
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The pancake bond: on the border of covalent and intermolecular†
Krešimir
Molčanov
 *a and
Petra
Stanić
ab
*a and
Petra
Stanić
ab
aRuđer Bošković Institute, Bijenička 54, HR-10000 Zagreb, Croatia. E-mail: kmolcano@irb.hr
bIstrian University of Applied Sciences, Riva 4, 52100 Pula, Croatia
First published on 11th June 2025
Abstract
The two-electron multicentre (2e/mc) bond or pancake bond, a weak π-bond occurring between planar π-based organic radicals, is described and discussed. Its importance in chemistry (nature of the chemical bond) and materials science (its application in design of radical-based functional materials) is emphasized: the latest developments, concepts, and new directions of research; challenges and possible pitfalls are discussed.
Introduction
The two-electron multicentre (2e/mc) bond or pancake bond is an interaction which occurs between planar organic π-based radicals,1–4 and is geometrically similar to stacking of closed-shell aromatic and nonaromatic rings.3–8 However, stacking of two (or more) π-based open-shell electronic systems involves interaction of electronic spins (magnetic exchange or coupling of spins),9 a very strong and usually attractive component of total interaction, which is absent in closed-shell systems. Other components involve dispersion, (local) dipolar and electrostatic interactions.2,4 Pairing of spins implies mixing of molecular π orbitals, and quantum chemical models indicate a considerable covalent component, which may exceed −15 kcal mol−1 (ref. 1, 2 and 10) (Fig. 1). This component makes the pancake bond considerably stronger than stacking of closed-shell rings; in fact, its energy is comparable to the strongest noncovalent interactions, hydrogen bonds and halogen bonds,3,4 which are known to have a partial covalent character.11,12 These three interactions can be considered to have a dual character, both covalent and non-covalent, and thus occupy a ‘grey zone’ between intramolecular (chemical bond) and intermolecular (supramolecular) worlds (Table 1). What puts the pancake bond apart from the other two interactions is the distribution of the bonding electron pair: it is not localised, but dispersed between multiple atoms of two contiguous rings.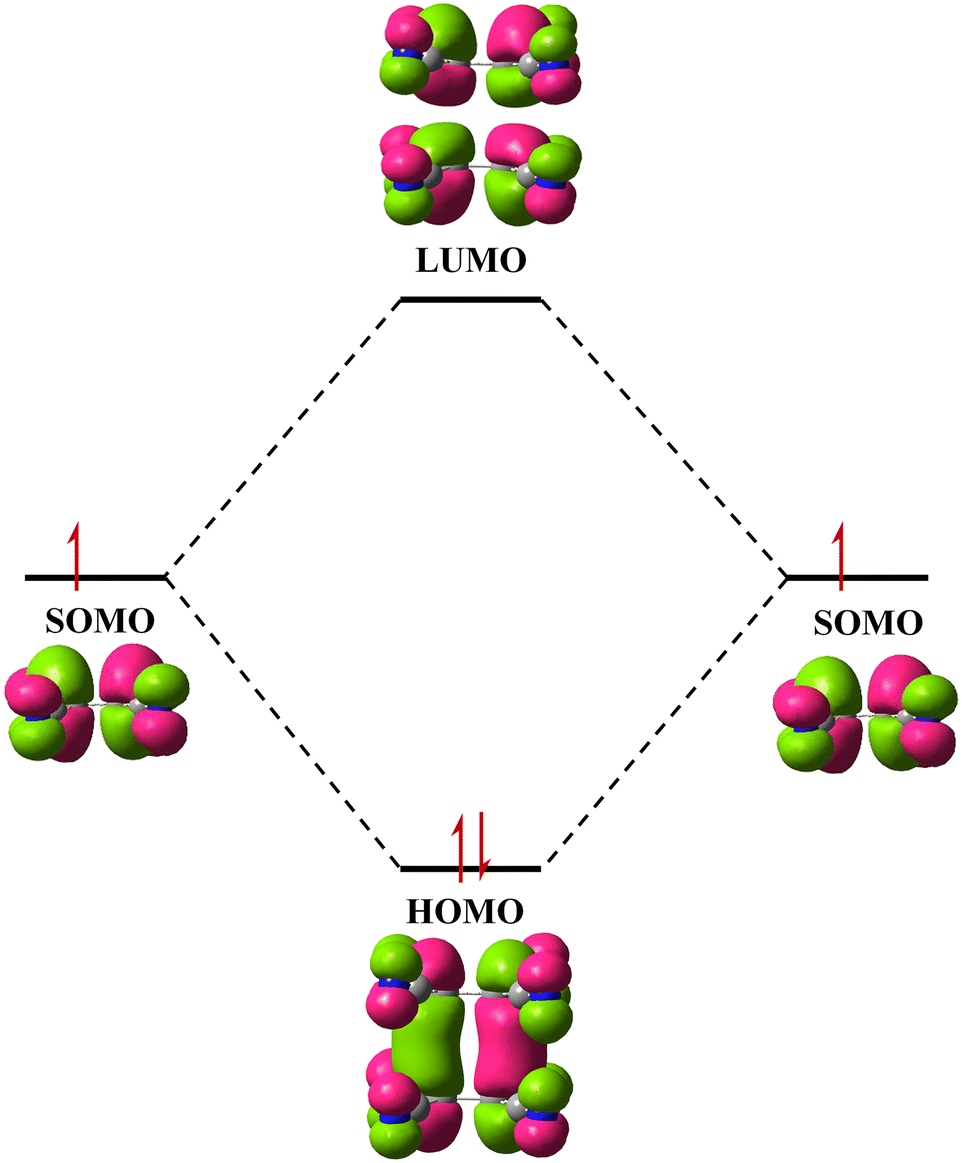 | ||
| Fig. 1 A schematic representation of combination of two SOMOs of two isolated TCNE˙− radicals into a HOMO of a dimer. | ||
| Hydrogen bond | Halogen bond | Pancake bond | |
|---|---|---|---|
| E/kcal mol−1 | >10 | >10 | >10 |
| Directionality | Strong | Strong | Strong |
| Localisation | Localised | Localised | Not localised |
| Interesting properties | Proton transfer, ferroelectricity | Halogen transfer | Charge transfer, magnetism, conductivity |
| Common structural motif | Low-barrier hydrogen bond, Zundel ion, HF⋯HF | Halonium ions | Radical dimers and trimers, equidistant radicals, charge-transfer compounds |
Multicentre bonds with multiple atoms sharing a single electron pair are relatively well known; they are common in boranes13 and metal clusters.14 However, these compounds involve short atom–atom distances which can be interpreted as chemical bonds, while in pancake bonding, interatomic distances are much longer and are usually interpreted as close intermolecular contacts.
