
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hierarchical iodine(I) complexes of nicotinamide†
Sharath
Mohan
,
Arto
Valkonen
 ,
Kari
Rissanen
and
Jas S.
Ward
*
,
Kari
Rissanen
and
Jas S.
Ward
*
Department of Chemistry, University of Jyvaskyla, 40014 Jyväskylä, Finland. E-mail: james.s.ward@jyu.fi
First published on 24th April 2025
Abstract
The incorporation of the 3-substituted pyridine derivative nicotinamide (1) into an iodine(I) complex gave the hierarchical iodine(I) complex, [I(nicotinamide)2]PF6 (3), synthesised through cation exchange with I2 of the respective silver(I) complex, [Ag(nicotinamide)2]PF6 (2). Complex 3 was characterised in both solution (1H, 1H–15N HMBC), and extensively in the solid state by single-crystal X-ray diffraction (SCXRD) due to the various polymorphs and solvates observed. As only the second example of a hierarchical halogen(I) complex, analysis of the different hydrogen-bonding networks present in the solid-state variants of 3 allowed the relationship between the halogen and hydrogen bonding to be explored in more depth. The presence of the hydrogen bonds in the non-solvated solid-state structure of 3 (3_NS) was found to impart increased resilience to H2O and acetic acid, all without diminishing its reactivity as an iodination reagent in comparison to Barluenga's reagent ([I(pyridine)2]BF4), as demonstrated via solution and mechanochemical reactivity studies with antipyrine.
Introduction
Halogen bonding, defined as the attraction between an electrophilic halogen atom and a neutral or anionic nucleophile,1 has proven itself a non-covalent interaction of great utility over the last twenty years, second only to the ubiquitous hydrogen bonding. With demonstrated applications toward generating functional materials (porous,2–5 magnetic,6–8 phosphorescent,9–11 responsive,12–18 ion recognition)17,19–24 and in organocatalysis,25–28 halogen bonding has proven itself an industrious field of study.Barluenga's eponymous reagent, [I(pyridine)2]BF4, a classic iodination and soft oxidation reagent that has a myriad of reported uses,29–31 belongs to the halogen bonding sub-field of halogen(I) chemistry. Halogen(I) complexes comprise a formally X+ (X = Cl, Br, I) ion stabilised by two Lewis bases (L) of the form [L–X–L]+. Owing to its origin as a σ-hole interaction,32 the stability of the resulting halogen(I) complexes generally follow the trend I > Br > Cl,33 and display a high degree of linearity in their halogen bonding. These properties have led to the recent slew of halogen-bonded organic frameworks (XOFs),2–5 analogous to coordination polymers,34 that have been reported.
The exploration of alternative iodination reagents to [I(pyridine)2]BF4 has been modest, which is to be expected given its remarkable utility.3,35 However, the potential advantages offered by heteroleptic halogen(I) complexes like chiral iodine(I) carboxylates or iodine(I) pnictogenates,36–39e.g., enantioselective organic transformations, cannot be overlooked.
The interplay of non-covalent interactions, such as cooperativity between halogen and hydrogen bonding,40–42 has already been appreciated across a myriad of disciplines. Hierarchical materials are ones whose structural elements themselves have structure,43 such that the presence of multiple different bonding types within a species can give rise to a hierarchy, or order of preference, amongst those interactions. The recent appearance of the first example of a hierarchical iodine(I) complex, [I(3-acetaminopyridine)2]PF6,44 indicated that the halogen bonding of the iodine(I) centre could be preserved whilst benefitting from the comparative flexibility of the hydrogen bonding present. This hierarchical iodine(I) complex was found to be capable of acting as an iodination reagent, but also demonstrated unprecedented reactivity toward common organic solvents like iPr2O and tBuOMe (Fig. 1),44,45 making it unsuited for general use. Nevertheless, hierarchical iodine(I) complexes represent the potential synergy of solid-state XOFs and soluble iodination reagents like Barluenga's reagent, by bridging this divide via their ability to construct hydrogen-bonded coordination polymers.46 The combination of these two extremes could potentially provide greater resilience to polar solvents like H2O, increasing their shelf life,35 counter-intuitively without losing or diminishing their reactivity as iodination reagents. These hydrogen-bonded halogen(I) reagents would also be conceptually compatible with the recent shift toward more environmentally-conscious mechanochemical practices that eschew the use of toxic solvents, which has been demonstrated to be viable for the field of halogen bonding.47
 | ||
