
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning carbonyl interactions in dibenzochalcogenophenes†
Lea
Höfmann
a,
Christoph
Wölper
b,
Alexander
Huber
a,
Hannah
Siera
a,
Constantin G.
Daniliuc
 c,
Gebhard
Haberhauer
a and
Jens
Voskuhl
*a
c,
Gebhard
Haberhauer
a and
Jens
Voskuhl
*a
aFaculty of Chemistry (Organic Chemistry), Center of Medical Biotechnology (ZMB) and Center for Nanointegration Duisburg-Essen (CENIDE), University of Duisburg-Essen, Universitätstr. 7, 45117 Essen, Germany. E-mail: jens.voskuhl@uni-due.de
bFaculty of Chemistry (Inorganic Chemistry), University of Duisburg-Essen, Universitätsstr. 7, 45117 Essen, Germany
cOrganisch-Chemisches Institut, University of Münster, Corrensstraße 40, 48149 Münster, Germany
First published on 3rd April 2025
Abstract
This study presents a series of functionalized dibenzochalcogenophenes (DBE; E: S, Se, Te) with a benzoyl moiety in the 3- and 4-position. Single crystal X-ray diffraction of all compounds was utilized to examine crystal packing and Hirshfeld analysis, focussing on chalcogen bonding. The data indicated an increasing number of chalcogen-driven contacts corresponding to the increasing size of the chalcogen within the structure, aligning with theoretical calculations and previous studies. For DBEs substituted with a benzoyl group at 4-position, conformational fixation could be induced by the carbonyl group, impeding the rotation of phenyl residues. Conversely, dibenzotellurophene with the benzoyl moiety in 3-position exhibited dimerization through chalcogen–carbonyl bonds based on its self-complementary structure, revealing a new interaction motif based on chalcogen bonding applicable for future use in supramolecular assembly.
Introduction
Oxygen and sulphur, two commonly used heteroatoms, are the primary chalcogens in organic chemistry due to their ubiquitous presence in biological structures.1 Heterocyclic dibenzofuran (DBF) and -thiophene (DBT) demonstrate excellent fluorescence and phosphorescence behaviour. Their straightforward synthesis, facile functionalisation and triplet emission ability enable multipurpose applications, e.g. in organic light-emitting diodes (OLEDs).2,3 Auspicious dibenzochalcogenophene (DBE) derivatives have been previously utilized to produce white light emission, taking advantage of their dual emission behaviour.2,4,5 Their heavier analogues, dibenzoselenophene (DBSe) and dibenzotellurophene (DBTe), were first investigated by Zander et al. in 1989.6 By increasing the size of the chalcogen, the intersystem crossing (ISC) rates rise, leading to higher phosphorescence quantum yield values.6 In previous studies, Jiang et al. synthesized several derivatives of DBTe, which are capable of room-temperature phosphorescence.7 For DBSe, only co-crystalline mixtures are reported that display a bathochromic shift when an electron-rich aromatic system is added.8Despite their photophysical properties, organic compounds containing heavy chalcogens are uncommon in organic chemistry. The scarcity of synthetic information hinders the incorporation of heavy chalcogens into new organic motifs. In contrast to their lighter homologues, selenium and tellurium show higher polarizability, a highly desired attribute for supramolecular interactions.9–12 In contrast to halogen bonding, chalcogens possess two σ-holes along the C–E bonds, enabling them to construct multivalent networks. These chalcogen interactions are increasingly present in latest publications, beginning with chalcogen donors varying to nitrogen, leading to binding energies almost up to those of hydrogen bonding.13,14 Therefore, chalcogenazoles offer promising starting motifs capable of dimeric or multivalent assembly.15 Even ionic donor motifs like N-oxides resulted in promising chalcogen donors building tetra- and hexameric aggregates.12,16 Due to their ongoing investigation and improvement of binding motifs through chalcogen bonding, they have already been successfully applied in organocatalysis or as building blocks for supramolecular frameworks.10,11,17
In addition to nitrogen donor moieties, current results present carbonyl groups as equally strong interaction partners.14,18 Aiming to investigate this carbonyl–chalcogen bonding in detail, this study focuses on a series of carbonyl-group containing DBEs. By selecting two different substitution patterns, the intra- and intermolecular impact of this secondary bonding type is compared. Previous studies in this context have primarily concentrated on 2-substituted, lighter DBEs.2,5,19
However, our interest lies in the 3- and 4-positions, which offer the potential for chalcogen–carbonyl bonds in the crystalline state. The 4-benzoyl DBEs (4a–c) serve as reference systems due to their structural capability to form intramolecular chalcogen-carbonyl bonds that restrict the rotation of the benzoyl residue. Their 3-substituted regioisomers (12a–c) exhibit self-complementarity, suggesting the potential to form dimers or networks bonded by carbonyl-chalcogen interactions (Fig. 1).