The colourful term ‘pancake bond’ was first coined by Mulliken in the 1960s,15 to denote then-unspecified attractive interactions occurring between large planar molecules, such as porphyrines,16,17 which form stacks similar to stacks of pancakes (Fig. 2). However, research soon moved to stacking of small aromatic compounds in the solid state,‡ which became known under a misleading name ‘π–π interactions’.§ The term pancake bond remained seldom used, until Fukui et al. used it to describe interactions between π-based planar radicals.18–20 As solid-state studies of stable radicals gained popularity due to possible applications in organic magnets,9,10,21–25 electronics10,26–28 or optoelectronics,29–31 so did the name of the little-studied interaction.
 | ||
| Fig. 2 A stack of pancakes resembling a stack of planar radicals. The electrons act similarly to jam in the figure, holding the radicals together. Pancakes and photo by P. Stanić. | ||
The most intensively studied prototype of pancake-bonded systems is a dimer of tetracyanoethylene (TCNE) radical anions.32–36 This dimer is characterised by unusually short C⋯C distances of <2.9 Å. Many of its salts have bulk diamagnetic properties, which indicates paired spins. Novoa and Miller33 showed that two unpaired electrons from two contiguous TCNE radicals form a bonding pair, thus forming a weak 4-centre two-electron π-bond (Fig. 1). Such a picture is consistent with both bulk diamagnetism and the existence of close dimers. This type of weak π-bond somewhat stretches the ‘classical’ definition of the chemical bond.37 Since TCNE is an acyclic molecule, the use of the term ‘pancake bonding’ should be extended to all planar radicals, not only to rings.
Pancake-bonded systems of π-based radicals involve stacking with very short intermolecular distances. Typical interplanar separations and close atom–atom distances are considerably shorter than the sum of van der Waals radii (C⋯C distances are in almost all cases shorter than 3.30 Å), and the molecular mean planes are parallel or nearly parallel.2–4 Also, the arrangement of the radicals is such to maximize orbital overlap; they most often stack face-to-face or have relatively small offsets to minimise electrostatic repulsion.3,4,38 An interesting feature of pancake bonded systems in the solid state is bulk diamagnetic or antiferromagnetic properties, which are a result of spin coupling.3,4 However, the same compounds in the amorphous state or in solution (lacking long-range order) produce strong EPR signals, indicating unpaired electrons. Collapse of the crystal structure (for example, by heating of the crystals) is also followed by a sudden increase of magnetic susceptibility, due to uncoupling of the electrons.
Some very stable radicals, such as 7,7,8,8-tetracyanoquinodimethane (TCNQ)39 and variously substituted dithiadiazolyls (DTDA),40 are present in quite a large number of published crystal structures, allowing a more thorough statistical database survey.40
This short review provides a highlight in the importance of pancake bonding in chemistry and (increasingly) in materials science. The emphasis is on the latest developments, concepts and new directions of research; challenges and possible pitfalls are also discussed. For a more detailed review, describing the very concept of pancake bonding, and more thorough theoretical description, the reader is pointed to a recent review by Kertesz.2
Pancake bond order
The prototype of the pancake bond is a dimer of closely interacting radicals, which is found in numerous compounds, such as neutral phenalenyls41,42 and dithiadiazolyls,10,40,43 cations such as perylene38 and fused-ring acenes,44 and anions such as semiquinones45 and TCNQ.39 When two radicals, each with a single unpaired electron, make close contact, these two electrons couple, forming a single bonding pair (Fig. 1). This corresponds to a single covalent bond. However, there are not necessarily two bonding electrons in a pancake bond.Kertesz defined formal pancake bond order as “the number of SOMO-based bonding electron pairs (minus antibonding pairs, if any) in the dimers”2 or more simply as 1/2(Nbonding electrons − Nantibonding electrons).41 Therefore, if there are two or more (nearly) degenerate SOMOs, two or more (nearly) degenerate HOMOs are formed, resulting in double or multiple pancake bonds (Fig. 3). Such bonds were found in triangulene radicals;2,41,46,47 however, they are not necessarily stronger than single ones due to increased Pauli repulsion (due to the shortening of the contact distances), which offsets higher SOMO–SOMO interaction energy.2
 | ||
| Fig. 3 A schematic representation of orbitals in a double pancake bond: each biradical has two degenerate SOMOs which combine into two bonding HOMOs. | ||
There are multiple examples of pancake bonds with an order lower than 1, many of which have been experimentally studied within the last several years. Dimers of a neutral phenalenyl (PLY) radical and its closed-shell cation have only a single bonding electron, and therefore its bond order is 1/2 (ref. 2 and 48) (Fig. 4a). This type of open-shell dimer is sometimes referred to as a π-mer49,50 and a half-pancake bond.51 Another example is a close contact between a TCNQ radical anion and a neutral TCNQ molecule found in its salt with 1,4-dimethyl-1,4-diazabicyclo[2.2.2]octanium (dabco)52 (Fig. 4b), which has two bonding electrons and one antibonding electron. A bond order of 1/2 is also found in tetramers of TCNQ radical anions with a formal charge of −1/2:52,53 such tetramers share two bonding electrons (and thus have a total charge of −2). Trimers of semiquinone54 and TCNQ radical anions39,52,53 have a total charge of −2 (formal charge of a single radical moiety is −2/3) and therefore a bond order of 2/3.
