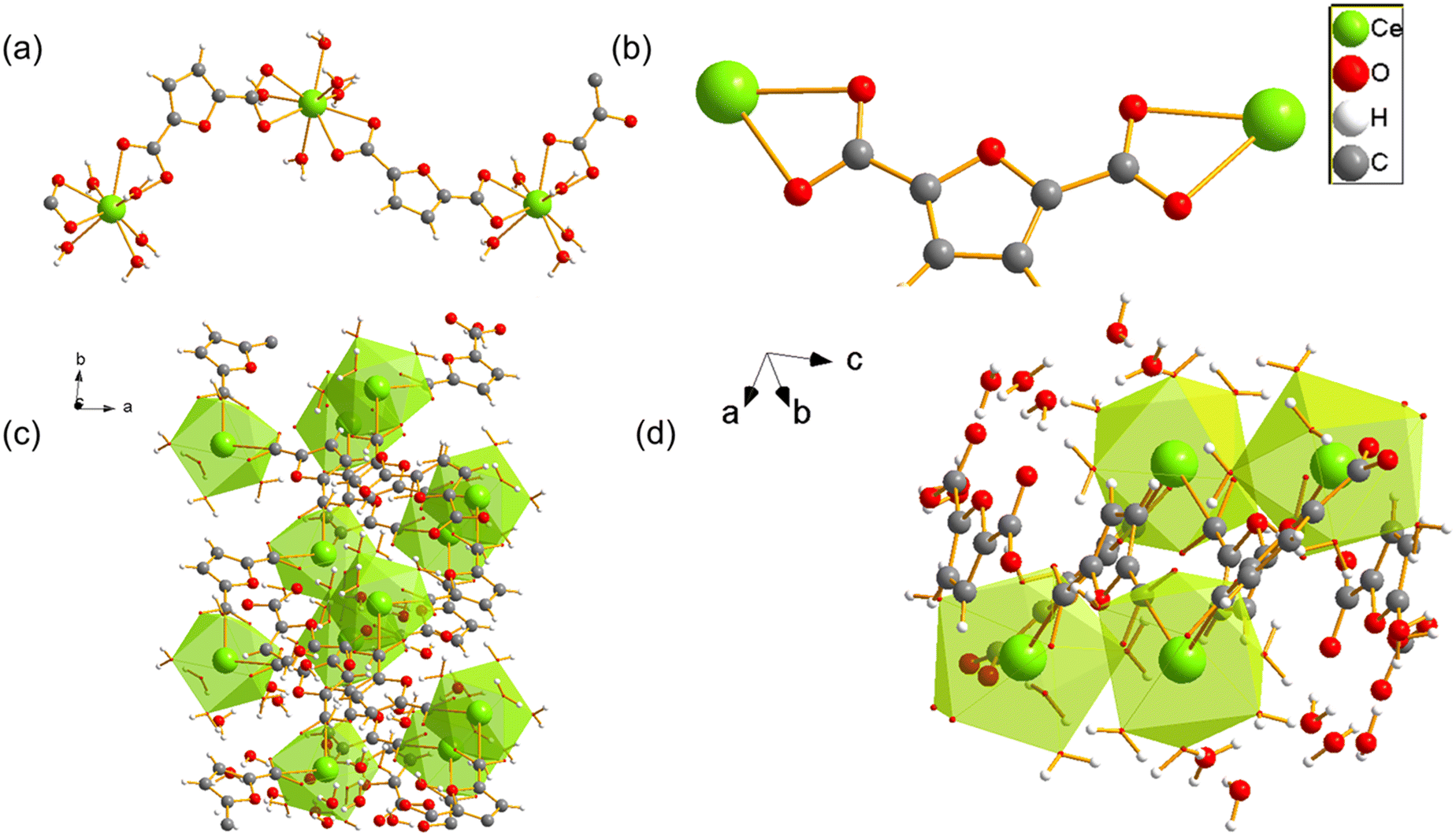 | ||
| Fig. 5 (a) Crystal structure of cerium UOW-2(Ce) viewed along a axis and (b) the binding mode of the FDC ligand, (c) the overall structure of UOW-2(Ce), (d) view of UOW-2(Ce) along [1−11]. | ||
In situ infrared (IR) spectroscopy measurements were conducted during the reaction of UOW-11 with water and it was observed that over time that peaks at 1360 and 1590 cm−1 corresponding to carboxylate stretching vibrations underwent noticeable changes (Fig. 6a). The differences can be explained by the distinct binding modes of FDC between the two structures (Fig. 6a inset).