| Fig. 1 The first example of a hierarchical iodine(I) complex, [I(3-acetaminopyridine)2]PF6, and its surprising reactivity toward tBuOMe and iPr2O.44,45 | ||
Results and discussion
For all its utility and H2O tolerance, Barluenga's reagent does have its weaknesses in that it degrades over time, especially in the presence of H2O.35 Therefore, there is merit in exploring alternative iodine(I) reagents that might have better shelf-lives, as long as the superb reactivity of Barluenga's reagent can also be replicated. Nevertheless, the incorporation of hydrogen bonding into iodine(I) complexes to provide improved longevity may be somewhat of a dichotomy: the newly introduced hydrogen-bond donors and acceptors would enable stabilising hydrogen-bond networks to form, but would also be expected to make the complex more hydrophilic and prone to attracting H2O as a consequence. However, the increased hydrophilicity from incorporating hydrogen-bond-capable functional groups would likely be negligible in the sense that the Barluenga-type [N–I–N]+ iodine(I) complexes are already charged salts suffering from the same drawback. Therefore, the introduction of such functional groups would predominantly have a positive impact toward the goal of increased longevity.Given that reactivity and stability are traditionally viewed to have an inverse correlation, with highly reactive compounds being highly unstable, and vice versa, it may at first seem counter-intuitive to increase the stability of a complex whilst preserving its reactivity. In comparison to their more even-keeled 4-substituted analogues,36,48–50 iodine(I) complexes incorporating 3-substituted pyridines are comparatively unwieldy with diminished stabilities.45 This is of course unsurprising, given that substitution at the 3-position would not resonance-enhance the Lewis basicity of a 3-substituted pyridine as with the 4-position. Therefore, the reactivity of an iodine(I) complex incorporating a 3-substituted pyridine could be relied upon to demonstrate iodination reactivity approaching or surpassing that of Barluenga's reagent, if only it could be preserved in the solid state to enable its long-term storage. To test the hypothesis, the 3-substituted pyridine, nicotinamide (pyridine-3-carboxamide; 1), was selected.
The iodine(I) complex, [I(1)2]PF6 (3; Fig. 2), was synthesised in a one pot reaction from the addition of 1 and AgPF6 in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry, followed by addition of one equivalent of I2. The 2-coordinate silver(I) intermediate, [Ag(1)2]PF6 (2),51 could also be isolated and converted to 3. The presence of the amide made the complex negligibly soluble in CH2Cl2, unfortunate given that CH2Cl2 has long been a staple of iodine(I) solution studies, and necessitated the use of polar solvents (MeCN, MeOH, acetone). The stronger polar solvents dioxane, N,N-dimethylformamide, and dimethylsulfoxide were found to immediately destroy the iodine(I) centre, as evidenced by the strong colours of the resulting solutions upon dissolving the colourless solid of 3.
:1 stoichiometry, followed by addition of one equivalent of I2. The 2-coordinate silver(I) intermediate, [Ag(1)2]PF6 (2),51 could also be isolated and converted to 3. The presence of the amide made the complex negligibly soluble in CH2Cl2, unfortunate given that CH2Cl2 has long been a staple of iodine(I) solution studies, and necessitated the use of polar solvents (MeCN, MeOH, acetone). The stronger polar solvents dioxane, N,N-dimethylformamide, and dimethylsulfoxide were found to immediately destroy the iodine(I) centre, as evidenced by the strong colours of the resulting solutions upon dissolving the colourless solid of 3.
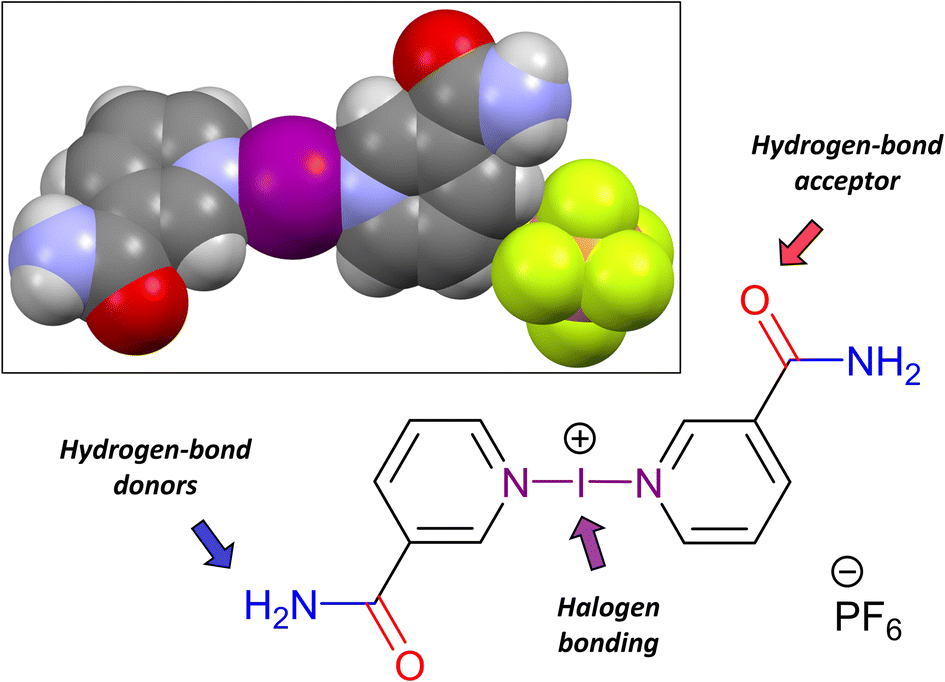 | ||
| Fig. 2 The hierarchical nature of 3, illustrating its capacity to form hydrogen bonds separate from the iodine(I) halogen bonding. Inset: Spacefill representation of 3_H2O. | ||
The 15N NMR spectroscopy studies (in CD3CN), determined via1H–15N HMBC experiments, demonstrated the expected coordination shifts (ΔN) of the pyridinic nitrogen chemical shifts progressively moving to lower frequency that were previously observed for other iodine(I) complexes.36,39,44,52,53 Only a small coordination shift of 8.2 ppm was observed for coordination of the free ligand 1 (−65.7 ppm) to form the silver(I) complex 2 (−73.9 ppm), though this was undoubtedly diminished due to the coordinating nature of the CD3CN to the silver(I) centre. Nevertheless, a much more characteristic coordination shift of 107.4 ppm was found between free ligand 1 and iodine(I) complex 3 (−173.1 ppm). The 1H NMR spectra were less informative, but the characteristic shifting of the peaks in 3 to higher frequencies compared to 1 was also a good indicator of the formation of an iodine(I) derivative.