 | ||
| Fig. 4 A schematic representation of orbitals in a bond with an order of 1/2: a) a single bonding electron in a dimer of PLY·PLY+,48 and b) two bonding electrons and one antibonding electron in a close contact of TCNQ·TCNQ˙−.51 | ||
Another definition of “intermolecular bond order” (IBO) was proposed by Hernández-Trujillo45 as a sum of bond orders (BOs) between all nonequivalent pairs of atoms in each molecule. The BOs are calculated using (i) the delocalization indices defined by the Bader space partition (QTAIM)55 and (ii) the Wiberg indices based on the natural atomic orbital (NAO) analysis.56,57 The IBO is then computed as

There are also extended π-systems with multiple rings and multiple unpaired electrons, such as hexaazatrinaphthylene triradical anions.58 Such radicals are capable of forming two or more pancake bonds. However, these are not double or triple pancake bonds, but two or three separate single ones.
It should be noted that the charge of the radicals does not affect the bond order. However, it affects the total energy of the pancake bond, since stacking of two radicals of the same charge creates strong electrostatic repulsion. Nevertheless, in crystals of the charged species, these repulsions are compensated by crystal field effects (i.e. surrounding counterions), so the net interaction in crystal packing is attractive.45
Extended arrays of pancake bonds
Numerous studies have shown that infinite stacks of equidistant radicals (Fig. 5 right) result in bulk antiferromagnetic properties and (semi)conductivity along the stacks.2,3,4,9,10,59 This not only implies long-range ordering, but also indicates that interactions between individual radicals also have a partial covalent character, i.e. in such systems, radicals also form pancake bonds.2,45 However, due to the long-range ordering, these pancake bonds must extend along the stacks. This explains electric conductivity: electrons can jump between the radicals thanks to a low energy barrier, which is low due to mixing of the orbitals.45 Thus, such stacks of equidistant radicals may be regarded as a sort of pancake-bonded polymers. | ||
| Fig. 5 A stack of pancake-bonded dimers (left) of tetrachlorosemiquinone radical anions transforms into a stack of equidistant tetrachlorosemiquinone radical anions (right).68 A spin-Peierls transformation requires adjustment of positions of one radical anion in each dimer (marked by a red arrow). | ||
While bulk properties, such as electric conductivity and magnetism, are readily measurable, quantum chemical modelling of infinite stacks is more difficult. Modelling interactions between two radicals with a geometry taken from the crystal structure disregards crystal field effects and provides only a partial answer. For infinite stacks of tetrachlorosemiquinone radical anions (Fig. 5 right), such a model estimates the covalent component of total interaction to −2.9 kcal mol−1 (much smaller than −9.4 kcal mol−1 in pancake-bonded dimers, but nevertheless significant) and an IBO of 0.26.45 A more thorough approach involves periodic computations; however, only a handful of such studies featuring infinite stacks have been published.60,61
More complex extended patterns are found in salts of TCNQ radicals, which often have a partial charge, −1/2 or −2/3.39 The most commonly occurring motifs here are trimers (with a total charge of −2, bond order 2/3), tetramers (with a total charge of −2, bond order 1/2) and dimers (with a total charge of −1, bond order 1/2); these usually form 1D stacks or 2D “brick-wall” arrays39 (Fig. 6). Distances between the oligomeric motifs are sometimes too small to be considered as non-bonding contacts, but have geometries similar to weaker pancake bonding: interplanar separation distances cluster between 3.10 and 3.35 Å with an angle between mean planes of 0–5°.52,53 Bulk magnetism and conductivity measurements, as well as EPR spectroscopy, are consistent with magnetic interactions and conductivity in 1D or 2D (Fig. 6).52,53 This indicates that pancake bonds may extend in 1D and 2D, forming infinite arrays. Similar 2D ordering was found in dithiazolyls,62 while in bisdithiazolyls, ladder-like 1D63 and 3D64,65 ordering was observed. In some bisdiselenazolyl radicals, it leads to 2D ferromagnetic ordering at very low temperatures (<20 K).66
 | ||
| Fig. 6 A 2D array of TCNQ radical anions (arranged in trimers with a formal charge of each moiety of −2/3) in its salt with N,N′-dimethyl-4,4′-bipyridium cations.53 Each trimer is shown in a different colour. Magnetic interactions between individual trimers extend in 1D along a stack (red arrow, indicating direction [001]) and laterally between the stacks (green arrow, indicating direction [101]). | ||
However, in the study of extended arrays of pancake bonds theory has yet to catch up with experiment. X-ray charge density and preliminary isolated-cluster quantum chemical models are in agreement with the bulk measurements.67 A more consistent picture would be provided by periodic computations, which are still in progress; however, preliminary results support the existence of extended arrays of pancake bonds.67
Moieties with a partial radical character
The existence of partially charged radicals discussed in previous sections implies that these moieties, having a partial charge, also have a partial radical character. This is achieved by more than two radicals sharing a single bonding pair.30,53,54,69 There are also systems with a partial charge transfer between electron donor and electron acceptor moieties. In such charge-transfer systems, both donor and acceptor molecules have a partial charge and therefore a partial radical character. The difference between a salt and a charge-transfer system is only in the degree of charge transfer (ρ), which is somewhat arbitrarily set to 0.5.70,71 Thus, systems with ρ lower than 0.5 are considered as ‘quasi-neutral’ charge-transfer systems, while those with ρ higher than 0.5 are considered as ‘quasi-ionic’.70,71 There are quite a few systems which can switch from quasi-neutral to quasi-ionic by an external factor such as temperature or pressure.70–74Planar donors and acceptors usually form mixed stacks with alternating donor and acceptor moieties (Fig. 7).70,75,76 Since the moieties have a partial radical character, their stacking interactions may also involve a small degree of covalent contribution, i.e. pancake bond. In a charge-transfer compound, donor and acceptor moieties have different energies of their HOMOs (Fig. 8); the lower energy difference, Δ, means the higher probability for electron transfer and formation of a charge-transfer compound. However, as Fig. 8 suggests, this also means formation of a HOMO which should extend between both moieties. This is also in agreement with the bulk (semi)conductivity of such compounds70,75 and stacking geometry (typically, interplanar separations between the stacked moieties are 3.2 Å or shorter). Measurements of electric conductivity and quantum chemical modeling of isolated clusters in a series of charge-transfer compounds of the donor N,N,N′,N′-tetramethyl-p-phenylenediamine (TMPD) with quinoid and TCNQ acceptors support the existence of pancake bonding in such systems.76 However, more rigorous quantum crystallographic studies have not yet been published.