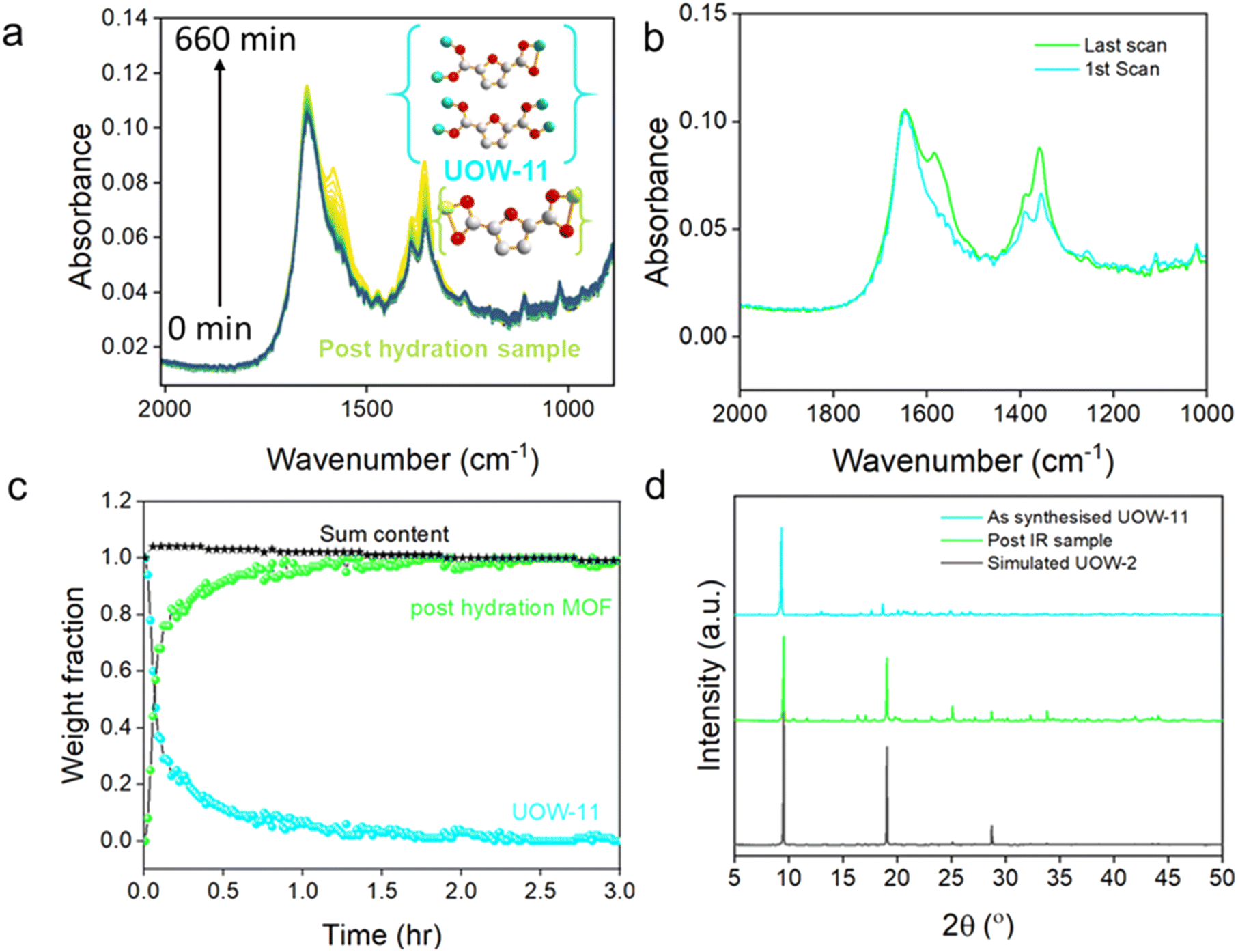 | ||
| Fig. 6 (a) Time dependent in situ IR measurements for UOW-11 expose to water, inset binding modes for UOW-11 and the post-hydration product UOW-2(Ce). (b) IR spectra of the 1st and last scan representing the initial and final phase. (c) Crystallisation curve of weight fraction with time of UOW-11 and UOW-2(Ce) generated from in situ IR measurements along with the sum of two curves showcasing complete structure transformation, and (d) post-IR XRD comparison. | ||
To further analyse the changes during the reaction, the IR spectra from the first and last scans were compared, highlighting significant differences at 1360 and 1590 cm−1, corresponding to key functional group transformations (Fig. 6b). By analysing the IR spectra through a linear combination of the two components, using the initial and final spectra, a crystallisation curve was determined to quantify the evolution of the weight fractions of the two materials over time (Fig. 6c). The resulting plot reveals that the transformation from the initial structure (UOW-11) to the final post-hydration UOW-2(Ce) begins immediately upon exposure to water. Notably, the crossover point where both phases coexist in nearly equal weight fractions occurs during the early stages of the transformation process, within the first few minutes. The crossover of the two curves at ∼50% indicates that these are the only two phases detected. The majority of conversion is complete after 1.5 hours, while complete conversion to the post-hydration material UOW-2(Ce) is achieved by around 3 hours.