The structure of 3 was definitively confirmed by single-crystal X-ray diffraction (SCXRD), with multiple polymorphs and solvates of 3 being observed in the solid state (Fig. 2, inset), originating from a variety of different solvents and anti-solvents, and by doping the crystallisations. The bond angles of 3 for all seven unique examples lied within the range 177.18(10)° to a symmetry-defined 180°, as expected for the linear-directed halogen bonding of the iodine(I) centre. This range was also comfortably within the range of angles previously reported for analogous [N–I–N]+ iodine(I) complexes (approximately 173.6–180°, excluding disordered or co-crystallised structures).54 Similarly, as would be expected for chemically-equivalent structures of 3, the I–N bond lengths were all found within the narrow range of 2.239(6)–2.278(6) Å (∼2.26 ± 0.02 Å), which was again within the known range for this value (2.22–2.32 Å, excluding disordered and legacy structures).54 Interestingly, excluding the two variants of 3 that possessed symmetry-equivalent ligands (3_H2O and 3·4(1)), all but one structure (3·3(MeCN)) demonstrated subtle asymmetry in their I–N bond lengths (Table S2†). Unfortunately, there are only seven prior solid-state examples of iodine(I) complexes incorporating 3-substituted pyridines, likely for the previously discussed reasons (vide supra), and only four examples that do not have symmetry-equivalent ligands: the two pairs of polymorphs for the iodine(I) complexes of 3-acetylpyridine and 3-acetaminopyridine;44,45 of those four examples, three demonstrate the same crystallographic asymmetry in their I–N bond lengths (Table S2†).
For these hierarchical iodine(I) complexes incorporating nicotinamide, what is perhaps most germane to the solid-state analysis is that of the hydrogen-bonded networks that are present. To streamline this discussion, the use of graph set notation will be used (Table S2†); an excellent summary of this notation (including worked examples) has been previously published for those unfamiliar with it or simply needing a refresher.55 The graph set notation of a hydrogen-bond interaction is of the general form, Gad(n), where G is the pattern designator (e.g., R = ring, C = chain, D = finite pattern), a and d are the number of hydrogen-bond acceptors and donors involved, respectively, and n is the degree of the pattern and shows the number of atoms involved.
Most of the solid-state structures of 3 revolve around the classic R22(8) motif (Fig. 3), which was present in five of the seven unique structures, and its reliable formation was one of the factors in selecting nicotinamide (1) with its C(O)NH2 substituent for this study. For the two structures that did not contain the R22(8) motif, they both instead assumed 1D-chains intermolecularly either along the length of the cation as in 3_NS (C(12); Fig. 4), or simply through the C(O)NH2 group only as in 3·acetone (C(4); or if viewed as a dimer, R22(24); Fig. 4). N.B. as per convention, the values of a and d are omitted if they equal 1, hence C11(12) is written as C(12) herein, and similarly the value of n is omitted if it equals 2 for a finite pattern (where G = D), therefore D11(2) is simply written as D. There is also the possibility of assigning ever-increasing hydrogen-bonded ring systems via the graph set notation, but this is counter-productive in this instance, so only the smallest unique ring systems have been assigned.