 | ||
| Fig. 7 A stack of alternating partially charged tetrachloroquinone (acceptor) and TMPD (donor) moieties in the co-crystal of (formally neutral) tetrachloroquinone and TMPD.76 Electron transfer is indicated by an arrow. | ||
 | ||
| Fig. 8 A schematic representation of orbitals in a charge-transfer complex of electron donor TMPD (left) and acceptor tetrachloroquinone (right) in their co-crystal.76 | ||
Transformations in the solid state: influence of temperature and pressure
Some radical stacks display reversible spin-Peierls phase transformations,9,77–79 transformations from infinite stacks of equidistant radicals to infinite stacks of pancake-bonded dimers, i.e. stacks with alternating short (<3.1 Å) and long (>3.3 Å) interplanar separations (Fig. 5).3,4 These transformations may be induced by an external factor (temperature, pressure, irradiation) and are marked by change of bulk properties from diamagnetic/insulating (stacks of dimers) to antiferromagnetic/semiconductive (equidistant stacks).3,4,9 In some particularly interesting compounds, bistability (an existence of two stable phases over a certain temperature and/or pressure range, marked by temperature hysteresis) was observed.9,23,76,80,81 One can hardly overemphasize the importance of switchability and/or bistability in organic conductors and magnetic materials.22,26–28,31,59,76,77,82 Therefore, research of phase transformations of radical systems in the solid state is an especially promising area.Generally, stacks of dimers (in a singlet ground state) are more stable due to their lower enthalpy, and therefore represent the low-temperature phase; stacks of equidistant radicals have a higher entropy and therefore represent the high-temperature phase.9 However, the energy difference between the two phases is small, so transformation from one type of stack to another requires only minor adjustments of positions of stacked radicals (red arrow in Fig. 5). The main contributor to the lower energy of the dimers is dimerisation enthalpy (ΔHdim), which in solution rarely exceeds −12 kcal mol−1;9 this figure is comparable to the estimated covalent component of the interaction2,4 and can be regarded as its approximation. This means that an entropy-driven gradual transition (i.e. second-order; red arrow in Fig. 9) is possible by thermal population of excited states. For such a mechanism, DSC studies of spin-Peierls phase transformations show only small, rather broad, maxima with energies often below 1 kcal mol−1.60 This type of transformation is reversible and no loss of crystallinity is noted after several cycles of transformations.68 This is consistent with very close energies of two phases and a very low energy barrier. Such transformations happen in a broad temperature range (therefore broad, diffuse DSC maxima) and their feature is a loss of long-range order near the transition temperature,60,68 which is manifested as poor diffraction in that temperature range. In a typical example, low-temperature and high-temperature phases diffract well, but near the phase transformation (in a range of 50–60 K), diffraction is much weaker and sometimes it is even difficult to determine the unit cell.60,68 The mechanism of this transformation was extensively studied by theoretical methods by Novoa et al.59,83–88 Due to a low energy barrier, a rapid switching between two degenerate states (caused by large-amplitude vibrations of stacked radicals), i.e. a dynamic equilibrium called pair-exchange dynamics (PED; blue arrow in Fig. 9),84,85 is possible. A result is a time-averaged structure similar to equidistant stacks.
 | ||
| Fig. 9 Stacks of pancake-bonded dimers (a and b) isomerise by a phase transformation with an energy barrier Eb. If Eb is low, a PED mechanism is possible (blue arrow), resulting in a time-averaged structure of equidistant stacks (c). Higher-energy equidistant stacks (c) can be considered as an excited state. A spin-Peierls transformation (red arrow) proceeds either by a first-order phase transformation (if ΔEs-p is high) or by a thermal population of the excited states (second-order, if ΔEs-p is low). | ||
However, besides dimerisation enthalpy, other packing forces may also be involved in stabilisation of the LT phase. A combination of high ΔHdim and stabilising crystal field effects (such as “tethering” of radicals by hydrogen bonding89) may result in an abrupt first-order solid-state transformation. If the energy barrier is sufficiently high, a temperature hysteresis is observed, so the compound is bistable.83,86
A vast majority of spin-Peierls transformations studied to date are temperature-dependent, but some are also pressure-dependent. However, only a few detailed high-pressure studies of pancake-bonded systems have been published so far;60,90–92 only one of them actually describes a pressure-driven spin-Peierls transformation.92 At ambient pressure, the studied salt of the 5,6-dichloro-2,3-dicyanosemiquinone (DDQ) radical anion has stacks of dimers with alternating short (2.92 Å) and long (3.45 Å) interplanar separations. At the pressure of 2.55 GPa, the two distances become equal; at higher pressure, they separate again, but both interplanar distances remain below 2.9 Å, suggesting that pancake bonding extends throughout the stack.92 A quantum crystallographic study of this compound indicates that with the increase of pressure, electron density in critical points between pancake-bonded moieties also increases.61 This may indicate an increase of the covalent character, and in that case the phase transformation would be followed by a rapid increase of electric conductivity. However, note that when there is no phase transformation, the increase of conductivity by application of pressure is rather modest: broadening of the bands is offset by an increase of the HOMO–LUMO gap.90,91
Another interesting pressure-driven phase transformation involves a quinhydrone, a co-crystal of 1,4-benzoquinone and 1,2-resorcinol, which form hydrogen-bonded chains and π-stacks. At pressures above 3 GPa, the two moieties transform into semiquinone radicals, while their π-stacking apparently involves pancake bonding.93
The study of pressure-driven spin-Peierls phase transformations is still in its infancy; it is experimentally demanding and few laboratories have necessary equipment and expertise to carry out the experiments (this is especially correct for electrical conductivity and magnetism measurements under high pressure64). Nevertheless, interesting new findings may be expected in the near future.