Following the IR measurements, the final product was analysed using powder X-ray diffraction (Fig. 6d), which shows a close match to the simulated pattern of the expected structure, with the major diffraction peaks well-aligned in position and relative intensity. Analysis using the Rietveld method using the single crystal structure as a starting model, Fig. S4 and Table S4, confirms that material after hydration is phase-pure UOW-2(Ce). Given the substantial structural differences between the two phases, the transformation is plausibly explained by a dissolution–recrystallisation mechanism rather than a direct solid-state transition. This interpretation is consistent with the rapid disappearance of the UOW-11 phase and the concurrent emergence of the post-hydration material. While this does not confirm a direct structural relationship between the two phases, the temporal overlap suggests that the original material may dissolve and provide a local environment conducive to the nucleation and fast crystallisation of the new structure phase. The transformation could be initiated by hydrolysis of metal ligand bonds of UOW-11, particularly the coordinated DMF, leading decomposition of the framework to release species into water for their reassembly into UOW-2(Ce).
The morphological transition from rod-like crystals in UOW-11 to platelet-like crystals in UOW-2(Ce) provides additional evidence for a change in crystal structure (see scanning electron microscopy images in Fig. S5, and an image of the crystal of UOW-2(Ce) studied crystallography in Fig. S6). The distinct change in crystal morphology is also consistent with a dissolution–crystallisation mechanism for the transformation of one phase to another, rather than a solid–solid transformation. The fact that we see no intermediate state in the in situ experiments likely implies that no significant concentration of an intermediate (whether a solid or solution) accumulates: this suggests that the dissolution–crystallisation occurs rapidly and perhaps only locally so that we only detect the initial and final states in the time-resolution of the IR experiment.