 | ||
| Fig. 3 The hydrogen bonding motifs observed for the C(O)NH2 substituents of the [I(1)2]+ cations in the various solid-state structures of 3 (hydrogen bonds as red dashed lines), annotated with their graph set notations. | ||
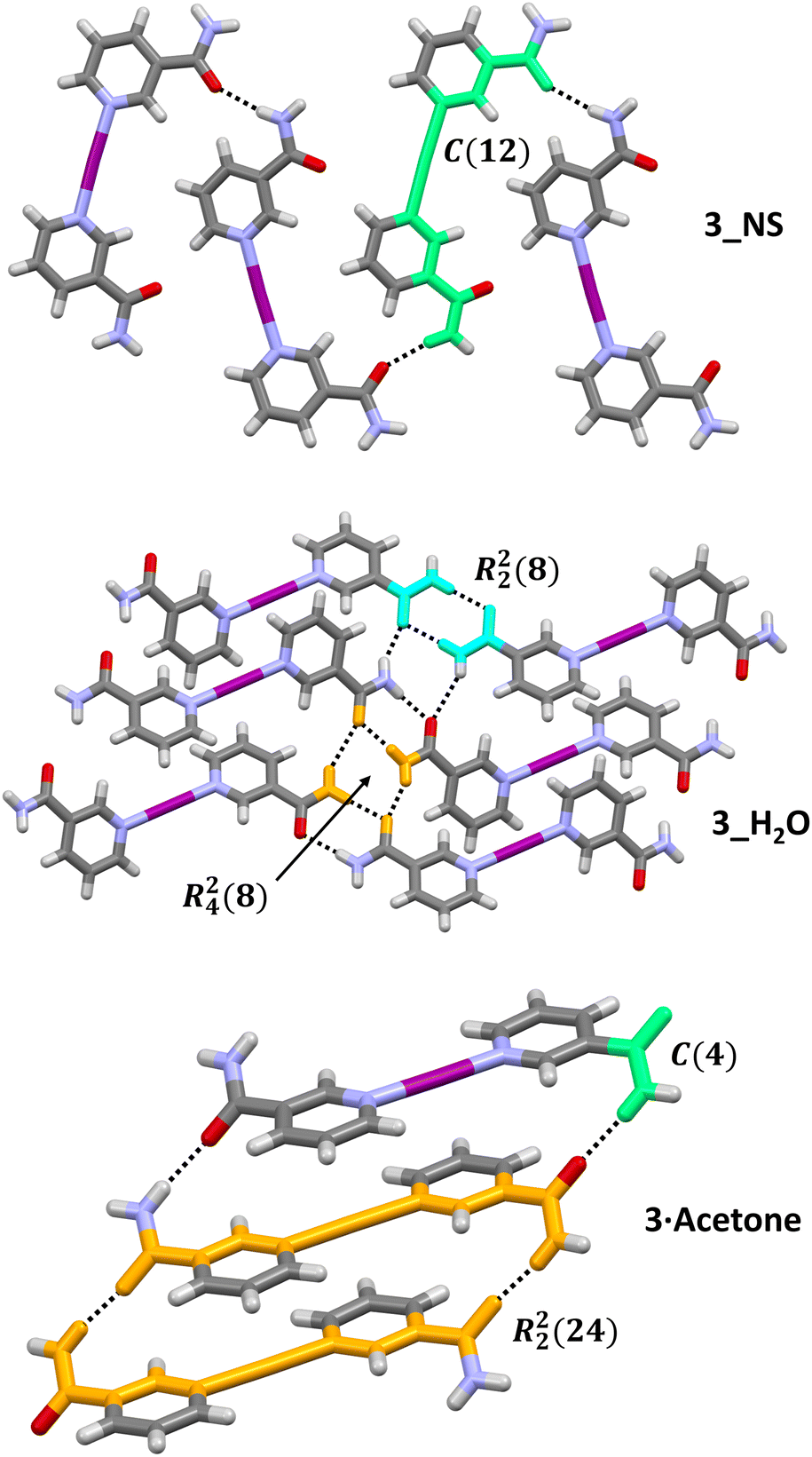 | ||
| Fig. 4 The hydrogen bonding networks (hydrogen bonds as black dashed lines), annotated with their graph set notations, formed by 3_NS, 3_H2O, and 3·acetone (anions and solvates have been omitted for clarity). Relevant fragments for the graph set notations have been block coloured for emphasis: chains (C) = green, rings (R) = turquoise and orange. | ||
The hydrogen-bonding motifs found for 3_NS and 3·acetone (Fig. 4) mirror those observed for the two different polymorphs of [I(acetaminopyridine)2]PF6, which were found to be in pseudo-syn and anti configurations in the solid state. Despite their apparent differences in hydrogen bonding and some of the structures also having solvates or co-crystals present, the structures all demonstrated hydrogen bonds of comparable strength. This was determined via the distances between the non-hydrogen donor (D) and acceptor (A) atoms involved in the hydrogen bonding, the positions of which (unlike hydrogen atoms) can be accurately determined by SCXRD. The intermolecular D(H)⋯A distances (Å) for all the non-guest (i.e., excluding solvates and co-crystallised guests) hydrogen bonds were found to lie within the range 2.849(2)–2.974(2) Å (Table S2†), with the exception of the slightly longer distance of 3.142(2) Å observed between two 1D-chains of C(O)NH2 dimers (bonded in an R24(8) motif) in 3_H2O (Fig. 4).