Crystal engineering: mixing different radicals in a stack
Design of radicals comprises synthetic modification, i.e. introduction of different functional groups to enhance stability (such as introduction of electronegative substituents to quinones) or introduce new properties by functionalisation (for example, adding a pyrene group to dithiadiazolyl radicals10,29). A more novel approach is a combination of two (or more) different radicals by the formation of co-crystals or salts which comprise a radical cation and a radical anion, to more easily enhance conductivity and tune magnetic interactions. The main goal of such crystal engineering is to obtain desirable ferro- or ferrimagnetic properties, which are rare among single-component organic radicals.10Combinations of dithiadiazolyl (DTDA) and diselenadiazolyl (DSDA) radicals with electron acceptors methyl benzodithiazolyl (MBDTA) and (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO)94 or TCNQ95 represent some of the first successful designs of mixed-radical stacks. The study of such systems indeed shows great potential; the first examples already include a bistable salt of the 3-methylpyridinium-1,2,3,5-dithiadiazolyl radical cation and TCNQ radical anion.79
Outlook
The importance of pancake bonding in chemistry can hardly be overstated – being both a covalent bond and an intermolecular interaction,2 it stretches the very concept of chemical bonding. Straddling the border of intra- and intermolecular, it creates a “grey zone” in between. The notion of a bonding electron pair dispersed between multiple atoms (with distances closer to van der Waals than to covalent radii) to form a weak unlocalised π-bond differs from the “classical” picture of the chemical bond,37 so detailed study of its nature may be expected to result in a broadened definition of the chemical bond.The modern study of the nature of pancake bonds involves systematic theoretical work,2,33,41,43,46,47,83–88 more recently joined by quantum crystallography4,45,54,61,96–100 and crystallography under non-ambient conditions.61,61,90–93
From the application perspective, studies on possibilities of tuning the charge and radical character of charge-transfer systems30,69,70 and crystal engineering of mixed-radical stacks29,79,94,95 seem especially promising. They will likely result in novel compounds with enhanced electric conductivity, tunable magnetism,87 ferroelectricity,74 multifunctionality22,101–103 and the possibility of designing bistable compounds for switching materials.80,81 Indeed, ferromagnetic coupling, and even switching between ferromagnetic and antiferromagnetic24,25,82,104,105 states, has been observed in some organic radicals. It is reasonable to expect that in the not-so-distant future, our knowledge of the formation of the pancake bond and reversible phase transformations related to its formation would be applied in the design of materials which could switch from insulating to conducting or from dia- or antiferromagnetic to ferro- or ferrimagnetic. A combination of magnetic, conductive and optical properties will ultimately lead to organic-based multifunctional materials.22,29,30
Design of novel charge-transfer compounds is also likely to benefit from the study of pancake bonding and its application. The main interest in research of charge-transfer compounds is that compared to the radicals, they are more stable and their preparation is usually simpler.
Multiple challenges remain, however, as the systematic study of the pancake bond and possibilities its design offers are novel. There remains much to be discovered: what actually drives the spin-Peierls transformations and how they can be affected (and eventually, engineered), the behaviour of pancake-bonded systems and phase transitions under non-ambient conditions, achieving 2D ordering, enhancing conductivity (lowering the band gap) without reducing the stability of the compound, etc.
Data availability
The data that support the structures and plots within this paper and other findings of this study are available from the corresponding authors upon reasonable request.Author contributions
K. M. – conceptualization, writing – original draft, review & editing. P. S. – preparation of figures, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Croatian Science Foundation (HrZZ), grant IP-2024-05-8711.Notes and references
- K. E. Preuss, Polyhedron, 2014, 79, 1–15 CrossRef CAS.
- M. Kertesz, Chem. – Eur. J., 2019, 25, 400–416 CrossRef CAS PubMed.
- K. Molčanov and B. Kojić-Prodić, IUCrJ, 2019, 6, 155–166 CrossRef PubMed.
- K. Molčanov, V. Milašinović and B. Kojić-Prodić, Cryst. Growth Des., 2019, 19, 5967–5980 CrossRef.
- C. A. Hunter and J. K. M. Sanders, J. Am. Chem. Soc., 1990, 112, 5525–5534 CrossRef CAS.
- J. W. G. Bloom and S. E. Wheeler, Angew. Chem., Int. Ed., 2011, 50, 7847–7849 CrossRef CAS PubMed.
- L. M. Salonen, M. Ellermann and F. Diedrich, Angew. Chem., Int. Ed., 2011, 50, 4808–4842 CrossRef CAS PubMed.
- C. L. Martinez and B. L. Iverson, Chem. Sci., 2012, 3, 2191–2201 RSC.
- A. Paul, A. Gupta and S. Konar, Cryst. Growth Des., 2021, 21, 5473–5489 CrossRef CAS.
- D. A. Haynes, CrystEngComm, 2011, 13, 4793–4805 RSC.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS.
- M. Eraković, D. Cinčić, K. Molčanov and V. Stilinović, Angew. Chem., Int. Ed., 2019, 58, 15702–15706 CrossRef PubMed.
- P. Melichar, D. Hynk and J. Fanfrlik, Phys. Chem. Chem. Phys., 2018, 20, 4666–4675 RSC.
- S. F. A. Kettle, Bonding in Cluster Compounds, in Physical Inorganic Chemistry, ed. S. F. A. Kettle, Springer, Berlin, 1996 Search PubMed.
- R. S. Mulliken and W. B. Person, Molecular Complexes, Wiley & Sons, New York, 1969, ch. 16 Search PubMed.
- A. E. Alexander, J. Chem. Soc., 1937, 1813–1816 RSC.
- J. Landauer and H. A. McConnel, J. Am. Chem. Soc., 1952, 74, 1221–1224 CrossRef CAS.