It is noteworthy that the Ce:FDC ratio in UOW-11 of 4:7 is different to the value 4:6 of UOW-2(Ce), suggesting that excess FDC, along with DMF and DMA, remain in solution after the conversion, again consistent with the process being solvent mediated, rather than a solid–solid transformation. Such conversions in MOF chemistry have been observed in situ previously: for example, the crystallisation of the iron(III) material MIL-53 that contains infinite chains is preceded by the formation and decay of MOF-235 that contains trimers of Fe(III) and occluded [FeCl4]−,15 while in the case of some Eu–carboxylate MOFs, an initially formed DMF-rich phase transforms with loss of some DMF and addition of water to a second structure, followed by collapse to Eu–formate via hydrolysis of the DMF.16
Conclusion
The synthesis of a novel cerium(III)-based metal–organic framework UOW-11 utilising the 2,5-furandicarboxylate (FDC) linker is reported, with its crystal structure elucidated through 3D electron diffraction. The material is the first example of a Ce(III) MOF constructed from only FDC as ligand. Notably, we observed a transformation of this material in aqueous conditions, leading to the formation of a second, distinct MOF framework. This structural conversion is accompanied by significant differences in the coordination environment of the FDC linker and the absence of dimethylammonium in the second phase, which was present in the initial MOF. Given these observations, we propose that the transformation mechanism is driven by a dissolution–recrystallisation process, rather than a simple structural rearrangement. This behaviour highlights the dynamic nature of cerium-based MOFs and underscores the influence of solvent interactions on their stability and structural evolution.Experimental section
Synthesis of UOW-11
UOW-11 was synthesised by a solvothermal method under mild conditions. In a typical reaction, 108 mg of Ce(NO3)3·6H2O and 78 mg of 2,5-furan dicarboxylic acid (FDCA) was mixed in a beaker. To that, 4 ml of DMF was added and the solution was stirred vigorously for 10 minutes. The solution was transferred to a Teflon-lined stainless-steel autoclave and heated to 120 °C at a ramp rate of 5 °C min−1 and held for 24 hours. The autoclave was cooled and the solid was recovered by suction filtration. The structure was solved by 3D electron diffraction (3D-ED, see below and SI).Scale-up reaction of UOW-11
To scale up the synthesis of UOW-11, the synthesis procedure was adapted by increasing the reagent quantities by a factor of ten. Specifically, 1.08 g of Ce(NO3)3·6H2O and 780 mg of 2,5-furan dicarboxylic acid (FDCA) were mixed in a beaker. Subsequently, 40 mL of DMF was added to the mixture, and the solution was stirred vigorously for 10 minutes to ensure complete homogenization. The resulting solution was transferred to an autoclave and heated at 120 °C for 24 hours with a ramp rate of 5 °C min−1. After completion of the reaction, the autoclave was allowed to cool to room temperature naturally. The solid product was collected by suction filtration, washed twice with ethanol, and dried at 60 °C (yield 65.3%). Phase purity was confirmed by powder X-ray diffraction (see below).Synthesis of hydration product
This material was synthesised by soaking UOW-11 in excess water (25 mL) at room temperature for 24 h. Crystals were obtained directly from this experiment and the structure was solved by single crystal XRD (see below).Experimental methods
PXRD patterns were recorded using a Siemens D5000 diffractometer equipped with graphite monochromated Cu Kα1,2, radiation (λ = 1.54184 Å) at room temperature, operating in Bragg–Brentano geometry. Data acquisition was performed with a step size of Δ2θ = 0.02° and a dwell time of 4 seconds per step. High-resolution synchrotron PXRD data were collected at beamline I11 of the Diamond Light Source (UK). Measurements were carried out in transmission mode using a position-sensitive detector (PSD), over the angular range 2° < 2θ < 150°, at a wavelength of 0.82311(±0.011584) Å as determined using a Si standard. Samples were loaded into 0.7 mm diameter borosilicate glass capillaries and measured at room temperature. Powder XRD data were analysed by fitting against the diffraction profiles using the GSAS software suite, employing the Pawley method to refine both the peak shape parameters and lattice parameters.17 TGA was performed on a Mettler Toledo TGA/DSC1 instrument under ambient atmospheric conditions. Samples were heated from 25 °C to 1000 °C at a constant heating rate of 10 °C min−1. XPS measurements were carried out at the Photoemission RTP facility, University of Warwick, using a Kratos Axis Ultra DLD spectrometer operating under a base pressure below 1 × 10−10 mbar. Samples were mounted on electrically conductive carbon tape and isolated from the sample bar using a layer of filter paper. A monochromated Al Kα X-ray source (hν = 1486.7 eV) was used to excite photoelectrons, with measurements taken at room temperature and a take-off angle of 90° relative to the surface plane. Core-level spectra were acquired with a pass energy of 20 eV (approximate resolution of 0.4 eV) over an analysis area of 300 × 700 μm2. The work function and binding energy scale were calibrated using the Fermi edge and Ag 3d5/2 peak from a polycrystalline silver standard. A low-energy electron beam charge neutraliser was employed to minimise surface charging, necessitating recalibration of the binding energy scale. Accordingly, the C–C/C–H component of the C 1s peak was referenced to 285.0 eV. Data analysis was performed with the CasaXPS software by using Shirley background subtraction and mixed Gaussian–Lorentzian (Voigt) peak fitting. The analyser transmission function was calibrated using clean metallic foils to ensure accurate detection efficiency across the full binding energy range.For the FT-IR in situ measurement 12.5 mg of Ce MOF was mixed with 30 μL of deionised water. This mixture was then transferred to a Bruker Vertex 70v FT-IR spectrometer fitted with a Si prism and a Pike technologies VeeMAX III mirror box. The in situ measurements were conducted using a 45° angle of incidence, with an acquisition time of 60 s between 4000–500 cm−1, with a 4 cm−1 resolution. The morphology of the MOFs was characterised by a Zeiss Supra 55-VP FESEM.