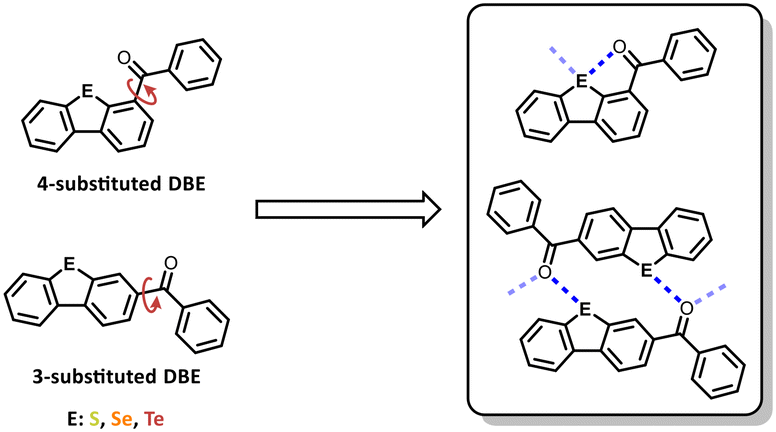 | ||
| Fig. 1 Presenting the compounds investigated in this study and the possible packing motifs driven by chalcogen carbonyl interactions. | ||
Results and discussion
Synthesis and spectroscopy
For the synthesis of the final compounds, known procedures were adapted and optimised.7,20,21 Because of the ongoing debate about impurities in DBT-related commercially available substances,22 we decided to synthesize all DBEs ourselves to rule out doping-caused results falsification (see Scheme 1). For every chalcogen, different procedures and purification methods were successfully applied, yielding the desired products. For the 4-substituted DBEs, the unsubstituted DBEs (3a, 3b) were obtained by two-step cyclisation of the corresponding dichalcogenide (2a, 2b) in good yield (S: 69%, Se: 66%).21 DBTe (3c) was synthesized using an iodonium salt, as reported by Jiang et al. (see ESI†).7ortho-Lithiation and acylation, using a Weinreb amide as an electrophile, was chosen to obtain the final products (4). | ||
| Scheme 1 Synthesis routes to the 4- and 3-substituted DBE compounds (4, 12; E: S (a), Se (b), Te(c)). i) S8 (2 eq.), CuI (0.2 eq.), Na2S (2 eq.), DMF, 100 °C, overnight; ii) Se (6 eq.), CuI (0.2 eq.), K3PO4 (6 eq.), DMSO, 120 °C, overnight; iii) PdCl2 (5 mol%), DMSO, 120 °C, overnight; iv) 1. nBuLi (1.5 eq.), TMEDA (1.1 eq.), THF, 0 °C; 2. N-methoxy-N-methylbenzamide (1.5 eq), 0 °C to room temperature; v) 1. Pd(dppf)Cl2 (6mol%), KOAc (3 eq.), (Bpin2)2 (1.2 eq.), 1,4-dioxane, reflux, 24 h; 2. 1,2-diiodobenzene (2 eq.), Pd(PPh3)4 (3mol%), Na2CO3 (4 eq.), Tol/EtOH/H2O, reflux, 28 h; vi) mCPBA (1.5 eq.), TfOH (3 eq.), DCM, room temperature, 1–5 h; vii) Te (3 eq.), 2-picoline (35 eq.), DMF, 120 °C, overnight. | ||
This was realised for DBT (3a) and DBSe (3b) at 0 °C lithiation temperature with good results (4a: 70%, 4b: 46%). However, the 4-benzoylated DBTe (4c) could not be obtained by this procedure from DBTe (3c). Even by varying the temperature (−78 °C, −40 °C, 0 °C), the desired product could not be isolated due to decomposition of the starting material. As a result, a different synthetic route was required.