A few interesting features emerged from analysing the seven crystallographically unique structures of 3 as a whole. The first was that all the solvated/co-crystallised structures of 3 were observed to be effectively co-planar with respect to the pair of Lewis bases, with the C(O)NH2 groups assuming an anti-arrangement. In contrast, the pair of Lewis bases in the two polymorphs of 3 with no solvates present were both found to be non-co-planar with angles between their planes of 21.9° (3_NS) and 43.8° (3_H2O), with their C(O)NH2 groups in pseudo-syn and pseudo-anti arrangements, respectively. These observations might suggest that the presence of solvates allows the preferred co-planar configuration for the pair of Lewis bases, but in the absence of a suitable solvated structure, 3 is compelled to diverge from this to construct a suitable hydrogen-bonding network in the solid state. The second observation was the deviation from planarity of the C(O)NH2 substituent to the pyridyl ring. The majority of the structures displayed negligible or minor deviations from planarity (<15°) in this respect, however, 3_H2O and 3·acetone both demonstrated larger deviations of 18.5° and 39.6/41.8°, respectively, which were essential to the formation of their hydrogen-bonding networks (Fig. 5).
 | ||
| Fig. 5 The view along the pyridyl–C(O)NH2 bond for 3_H2O and 3·acetone, annotated with the deviations of the C(O)NH2 substituents (relevant C(O)NH2 groups block coloured in turquoise for emphasis) from the plane of their respective pyridyl rings (anions and solvates omitted for clarity). | ||
This was similarly observed in the hydrogen bonding networks of 2, which have been previously discussed in detail by Aakeröy & Beatty, where the effects of anion substitution were also explored for the [Ag(1)2]+ cation.51 Three additional solvated structures of 2 were observed in the solid state and included herein for completeness: 2·2(MeCN), 2·3(MeCN), and 2·acetone; these silver(I) complexes demonstrated the same R22(8) motif between the C(O)NH2 groups of adjacent [Ag(1)2]+ cations, with all but one of the solvates, one of the three MeCN solvates in 2·3(MeCN), preferring to weakly coordinate to the silver(I) centre instead of hydrogen bonding to NH2. These solvated structures, though analogues of one another, were different from the non-solvated structure of 2 previously reported,51 which instead formed a 2D network comprised of C(4) chains and R44(32) rings in its respective hydrogen bonding of the C(O)NH2 group.
The hydrogen-bond networks of 3 actually proved themselves quite robust, such that the stoichiometric addition of hydrogen-bond-capable dopants to the crystallisations all failed to effect any changes, with the exception of 2-pyrrolidone. Doping with 2-pyrrolidone yielded the solid-state structure 3·0.65(MeCN), which contained a partially-occupied (65%) MeOH and other unknown solvates that ultimately had to be accounted for using Squeeze within Platon.56 The attempt to incorporate 1 as both the Lewis base and guest, via the use of 6 equivalents of 1 instead of 2, also found some success; the additional equivalents of 1 did co-crystallise with 3, but with no meaningful intermolecular interactions being observed between the two separate hydrogen-bonded networks (Fig. 6).
 | ||
| Fig. 6 The hydrogen-bonding networks formed by 3·4(1) (top; hydrogen bonds as black dashed lines; annotated with graph set notations; block coloured in turquoise and orange for emphasis; anions omitted for clarity). The packing of 3·4(1) (bottom), block coloured in blue (3) and orange (1), showing the two separate hydrogen-bonding networks present in alternating layers. Inset: The view along the crystallographic c-axis of 3·4(1) showing the separate layers formed by 3 (blue) and 1 (orange) in 3·4(1). | ||
Hierarchical halogen(I) complexes are perhaps best viewed as alternatives to halogen-bonded organic frameworks (XOFs), in that they offer many of the same advantages, such as increased stabilities due to the 3-dimensional nature of their extended solid-state structures, but without some of the drawbacks. Drawbacks of XOFs, like poor solubility, their reliance on multi-dentate ligands which are not straightforward to tailor the properties of,4,5 elevated temperatures and an inert atmosphere to enable their use as iodination reagents,3 or an inability to purify and crystallise their structures in the solid state,2,3 are generally not shared by halogen(I) complexes, hierarchical or otherwise.44,45,57
The original concept of preparing more resilient iodine(I) reagents was tested via the crystallisation of 3 using H2O or acetic acid as the anti-solvents, solvents which would readily degrade Barluenga's reagent. These crystallisations successfully yielded either the same solid-state structure previously observed using Et2O as the anti-solvent (3·MeCN_H2O), or a new polymorph (3_H2O) that again retained the iodine(I) moiety. However, grinding the crystals of 3_NS in a small amount of saturated aqueous NaHCO3 solution did still cause the decomposition of the iodine(I) within a few minutes.