- K. Fukui, K. Sato, D. Shiomi, T. Takui, K. Itoh, K. Gotoh, T. Kubo, K. Yamamoto, K. Nakasuji and A. Naito, Synth. Met., 1999, 103, 2257–2258 CrossRef CAS.
- K. Fukui, K. Sato, D. Shiomi, T. Takui, K. Itoh, T. Kubo, K. Gotoh, K. Yamamoto, K. Nakasuji and A. Naito, Mol. Cryst. Liq. Cryst., 1999, 334, 48–58 CrossRef.
- S. Suzuki, Y. Morita, K. Fukui, K. Sato, D. Shiomi, T. Takui and K. Nakasuji, J. Am. Chem. Soc., 2006, 128, 2530–2531 CrossRef CAS PubMed.
- J. S. Miller, Chem. Soc. Rev., 2011, 40, 3266–3296 RSC.
- S. Sanvito, Chem. Soc. Rev., 2011, 40, 3336–3355 RSC.
- I. Ratera and J. Veciana, Chem. Soc. Rev., 2012, 41, 303–349 RSC.
- J. Mahmood and J.-B. Baek, Chem, 2019, 5, 1012–1014 CAS.
- H. Phan, T. S. Herng, D. Wang, X. Li, W. Zeng, J. Ding, K. P. Loh, A. T. S. Wee and J. Wu, Chem, 2019, 5, 1223–1234 CAS.
- S. Horiuchi and Y. Tokura, Nat. Mater., 2008, 7, 357–366 CrossRef CAS PubMed.
- S. D. Bader and S. S. P. Parkin, Annu. Rev. Condens. Matter Phys., 2010, 1, 71–88 CrossRef CAS.
- D. A. Wilcox, V. Agarkar, S. Mukherjee and B. W. Boudouris, Annu. Rev. Chem. Biomol. Eng., 2018, 9, 83–103 CrossRef PubMed.
- Y. Beldjoudi, M. A. Nascimento, Y. J. Cho, H. Yu, H. Aziz, D. Tonouchi, K. Eguchi, M. M. Matsushita, K. Awaga, I. Osorio-Rozman, C. P. Constantinides and J. M. Rawson, J. Am. Chem. Soc., 2018, 140, 6260–6270 CrossRef CAS PubMed.
- R. Sato, T. Kawamoto and T. Mori, J. Mater. Chem. C, 2019, 7, 567–577 RSC.
- M. Gao, Z. Wang, X. Zhang, X. Hao and W. Qin, Nano Lett., 2019, 19, 9008–9012 CrossRef CAS PubMed.
- J. J. Novoa, P. Lafuente, R. E. Del Sesto and J. S. Miller, Angew. Chem., Int. Ed., 2001, 40, 2540–2545 CrossRef CAS PubMed.
- J. J. Novoa and J. S. Miller, Acc. Chem. Res., 2007, 40, 189–196 CrossRef PubMed.
- F. Mota, J. S. Miller and J. J. Novoa, J. Am. Chem. Soc., 2009, 131, 7699–7707 CrossRef CAS PubMed.
- J. Casado, P. M. Burrezo, F. J. Ramírez, J. T. López Navarrete, S. H. Lapidus, P. W. Stephens, P. W. Vo, J. S. Miller, F. Mota and J. J. Novoa, Angew. Chem., Int. Ed., 2013, 52, 6421–6425 CrossRef CAS PubMed.
- Z.-H. Cui, H. Lischka, T. Mueller, F. Plasser and M. Kertesz, ChemPhysChem, 2014, 15, 165–176 CrossRef CAS PubMed.
- L. Pauling, The nature of the chemical bond, Cornell University Press, Ithaca, NY, USA, 3rd edn, 1939 Search PubMed.
- R. Bhattacharjee, H. Jervis, M. E. McCormack, M. A. Petrukhina and M. Kertesz, J. Am. Chem. Soc., 2024, 146, 10465–10477 CrossRef CAS PubMed.
- F. H. Herbstein and M. Kapon, Crystallogr. Rev., 2008, 14, 3–74 CrossRef CAS.
- M. Strydom and D. A. Haynes, Cryst. Growth Des., 2024, 24, 6311–6325 CrossRef CAS.
- Z. Mou, Pancake bonding in organic radicals, Doctoral dissertation, Georgetown University, USA, 2017 Search PubMed.
- Z. Mou, T. Kubo and M. Kertesz, Chem. – Eur. J., 2015, 21, 18230–18236 CrossRef CAS PubMed.
- H. Z. Beneberu, Y.-H. Tian and M. Kertesz, Phys. Chem. Chem. Phys., 2014, 14, 10713–10725 RSC.
- R. Bhattacharjee, H. Lischka and M. Kertesz, ACS Mater. Au, 2025, 5, 365–376 CrossRef CAS PubMed.
- K. Molčanov, C. Jelsch, B. Landeros-Rivera, J. Hernández-Trujillo, E. Wenger, V. Stilinović, B. Kojić-Prodić and E. C. Escudero-Adán, Cryst. Growth Des., 2019, 19, 391–402 CrossRef.
- Z. Mou and M. Kertesz, Angew. Chem., Int. Ed., 2017, 129, 10188–10191 CrossRef PubMed.
- Z.-H. Cui, H. Lischka, H. Z. Beneberu and M. Kertesz, J. Am. Chem. Soc., 2014, 136, 12958–12965 CrossRef CAS PubMed.
- D. Small, V. Zaitsev, Y. Jung, S. V. Rosokha, M. Head-Gordon and J. K. Kochi, J. Am. Chem. Soc., 2004, 126, 13850–13858 CrossRef CAS PubMed.
- D.-H- Tuo, S. Tang, P. Jin, J. Li, X. Wang, C- Zhang, C.-F. Ao, Q.-Q. Wang and D.-X. Wang, CCS Chem., 2023, 5, 1343–1352 CrossRef CAS.