Therefore, we started with a benzophenone core (5), substituting the meta-position with 1,2-iodobenzene via Miyaura borylation and Suzuki–Miyaura coupling (6).20 By cyclisation of a diaryliodonium salt (7), the desired tellurium compound (4c) was isolated in moderate yield (33%). Notably, the cyclisation of the iodobiphenyl intermediate (7) can lead to two different regioisomers of the iodonium salt. By either ortho- or para-H abstraction, the 4- and 2-benzoyl substituted iodonium salt can be formed. After isolation, 2D NMR analysis showed only the desired position 4-regioisomer, indicating that this cyclisation proceeds highly regioselective (Fig. S5 and S6†). This may result from the coordination of the carbonyl oxygen to the iodine(III) atom during the formation of the new C–I bond.23
The 3-substituted regioisomers had to be obtained differently due to the lesser accessibility of this position at the DBE core. We started synthesizing 3-bromo DBEs 16a–c from diaryliodonium salts (15) but obtained low yields and purification problems (16a, 16b, see ESI†).7,24 Fortunately, we received a single crystal of 3-bromo-[b,d]-dibenzoselenophene (16b), which could be used for X-ray diffraction (see Table S4, Fig. S37†) to prove the correct substitution pattern. For 3-bromo substituted DBTe 16c, the bromine–lithium exchange was unsuccessful.
Due to these complications, another route, similar to the procedure for 4c, was chosen. Starting with a para-substituted benzophenone core (8), Suzuki–Miyaura coupling yielded the iodobiphenyl derivative (9).20 For sulphur and selenium, dichalcogenides (11a, 11b) were synthesized using the procedure of Nishino et al.21 These compounds were used as obtained for the palladium-catalysed cyclisation of the final compounds (12a, 12b).21 We observed that the same reaction using iodine and copper(I)-chloride, reported by Nishino et al., led to different side products and bad conversion.25
For the synthesis of the final tellurium compound 12c, the same procedure was applied as previously for 4c using a diaryliodonium salt (10), yielding the desired compound 12c in moderate yield (27%).7 The loss of yield in the synthesis of tellurium-containing substances is mainly caused by the compound's oxidation during column chromatography on SiO2.
All final compounds were isolated in high purity (>99%, verified by HPLC, Table S1, Fig. S29†) and fully characterised by 2D NMR and mass spectrometry.
UV/vis spectra were also measured for solutions in 2-methyltetrahydrofuran (10 μM, see Table S2, Fig. S30†). The different absorption behaviour of the final compounds correlates with the size of the chalcogen. Compound 4a shows an absorption band of 357 nm, whereas the absorption band of tellurium compound 4c is bathochromically shifted to 414 nm. This trend is analogous to the 3-substituted DBEs 12a–c, exhibiting a smaller shift to higher wavelengths with increasing size of chalcogen (12a: 344, 12b: 350, 12c: 379 nm). This is in agreement with previously observed absorption behaviour of chalcogen congeners.26
Crystal packing analysis
Slow evaporation from dichloromethane layered with n-hexane yielded single crystals of all final compounds. Additionally, we could crystallise the benzoyl diaryliodonium salts (7, 10) from acetone, verifying the regioselectivity of the cyclisation of iodonium salt 7 (Table S4, Fig. S38†). Complete crystal data can be found in The Cambridge Crystallographic Data Centre (2389699, 2389700, 2403909–2403915). This section focuses on the packing of the six final compounds in the crystal lattice, especially chalcogen–carbonyl interactions, aiming to gain a deeper understanding of the ground-lying parameters influencing this interaction. An in-depth crystal packing analysis was performed, measuring the distances between surrounding moieties in the crystal structure. The criterion for a contact is the sum of van der Waals radii. Furthermore, the quantum theory of atoms in molecules (QTAIM)27 and the interacting quantum atoms (IQA)28 analyses were used to analyse the contacts with respect to the interaction energies (see ESI,† Tables S5–S10). The analysis of the chalcogen–carbonyl interactions will be discussed in detail in the section about quantum chemical calculations (vide infra). The crystals of the sulphur- and selenium-containing substances (4a–b; 12a–b) are isomorphic, as can be seen by the dimension of the unit cell (Table S3†). The atom indexes will be used analogously to the indices of 4a/12a (see Fig. S31 and S34†).The 4-substituted compounds (4) serve as reference systems. They are not expected to build dimers or chains via chalcogen bonding. However, the substitution pattern enables the structure to construct an intramolecular chalcogen–carbonyl interaction, where the carbonyl oxygen lone-pair is directed to the σ-hole of the chalcogen. This contact has the potential to determine the conformation of the benzoyl moiety. In all three crystal structures of the 4-substituted DBEs (4a–c), we observed that only the conformation allowing a chalcogen–carbonyl interaction is present (Fig. S40†). The E⋯O distances are almost independent of the size of the chalcogen (2.74 Å (S), 2.74 Å (Se), 2.81 Å (Te)). With respect to increasing atom sizes, this indicates that the contact significantly impacts the molecular arrangement of the compounds, especially with heavier chalcogens, the energetic aspect of this will be discussed in the section regarding quantum chemical calculations (vide infra).