The reactivity of 3 was also probed, via the iodination of antipyrine, to determine if the increased resilience of 3 came at the cost of its diminished reactivity both in solution and the solid state. To investigate this, the non-solvated structure of 3_NS (reliably prepared from MeOH/Et2O) was utilised to ensure co-crystallised solvates were not affecting the results.
Reactions were repeated in triplicate following prior protocols for screening iodine(I) reagents in solution,35,39,58 or via mechanochemical grinding of 3_NS and antipyrine together, followed by a modified procedure to ensure quenching of unreacted 3 as a solid prior to dissolution during the subsequent aqueous work-up (ESI,† page S16).47 The average percentage yield of iodo-antipyrine of 92% in solution and 91% in the solid state, were both excellent. More importantly, these yields were effectively identical to those reported for Barluenga's reagent in solution using the same protocol, both without isolation of the product (>95% as determined by gas chromatography),58 or where the product was also isolated and its purity confirmed by 1H NMR spectroscopy (93%).35 These yields indicate that the reactivity of 3 has, despite its increased resilience to solvents like H2O, not been diminished whatsoever in comparison to the current gold-standard benchmark for iodination reagents, [I(pyridine)2]BF4.
Conclusions
In conclusion, the hierarchical iodine(I) complex [I(nicotinamide)2]PF6 (3), featuring a combination of halogen and hydrogen bonding through the incorporation of nicotinamide (1) as the stabilising Lewis base, was synthesised and characterised via NMR spectroscopy (1H and 1H–15N HMBC) and single-crystal X-ray diffraction (SCXRD). A variety of hydrogen-bonded motifs were demonstrated by the C(O)NH2 substituents of 3, yielding seven crystallographically unique solid-state structures, both with and without solvates, all with comparable halogen bonding. The various hydrogen-bonding motifs were distinguished and discussed using graph set notation, with five of the seven structures of 3 featuring the symmetric R22(8) motif. Despite the apparent differences in the packing of 3, the hydrogen bonding was found to be of comparable strength in all variants. The increased resilience of 3 in comparison to the benchmark iodination reagent, [I(pyridine)2]BF4 (Barluenga's reagent), was confirmed via its successful crystallisation even when H2O or acetic acid were used as the anti-solvents. Counter-intuitively, though by design, the increased resilience of 3 did not come at the cost of its reactivity as an iodination reagent, as confirmed via the iodination of antipyrine. The resulting percentage yields of the iodo-antipyrine product when utilising 3 as the iodine(I) reagent (both in solution and mechanochemically in the solid state via its non-solvated crystalline form, 3_NS), in comparison to when [I(pyridine)2]BF4 was used, were nearly identical (within 1–2%). These reactivity studies highlight 3 remains a potent iodination reagent, despite the increased solid-state resilience it possesses. This work demonstrates that careful design of the stabilising Lewis bases in halogen(I) complexes can offer dramatic and measurable improvements, even to already exceptional halogen(I) complexes such as Barluenga's reagent, and that further systematic studies are indeed warranted.Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for all crystal structures have been deposited at the Cambridge Crystallographic Data Centre (CCDC 2433034–2433043).Author contributions
SM performed the reactivity studies. JSW performed the syntheses and crystallographic studies, and was responsible for the conceptualisation, visualisation, and validation of the data. AV and JSW conceived the study. JSW wrote the original draft of the manuscript, and all authors contributed to the reviewing and editing process. KR and JSW acquired the funding.Conflicts of interest
There are no conflicts to declare.Acknowledgements
K. R. and J. S. W. gratefully acknowledge the Research Council of Finland (formerly the Academy of Finland, grant numbers 351121 and 356187, respectively) for funding.Notes and references
- G. R. Desiraju, P. S. Ho, L. Kloo, A. C. Legon, R. Marquardt, P. Metrangolo, P. Politzer, G. Resnati and K. Rissanen, Pure Appl. Chem., 2013, 85, 1711–1713 CrossRef CAS
.