- J. Joseph, M. Berville, J. Wytko, J. Weiss and H.-P. Jacquot de Rouville, Chem. – Eur. J., 2025, 31, e202403115 CrossRef CAS PubMed.
- B. G. Janesko and E. N. Brothers, Energy Fuels, 2021, 35, 15657–15662 CrossRef CAS.
- P. Stanić, K. Smokrović, N. Maltar-Strmečki, M. Herak, F. Meurer, M. Bodensteier, C. Hennig and K. Molčanov, Cryst. Growth Des., 2024, 24, 9365–9378 CrossRef.
- K. Molčanov, V. Milašinović, B. Kojić-Prodić, N. Maltar-Strmečki, J. You, A. Šantić, L. Kanižaj, V. Stilinović and L. Fotović, IUCrJ, 2022, 9, 449–467 CrossRef PubMed.
- K. Molčanov, Z. Mou, M. Kertesz, B. Kojić-Prodić, D. Stalke, S. Demeshko, A. Šantić and V. Stilinović, Chem. – Eur. J., 2018, 24, 8292–8297 CrossRef PubMed.
- R. F. W. Bader and M. E. Stephens, J. Am. Chem. Soc., 1975, 97, 7391–7399 CrossRef CAS.
- K. B. Wiberg, Tetrahedron, 1968, 24, 1083–1096 CrossRef CAS.
- J. P. Foster and F. Weinhold, J. Am. Chem. Soc., 1980, 102, 7211–7218 CrossRef CAS.
- L. Barluzzi, S. P. Ogilvie, A. B. Dalton, P. Kaden, R. Gericke, A. Mansikkämaki, S. R. Giblin and R. A. Layfield, J. Am. Chem. Soc., 2024, 146, 4234–4241 CrossRef CAS PubMed.
- T. Francese, J. Ribas-Arino, J. J. Novoa, R. W. A. Havenith, R. Broer, C. de Graaf and M. Deumal, Phys. Chem. Chem. Phys., 2018, 20, 20406–20416 RSC.
- P. Stanić, T. Poręba, L. Androš Dubraja, A. Krawczuk and K. Molčanov, Cryst. Growth Des., 2023, 23, 3284–3296 CrossRef.
- V. Milašinović, K. Molčanov, A. Krawczuk, N. E. Bogdanov, B. A. Zakharov, E. V. Boldyreva, C. Jelsch and B. Kojić-Prodić, IUCrJ, 2021, 8, 644–654 CrossRef PubMed.
- A. Mailman, C. M. Robertson, S. M. Winter, P. A. Dube and R. T. Oakley, Inorg. Chem., 2019, 58, 6495–6506 CrossRef CAS PubMed.
- K. Lekin, J. W. L. Wong, S. M. Winter, A. Mailman, P. A. Dube and R. T. Oakley, Inorg. Chem., 2013, 52, 2188–2198 CrossRef CAS PubMed.
- K. Irie, K. Shibayama, M. Mito, S. Takagi, M. Ishizuka, K. Lekin and R. T. Oakley, Phys. Rev. B, 2019, 99, 014417 CrossRef CAS.
- N. J. Yutronkie, D. Bates, P. A. Dube, S. M. Winter, C. M. Robertson, J. L. Brusso and R. T. Oakley, Inorg. Chem., 2019, 58, 419–427 CrossRef CAS PubMed.
- C. M. Robertson, S. M. Winter, J. A. K. Howard, M. R. Probert and R. T. Oakley, Chem. Commun., 2021, 57, 10238–10241 RSC.
- P. Stanić, A. Krawczuk, F. Meurer, C. Hennig, I. Nikšić-Franjić, M. Bodensteiner and K. Molčanov, Charge density study of stacked arrays of partially charged pancake-bonded TCNQ radicals, manuscript in preparation.
- K. Molčanov, B. Kojić-Prodić, D. Babić, D. Pajić, N. Novosel and K. Zadro, CrystEngComm, 2012, 14, 7958–7964 RSC.
- T. Fujino, R. Kameyama, K. Onozuka, K. Matsuo, S. Dekura, T. Miyamoto, Z. Guo, H. Okamoto, T. Nakamura, K. Yoshimi, S. Kitou, T.-H. Arima, H. Sato, K. Yamamoto, A. Takahashi, H. Sawa, Y. Nakamura and H. Mori, Nat. Commun., 2024, 15, 3028 CrossRef CAS PubMed.
- H. Jiang, P. Hu, K. K. Zhang, Y. Long, W. Hu and C. Kloc, J. Mater. Chem. C, 2018, 6, 1884–1902 RSC.
- M. Masino, N. Castagnetti and A. Girlando, Crystals, 2017, 7, 108 CrossRef.
- A. Filhol, G. Bravic, J. Gaultier, D. Chasseau and C. Vettier, Acta Crystallogr., Sect. B, 1981, 37, 1225–1235 CrossRef.
- A. Girlando, A. Painelli, S. A. Bewick and Z. G. Soos, Synth. Met., 2004, 141, 129–138 CrossRef CAS.
- S. Horiuchi, K. Kobayashi, R. Kumai, N. Minami, K. Kagawa and Y. Tokura, Nat. Commun., 2015, 6, 7469 CrossRef PubMed.
- S. V. Rosokha and J. K. Kochi, Acc. Chem. Res., 2008, 41, 641–653 CrossRef CAS PubMed.
- P. Stanić, I. Nikšić-Franjić, L. Pavić and K. Molčanov, Cryst. Growth Des., 2023, 23, 4460–4471 CrossRef.
- M. E. Itkis, X. Chi, A. W. Cordes and R. C. Haddon, Science, 1999, 296, 1443–1445 CrossRef PubMed.
- A. Dragulescu-Andrasi, A. S. Filatov, R. T. Oakley, X. Li, K. Lekin, A. Huq, C. Pak, S. M. Greer, J. McKay, M. Jo, J. Lengyel, I. Hung, E. Maradzike, A. E. DePrince, S. A. Stoian, S. Hill, Y.-Y. Hu and M. Shatruk, J. Am. Chem. Soc., 2019, 141, 17989–17994 CrossRef CAS PubMed.