Compounds 4a–b and 12a–b crystallise in monoclinic space group P21/n with one molecule in the asymmetric unit (Table S3†). The packing of 4a–b is mainly determined by π⋯π stacking interactions (3.61–3.69 Å) of all rings building strand-like structures accompanied by a weak non-classical hydrogen bond (O1⋯H15/26). Additionally, a weak E⋯O contact above the sum of van der Waals radii can be observed (S1/Se1⋯O1, see Fig. S41†). These structures are linked to the surroundings by hydrogen bonding of the carbonyl oxygen and chalcogen with the core hydrogens (O1⋯H5, O1⋯H8, S1⋯H9, Se1⋯H11, Fig. S42†).
Compound 4c crystallises in the monoclinic space group C2/c with one molecule in the asymmetric unit (Table S3†). Therefore, the packing motifs differ from those of the lighter chalcogens. π⋯π stacking also occurs as an interaction between two molecules but with a twisted arrangement, probably due to steric hindrance caused by the tellurium, so the π-systems only partially overlap. Additionally, this arrangement allows a Te⋯H contact with the benzoyl ring of the other molecule (Te1⋯H19, Fig. S45†). The packing also shows a motif determined by interactions of two planar strings of molecules. These two strings are connected via non-classical hydrogen bonding along the strand and Te⋯H contacts between the two strings (O1⋯H5, O1⋯H8, Te1⋯H3, Fig. 2). As a description, we refer to this packing motif as double-strand in the following text (see Fig. 2).
 | ||
| Fig. 2 Packing motif of a double-strand like in the crystal structure of compound 4c forming via double hydrogen bonding (red) and Te⋯H bonding (black). Displacement ellipsoids are shown at 50% probability. | ||
The layers are connected by self-complementary oxygen- and tellurium-donating hydrogen bonds (O1⋯H15, Te1⋯H16, Fig. S46†).
Similar to their regioisomers 4a–b, the packing of 3-substituted DBEs (12a–b) shows mostly π⋯π stacking of the whole aromatic system, but with a longer distance between the centres of the rings (3.95 Å (S), 3.98 Å (Se), Fig. S48†). For 12b, a Se⋯Se contact is also observed. In the plane, hydrogen bonding with the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O moiety is the central motif, accompanied by weak CH⋯π interaction (Fig. S49†).
O moiety is the central motif, accompanied by weak CH⋯π interaction (Fig. S49†).
The last compound, 12c, crystallises in monoclinic space group P21/c with two independent molecules in the asymmetric unit (Table S3†). The molecules are addressed by their corresponding residues from the CIF file (12c_1 & 12c_2, CCDC 2403914). These two molecules are in contact through π⋯π interactions (Fig. S53†). These stacked molecules interact with a moiety generated via inversion. This motif is held by complementary Te⋯O contacts (3.34 Å/3.24 Å), showing the expected dimerization (Fig. 3).
 | ||
| Fig. 3 Packing motif of dimerization in the crystal structure of compound 12c by chalcogen carbonyl interaction (black) and hydrogen bonding (red). Displacement ellipsoids are shown at 50% probability. _X indicates which residues from the asymmetric unit are interacting. | ||
This dimeric arrangement is the main component of the crystal structure of 12c. The chalcogen–carbonyl interaction is accompanied by two non-classical hydrogen bonds (O1⋯H12), which could also be responsible for the packing motif. For the hydrogen bond, a shift of the molecules, so only the O⋯H bond without Te⋯O proximity is possible and could be energetically more favourable due to the closer distance of hydrogen bond donor and acceptor with less repulsion of the remaining molecules. Thus, the found arrangement indicates that the chalcogen bond is an interaction of higher importance.