- G. Gong, S. Lv, J. Han, F. Xie, Q. Li, N. Xia, W. Zeng, Y. Chen, L. Wang, J. Wang and S. Chen, Angew. Chem., Int. Ed., 2021, 60, 14831–14835 CrossRef CAS PubMed
- N. Xia, J. Han, F. Xie, G. Gong, L. Wang, J. Wang and S. Chen, ACS Appl. Mater. Interfaces, 2022, 14, 43621–43627 CrossRef CAS PubMed
- X. Bai, Z. Tian, H. Dong, N. Xia, J. Zhao, P. Sun, G. Gong, J. Wang, L. Wang, H. Li and S. Chen, Angew. Chem., Int. Ed., 2024, 63, e202408428 CrossRef CAS PubMed
- Q. Zhao, S. Lin, P. Sun, Y. Lu, Q. Li, Z. Tian, X. Bai, J. Wang, L. Wang and S. Chen, Adv. Funct. Mater., 2025, 2421755 CrossRef
- M. Fourmigué and P. Batail, Chem. Rev., 2004, 104, 5379–5418 CrossRef PubMed
- L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118–7195 CrossRef CAS PubMed
- S. Saha, S. Khamrui and K. Biradha, J. Am. Chem. Soc., 2024, 146, 26556–26566 CrossRef CAS PubMed
- L. Xiao, Y. Wu, J. Chen, Z. Yu, Y. Liu, J. Yao and H. Fu, J. Phys. Chem. A, 2017, 121, 8652–8658 CrossRef CAS PubMed
- Z. Yang, C. Xu, W. Li, Z. Mao, X. Ge, Q. Huang, H. Deng, J. Zhao, F. L. Gu, Y. Zhang and Z. Chi, Angew. Chem., Int. Ed., 2020, 59, 17451–17455 CrossRef CAS PubMed
- J. Zhou, L. Stojanović, A. A. Berezin, T. Battisti, A. Gill, B. M. Kariuki, D. Bonifazi, R. Crespo-Otero, M. R. Wasielewski and Y.-L. Wu, Chem. Sci., 2021, 12, 767–773 RSC
- L. Maugeri, E. M. G. Jamieson, D. B. Cordes, A. M. Z. Slawin and D. Philp, Chem. Sci., 2017, 8, 938–945 RSC
- H. Wang, H. K. Bisoyi, L. Wang, A. M. Urbas, T. J. Bunning and Q. Li, Angew. Chem., Int. Ed., 2018, 57, 1627–1631 CrossRef CAS PubMed
- H. Wang, H. K. Bisoyi, B.-X. Li, M. E. McConney, T. J. Bunning and Q. Li, Angew. Chem., Int. Ed., 2020, 59, 2684–2687 CrossRef CAS PubMed
- A. Docker, X. Shang, D. Yuan, H. Kuhn, Z. Zhang, J. J. Davis, P. D. Beer and M. J. Langton, Angew. Chem., Int. Ed., 2021, 60, 19442–19450 CrossRef CAS PubMed
- A. Kerckhoffs, I. Moss and M. J. Langton, Chem. Commun., 2023, 59, 51–54 RSC
- N. Kim, V. S. Jeyaraj, J. Elbert, S. J. Seo, A. V. Mironenko and X. Su, JACS Au, 2024, 4, 2523–2538 CrossRef CAS PubMed
- Q. Jin, Y. Hai, L.-J. Liu, T.-G. Zhan and K.-D. Zhang, Org. Chem. Front., 2025, 12, 1217–1226 RSC
- M. S. Taylor, Coord. Chem. Rev., 2020, 413, 213270 CrossRef CAS
- A. Docker, C. H. Guthrie, H. Kuhn and P. D. Beer, Angew. Chem., Int. Ed., 2021, 60, 21973–21978 CrossRef CAS PubMed
- R. Hein and P. D. Beer, Chem. Sci., 2022, 13, 7098–7125 RSC
- H. M. Tay, Y. C. Tse, A. Docker, C. Gateley, A. L. Thompson, H. Kuhn, Z. Zhang and P. D. Beer, Angew. Chem., Int. Ed., 2023, 62, e202214785 CrossRef CAS PubMed
- K. M. Bąk, S. C. Patrick, X. Li, P. D. Beer and J. J. Davis, Angew. Chem., Int. Ed., 2023, 62, e202300867 CrossRef PubMed
- Y. C. Tse, A. Docker, I. Marques, V. Félix and P. D. Beer, Nat. Chem., 2025, 17, 373–381 CrossRef CAS PubMed
- H. Nakatsuji, Y. Sawamura, A. Sakakura and K. Ishihara, Angew. Chem., Int. Ed., 2014, 53, 6974–6977 CrossRef CAS PubMed
- Y. Lu, H. Nakatsuji, Y. Okumura, L. Yao and K. Ishihara, J. Am. Chem. Soc., 2018, 140, 6039–6043 CrossRef CAS PubMed
- A. C. Keuper, K. Fengler, F. Ostler, T. Danelzik, D. G. Piekarski and O. García Mancheño, Angew. Chem., Int. Ed., 2023, 62, e202304781 CrossRef CAS PubMed
- D. Pal, T. Steinke, L. Vogel, E. Engelage, S. Heinrich, D. Kutzinski and S. M. Huber, Adv. Synth. Catal., 2023, 365, 2718–2723 CrossRef CAS