3D electron diffraction
For the 3D-ED experiment, the sample was dispersed dry onto a copper-supported holey amorphous carbon TEM grid and loaded at 175 K via a high-tilt Gatan Elsa specimen holder into a Rigaku XtaLAB Synergy-ED electron diffractometer, operated at 200 kV and equipped with a Rigaku HyPix-ED hybrid pixel array area detector. Data were collected on various crystallites of size ∼0.5–1.0 μm in their dimensions as single-rotation scans collecting 0.25° frames using CrysAlisPRO system (CCD 1.171.43.97a 64-bit (release 21-11-2023))18 using continuous rotation electron diffraction with a selected area aperture of 2 μm apparent diameter. Further experimental details are provided in Table S1.All single crystal component datasets were individually indexed and integrated, prior to the merging of two suitable datasets for scaling using CrysAlisPRO (version 1.171.44.113a);18 no absorption corrections were applied. Images of the crystals from which these data are obtained are shown in Fig. S4. The structure was solved using ShelXT19 and refined using Olex2.refine in the kinematic approximation, applying an extinction correction to broadly account for multiple scattering, alongside judicious rejection of outlying reflections, as implemented in Olex2 (version 1.5-ac7-018, compiled 2025.08.06 svn.rb7424aed for Rigaku Oxford Diffraction, GUI svn.r7314)20,21 and using published scattering factors.22
All thermal parameters of Ce, FDCA, and DMA non-H atoms are refined anisotropically, while disorder in the DMF adducts resulted in various sites being refined isotropically. Hydrogen atoms were placed geometrically with neutron average bond distances and riding isotropic displacement parameters; CH3 hydrogens for two of the three DMF sites (all disordered) could not be refined stably and so are omitted from the final model in light of their negligible merit and import to the refinement and discussion. To improve resilience of the model to dynamical and twin effects, distance similarity restraints were placed across all chemically equivalent covalent bonds, with select further restraints places on the DMF moieties. A global rigid bond restraint and select isotropicity and proximal similarity restraints are further applied to improve the robustness of the displacement parameters, as well as select equivalent constraints at select DMF atoms. Complete experimental and refinement information are contained within the deposited CIF along with structure factors and embedded. RES file; CIF deposited in the CSD with Deposition Number 2463394.
Single crystal XRD
A suitable crystal was selected and mounted on an XtaLAB Synergy, Dualflex, HyPix-Arc 100 diffractometer. The crystal was kept at 100.15 K during data collection. The structure was solved with the ShelXT19 solution program using the intrinsic phasing method and by using Olex2 (ref. 21) as the graphical interface. The model was refined with ShelXL 2019/3 (ref. 23) using full matrix least-squares minimization on F2.Conflicts of interest
There are no conflicts to declare.Data availability
Supplementary information is available. See DOI: https://doi.org/10.1039/D5CE00689A.CCDC 2454321 (for UOW-2(Ce)) and 2463394 (for UOW-11) contain the supplementary crystallographic data for this paper.24a,b Other supporting data are available at https://wrap.warwick.ac.uk/192002/.
Acknowledgements
S. D. expresses gratitude to the University of Warwick for a Chancellor's International Scholarship. M. W. acknowledges financial support from the EPSRC-funded Warwick Analytical Science Centre (EP/V007688/1). The research benefited from the equipment made available by the University of Warwick Research Technology Platforms, and we thank the EPSRC for funding (EP/X014606/1, a National Electron Diffraction Facility for Nanomaterial Structural Studies). We thank Diamond Light Source for beamtime at I11 (Block Allocation Grant CY25166). We also thank Ahmad Hamadi for measuring SEM images of the samples.References
- H. Molavi, Coord. Chem. Rev., 2024, 527, 216405 CrossRef.