Additionally, the crystal structure differs from the isomorphic S- & Se-analogue (12a–b), so the chalcogen is the main difference, which results in a new packing pattern because chalcogen bonding becomes more favourable. Furthermore, this packing motif held by chalcogen-carbonyl interactions forms the primary interaction, which can be seen as the repeating unit in the crystal structure (Fig. S54†). The main units are connected by π⋯π/CH⋯π and O⋯H interactions (see Fig. S52 and S53†). However, with respect to the van der Waals radii, most of these contacts almost surpass the sum (see Table S10†), highlighting the dimer as the strongest contact in the packing pattern.
Compound 4c could also be capable of building chain or dimer-like structures based on chalcogen bonding. This behaviour can be observed by Te⋯H contacts. However, no intermolecular Te⋯O interaction could be found. 12c clearly shows self-complementary chalcogen–carbonyl contacts as the central motif of the packing.
The electrostatic potential surfaces (ESP, Fig. S69 and S70†) visualise the critical contrast between 4- and 3-substituted DBEs (4, 12). The 4-substituted DBEs σ-hole is less electron deficient than those of their corresponding regioisomers. For 4c, only a light blue area can be observed. Almost the same intensity is indicated for 12b. The ESP of the tellurium compound 12c shows a dark blue area relating to the σ-hole along the C–Te bond (Fig. S70†). This difference in the electron density distribution explains the variable packing behaviour with respect to the substitution pattern.
Hirshfeld surface analysis
We then performed Hirshfeld surface (HS) analysis using CrystalExplorer17 to further analyse the crystal packings.29 This section discusses the HS of target compounds 4a–c and 12a–c and the distribution of interactions with respect to the element via Fingerprint plots (FPPs) of occurring interactions, supporting the found contacts by in-depth crystal packing analysis.In the HS of 4-substituted DBEs containing sulphur and selenium (4a, 4b), the interaction sites are located on the core hydrogens H5 and H8, which were previously mentioned as acceptors for hydrogen bonding by O1 and S1 (Fig. S55 and S57†). Tellurium-containing compound 4c shows interaction points at the oxygen (O1) and the phenyl moiety (Fig. S59†), also in agreement with the identified non-classical hydrogen bond. The tellurium atom shows no close distance to surrounding surfaces (no red area on the HS) mostly because the Te⋯H distance is slightly above the sum of their van der Waals radii. So, this highlights the hydrogen bonding as the main interaction in the crystal packing patterns of 4-substituted DBEs (4a–c).
Fig. 4 shows histograms for chalcogen- and oxygen-participating contacts evaluated by FPP analysis (full data in ESI, Fig. S68†).
 | ||
| Fig. 4 Histogram with O- and E-participating interactions in the crystal structures of 4a–c and 12a–c by analysis of the Hirshfeld surfaces using CrystalExplorer17.29 For 12c, two different Hirshfeld surfaces for both molecules in the asymmetric unit could be obtained. Their corresponding residue index indicates them. | ||
For all structures of 4, the packing contains mostly non-classical hydrogen bonds besides C⋯C, C⋯H and H⋯H contacts. In contrast to 4a, selenium- and tellurium-containing compounds 4b and 4c feature an increase from 9.7% to 12.4% in chalcogen bonding. Additionally, tellurium congener 4c exhibits E⋯E contacts (0.1%) and a higher amount of E⋯H interactions (4b: 7.8%, 4c: 9.33%), leading to a decrease of H⋯H contacts (4b: 44.0%, 4c: 41.2%, Table S13†). A slightly increasing percentage of E⋯O contacts from sulphur (4a, 0.5%) to tellurium (4c, 0.7%) indicates the benefit of binding with the increasing size of chalcogen, eventually leading to better conformational fixation of the benzoyl moiety by the observed intramolecular bonding between the chalcogen and carbonyl oxygen (vide infra).