- J. Barluenga, J. M. González, M. A. Garcia-Martin, P. J. Campos and G. Asensio, J. Chem. Soc., Chem. Commun., 1992, 1016–1017 RSC
- J. Ezquerra, C. Pedregal, C. Lamas, J. Barluenga, M. Pérez, M. A. García-Martín and J. M. González, J. Org. Chem., 1996, 61, 5804–5812 CrossRef CAS
- G. Espuña, G. Arsequell, G. Valencia, J. Barluenga, M. Pérez and J. M. González, Chem. Commun., 2000, 1307–1308 RSC
- T. Clark, WIREs Comput. Mol. Sci., 2013, 3, 13–20 CrossRef CAS
- J. Pancholi and P. D. Beer, Coord. Chem. Rev., 2020, 416, 213281 CrossRef CAS
- B. F. Hoskins and R. Robson, J. Am. Chem. Soc., 1990, 112, 1546–1554 CrossRef CAS
- L. M. E. Wilson, K. Rissanen and J. S. Ward, New J. Chem., 2023, 47, 2978–2982 RSC
- J. S. Ward, G. Fiorini, A. Frontera and K. Rissanen, Chem. Commun., 2020, 56, 8428–8431 RSC
- S. Yu and J. S. Ward, Dalton Trans., 2022, 51, 4668–4674 RSC
- M. Mattila, K. Rissanen and J. S. Ward, Chem. Commun., 2023, 59, 4648–4651 RSC
- S. Mohan, K. Rissanen and J. S. Ward, Commun. Chem., 2024, 7, 159 CrossRef CAS PubMed
- A. S. Mahadevi and G. N. Sastry, Chem. Rev., 2016, 116, 2775–2825 CrossRef CAS PubMed
- R. K. Rowe and P. S. Ho, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2017, 73, 255–264 CrossRef CAS PubMed
- D. Givaudan, B. Biletskyi, A. Recupido, V. Héran, O. Chuzel, T. Constantieux, J.-L. Parrain and X. Bugaut, Eur. J. Org. Chem., 2024, 27, e202300261 CrossRef CAS
- R. Lakes, Nature, 1993, 361, 511–515 CrossRef
- J. S. Ward, CrystEngComm, 2022, 24, 7029–7033 RSC
- K. Rissanen and J. S. Ward, ACS Omega, 2023, 8, 24064–24071 CrossRef CAS PubMed
- Q. Shi, X. Zhou, W. Yuan, X. Su, A. Neniškis, X. Wei, L. Taujenis, G. Snarskis, J. S. Ward, K. Rissanen, J. de Mendoza and E. Orentas, J. Am. Chem. Soc., 2020, 142, 3658–3670 CrossRef CAS PubMed
- C. Schumacher, K.-N. Truong, J. S. Ward, R. Puttreddy, A. Rajala, E. Lassila, C. Bolm and K. Rissanen, Org. Chem. Front., 2024, 11, 781–795 RSC
- A.-C. C. Carlsson, K. Mehmeti, M. Uhrbom, A. Karim, M. Bedin, R. Puttreddy, R. Kleinmaier, A. A. Neverov, B. Nekoueishahraki, J. Gräfenstein, K. Rissanen and M. Erdélyi, J. Am. Chem. Soc., 2016, 138, 9853–9863 CrossRef CAS PubMed
-
J. S. Ward, K.-N. Truong, M. Erdélyi and K. Rissanen, in Comprehensive Inorganic Chemistry III, ed. J. Reedijk and K. R. B. T. Poeppelmeier, Elsevier, Oxford, 3rd edn, 2023, pp. 586–601 Search PubMed
- J. S. Ward, E. I. Sievänen and K. Rissanen, Chem. – Asian J., 2023, 18, e202201203 CrossRef CAS PubMed
- C. B. Aakeröy and A. M. Beatty, Chem. Commun., 1998, 1067–1068 RSC
- D. von der Heiden, K. Rissanen and M. Erdélyi, Chem. Commun., 2020, 56, 14431–14434 RSC
- S. Yu, J. S. Ward, K.-N. Truong and K. Rissanen, Angew. Chem., Int. Ed., 2021, 60, 20739–20743 CrossRef CAS PubMed
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed
- J. Bernstein, R. E. Davis, L. Shimoni and N.-L. Chang, Angew. Chem., Int. Ed., 1995, 34, 1555–1573 CrossRef CAS
- A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9–18 CrossRef CAS PubMed
- P. Kumar, J. M. Rautiainen, J. Novotný, J. S. Ward, R. Marek, K. Rissanen and R. Puttreddy, Chem. – Eur. J., 2024, 30, e202303643 CrossRef CAS PubMed
- P. J. Campos, J. Arranz and M. A. Rodríguez, Tetrahedron Lett., 1997, 38, 8397–8400 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, NMR characterisation, and SCXRD. (CCDC 2433034–2433043). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5ce00339c |
| This journal is © The Royal Society of Chemistry 2025 |