- Y. Li, T. Hao, Y.-P. Han, H.-Z. Li, Y. Yan, Q.-H. Li, S. Chen, F. Wang and J. Zhang, ACS Mater. Lett., 2025, 2337–2342 CrossRef CAS.
- F. Nouar, M. I. Breeze, B. C. Campo, A. Vimont, G. Clet, M. Daturi, T. Devic, R. I. Walton and C. Serre, Chem. Commun., 2015, 51, 14458–14461 RSC.
- M. Lammert, M. T. Wharmby, S. Smolders, B. Bueken, A. Lieb, K. A. Lomachenko, D. D. Vos and N. Stock, Chem. Commun., 2015, 51, 12578–12581 RSC.
- J. Gosch, D. M. Venturi, E. S. Grape, C. Atzori, L. Donà, F. Steinke, T. Otto, T. Tjardts, B. Civalleri, K. A. Lomachenko, A. K. Inge, F. Costantino and N. Stock, Inorg. Chem., 2023, 62, 5176–5185 CrossRef CAS PubMed.
- J. Jacobsen, A. Ienco, R. D'Amato, F. Costantino and N. Stock, Dalton Trans., 2020, 49, 16551–16586 RSC.
- J. Shen and Z. Fan, Spectrochim. Acta, Part A, 2023, 302, 123070 CrossRef CAS PubMed.
- J. Jacobsen, A. Ienco, R. D'Amato, F. Costantino and N. Stock, Dalton Trans., 2020, 49, 16551–16586 RSC.
- S. Das, G. Cibin and R. I. Walton, ACS Sustainable Chem. Eng., 2024, 12, 5575–5585 CrossRef CAS.
- A. C. Dreischarf, M. Lammert, N. Stock and H. Reinsch, Inorg. Chem., 2017, 56, 2270–2277 CrossRef CAS PubMed.
- A. D. Burrows, K. Cassar, R. M. W. Friend, M. F. Mahon, S. P. Rigby and J. E. Warren, CrystEngComm, 2005, 7, 548 RSC.
- R. A. Vazirov, S. Y. Sokovnin, V. G. Ilves, I. N. Bazhukova, N. Pizurova and M. V. Kuznetsov, J. Phys.: Conf. Ser., 2018, 1115, 032094 CrossRef.
- S. Das, J. Zhang, T. W. Chamberlain, G. J. Clarkson and R. I. Walton, Inorg. Chem., 2022, 61, 18536–18544 CrossRef CAS PubMed.
- X. Du, R. Fan, L. Qiang, P. Wang, Y. Song, K. Xing, X. Zheng and Y. Yang, Cryst. Growth Des., 2017, 17, 2746–2756 CrossRef CAS.
- F. Millange, M. Medina, N. Guillou, G. Férey, K. M. Golden and R. I. Walton, Angew. Chem., Int. Ed., 2010, 49, 763–766 CrossRef CAS PubMed.
- H. Dai, Y. Zhang, X. Jin, W. Yang, Y. Luo, K. Yang, Y. Fu and W. Xu, Inorg. Chem., 2024, 63, 18058–18072 CrossRef CAS PubMed.
- B. H. Toby and R. B. Von Dreele, J. Appl. Crystallogr., 2013, 46, 544–549 CrossRef CAS.
- Rigaku Oxford Diffraction, CrysAlisPro Software system, version 1.171.38.46, Rigaku Corporation, Oxford, UK, 2017 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- L. J. Bourhis, O. V. Dolomanov, R. J. Gildea, J. A. K. Howard and H. Puschmann, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 59–75 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- A. Saha, S. S. Nia and J. A. Rodríguez, Chem. Rev., 2022, 122, 13883 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- (a) CCDC 2454321: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2ncxnm; (b) CCDC 2463394: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2npcb2.
| This journal is © The Royal Society of Chemistry 2025 |