- A. I. Taponen, A. Ayadi, M. L. Lahtinen, I. Oyarzabal, S. Bonhommeau, M. Rouzieres, C. Mathoniere, H. M. Tounonen, R. Clerac and A. Mailman, J. Am. Chem. Soc., 2021, 143, 15912–15917 CrossRef CAS PubMed.
- W. Fujita and K. Awaga, Science, 1999, 286, 261–262 CrossRef CAS PubMed.
- R. G. Hicks, Nat. Chem., 2011, 3, 189–191 CrossRef CAS PubMed.
- S. Horiuchi, K. Kobayashi, R. Kumai, N. Minami, F. Kagawa and Y. Tokura, Nat. Commun., 2015, 6, 7469 CrossRef PubMed.
- S. Vela, M. Deumal, M. Shiga, J. J. Novoa and J. Ribas-Arino, Chem. Sci., 2015, 6, 2371–2381 RSC.
- M. Fumanal, J. Jornet-Somoza, S. Vela, J. J. Novoa, J. Ribas-Arino and M. Deumal, J. Mater. Chem. C, 2021, 9, 10647–10660 RSC.
- S. Vela, F. Mota, M. Deumal, R. Suizu, Y. Shuku, A. Mizuno, K. Awaga, M. Shiga, J. J. Novoa and J. Ribas-Arino, Nat. Commun., 2014, 5, 5411 CrossRef PubMed.
- S. Vela, M. B. Reardon, C. E. Jakobsche, M. M. Turnbull, J. Ribas-Arino and J. J. Novoa, Chem. – Eur. J., 2017, 23, 2479–3489 CrossRef PubMed.
- M. Deumal, S. Vela, M. Fumanal, J. Ribas-Arino and J. J. Novoa, J. Mater. Chem. C, 2021, 9, 10624–10646 RSC.
- T. Francese, S. Vela, M. Deumal, F. Mota, J. J. Novoa, M. F. Camellone, S. Fabris, R. W. A. Havenith, R. Broer and J. Ribas-Arino, J. Mater. Chem. C, 2020, 8, 5437–5448 RSC.
- S. V. Rosokha, J. Lu, T. Y. Rosokha and J. K. Kochi, Phys. Chem. Chem. Phys., 2009, 11, 324–332 RSC.
- A. Mailman, J. W. L. Wong, S. M. Winter, R. C. M. Claridge, C. M. Robertson, A. Assoud, W. Yong, E. Steven, P. A. Dube, J. S. Tse, S. Desgreniers, S. A. Secco and R. T. Oakley, J. Am. Chem. Soc., 2017, 139, 1625–1635 CrossRef CAS PubMed.
- W. Yong, K. Lekin, R. P. C. Bauer, J. S. Tse, S. Desgreniers, R. A. Secco, N. Hirao and R. T. Oakley, Inorg. Chem., 2019, 58, 3550–3557 CrossRef CAS PubMed.
- N. E. Bogdanov, V. Milašinović, B. A. Zakharov, E. V. Boldyreva and K. Molčanov, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2020, 76, 285–291 CrossRef CAS PubMed.
- H. Q. Le, M. Rusek and A. Katrusiak, J. Phys. Chem. C, 2023, 127, 4310–4318 CrossRef CAS.
- M. A. Nascimento, E. Heyer, J. J. Clarke, H. J. Cowley, A. Alberola, N. Stephaniuk and J. M. Rawson, Angew. Chem., 2019, 131, 1385–1389 CrossRef.
- A. I. Taponen, A. Ayadi, N. Svahn, M. L. Lahtinen, M. Rouzieres, R. Clerac, H. M. Tounonen and A. Mailman, Cryst. Growth Des., 2022, 22, 7110–7122 CrossRef CAS.
- A. B. Voufack, A. B. Dippenaar, C. Esterhuysen, D. A. Haynes, M. Souhassou, C. Lecomte and N. Claiser, Cryst. Growth Des., 2024, 24, 8736–8747 CrossRef CAS.
- S. Domagała, K. Kosc, S. W. Robinson, D. A. Haynes and K. Wożniak, Cryst. Growth Des., 2014, 14, 4834–4848 CrossRef.
- S. Domagała and D. A. Haynes, CrystEngComm, 2016, 18, 7116–7125 RSC.
- D. A. Haynes, Acta Crystallogr., Sect. A: Found. Adv., 2019, 75, e360 Search PubMed.
- P. Stanić, I. Nikšić-Franjić and K. Molčanov, Cryst. Growth Des., 2023, 23, 4571–4579 CrossRef.
- S. Ren and M. Wuttig, Adv. Mater., 2012, 24, 724–727 CrossRef CAS PubMed.
- W. Qin, X. Chen, H. Li, M. Gong, G. Yuan, J. C. Grossman, M. Wuttig and S. Ren, ACS Nano, 2015, 9, 9373–9379 CrossRef CAS PubMed.
- Y.-B. Huang, J. Liang, X.-S. Wang and R. Cao, Chem. Soc. Rev., 2017, 46, 126–157 RSC.
- R. Suizu, Y. Shuku, V. Robert, P. Roseiro, N. Ben Amor, Z. Khawar, N. Robertson and K. Awaga, Dalton Trans., 2023, 53, 1961–1965 RSC.
- M. Gao, Z. Wang, S. Ren, X. Hao and W. Qin, Cell Rep. Phys. Sci., 2021, 2, 100442 CrossRef CAS.
Footnotes |
| † Dedicated to the memory of Dr. Biserka Kojić-Prodić. |
| ‡ Probably because the state-of-the art of the time allowed study of only small organic molecules in the solid state. Crystallographic study of larger systems and radicals became possible a few decades later. |
| § As shown by many studies [ref. 3–8], interactions of two π-electron clouds of two contiguous aromatic rings are repulsive, rather than attractive. It is other components, mainly σ–π and dipolar, which make the total interaction weakly attractive. |
| This journal is © The Royal Society of Chemistry 2025 |