Analogously, in the 3-substituted lighter DBEs (12a–b), participation of chalcogen interactions is found to be almost the same; 12b only differs significantly in a slightly increased number of E⋯H contacts (12a: 8.7%, 12b: 9.5%). HS analysis of 12c resulted in two independent surfaces and FPPs for both residues (12c_1 & 12c_2). Compound 12c_1 shows about 1% more E⋯H and O⋯C contacts. C⋯C interactions of 12c_2 are almost only half of the value of the other residue, but C⋯H contacts increase by about 8% (Table S13†). These differences can be explained by their different interaction motifs with surrounding dimers (Fig. S52 and S53†). Correlating the tellurium compound 12c with its lighter derivatives (12a–b), it becomes evident that only this structure displays E⋯O contacts with 1.5% for both residues, highlighting the observed chalcogen-driven dimer formation. In the respective HS, near contacts are also visualised at the location of the σ-hole of the tellurium (Fig. S65†). In agreement with their ESPs, the 3-substituted DBTe (12c) displays better acceptor properties in the HS than its 4-substituted analogue (4c), verified by the increase of tellurium-driven contacts in the FPP (Fig. S66 and S67†).
Quantum chemical calculations
Quantum chemical calculations were performed to investigate the bonding properties in monomers 4-I and 4-II as well as in dimers 12·12 (E = S, Se and Te). The geometric parameters for monomers 4 & 12 were calculated using density functional theory (Fig. 5A). The functional used was B3LYP30 in combination with the dispersion correction D3BJ.31 The basis sets applied were def2-TZVP for the light elements (H, C, O, S and Se) and aug-cc-pVTZ-PP for tellurium. This method is abbreviated in the following as B3LYP-D3BJ/def2-TZVP,aug-cc-pVTZ-PP. No symmetry restriction was used to calculate the structures of the monomers. A comparison of the energies between conformers 4-I and 4-II reveals that for the heavier chalcogens (S, Se and Te), conformer 4-II, showing an intramolecular chalcogen bond, is energetically favoured. Interacting quantum atoms (IQA)28 analysis using B3LYP-D3BJ/TZ2P was employed to study the stabilising interactions in 4-I and 4-II. Considering the covalent part of the interaction energies of the CO⋯E bonds (EE⋯O) computed using IQA, it becomes obvious that EE⋯O strongly increases with increasing size of the chalcogen atom and thus causes the stabilisation of conformer 4-II for higher chalcogens (Fig. S71†). This is in agreement with the conformations found in the crystal structures (Fig. S40†). | ||
| Fig. 5 A) Structures of the two conformations (I, II) with possible interactions for 4 and 12 (green: E⋯C, red: O⋯H, blue: E⋯O). B) Binding energies of the dimers 12·12 (E = S (yellow), Se (red) and Te (blue)) as functions of chalcogen–oxygen distance (dE⋯O) calculated at B3LYP-D3BJ/def2-TZVP,aug-cc-pVTZ-PP level of theory with symmetry restriction (Ci). C) The energy terms ΔEoi (total orbital interactions), ΔEDisp (dispersion energy) and ΔEtotal (total bonding energy) of the dimers 12·12 calculated by an energy decomposition analysis (EDA, B3LYP-D3BJ/TZ2P). The sum of the covalent parts of the interaction energy between the atoms of the E⋯O and O⋯H bonds of the dimers 12·12 calculated by an interacting quantum atoms analysis (IQA, B3LYP-D3BJ/TZ2P). The energies are given in kcal mol−1. The geometries of the dimers 12·12 stem from calculations (calc; B3LYP-D3BJ/def2-TZVP,aug-cc-pVTZ-PP) with symmetry restriction (Ci) and from X-ray structure analyses (single point calculations, X-ray). | ||
In contrast, the energy difference for stereoisomers 12-I and 12-II, which cannot form intramolecular carbonyl–chalcogen bonds, is almost zero.
In the next step, quantum chemical calculations were used to investigate whether, in addition to the dimers of the tellurium compound (12c·12c) found in solid-state, the sulphur- and selenium-containing compounds 12a and 12b can also form analogous dimers via CO⋯E bonds. For this purpose, a geometry optimisation of the structures for dimers 12·12 (E = S, Se and Te) was performed by means of B3LYP-D3BJ/def2-TZVP,aug-cc-pVTZ-PP, with the Ci point group used as a symmetry restriction. In addition, the distance between the oxygen atom of the carbonyl group and the chalcogen atom (dE⋯O) was fixed at values from 2.2 to 4.0 Å, while all other parameters were optimised. Here again, the optimisation was carried out retaining Ci point group. The resulting graphs are presented in Fig. 5B. There is a clear trend: as the size of the chalcogen atom increases (Te > Se > S), the distance dE⋯O for the dimer decreases (S: 3.32 Å; Se: 3.18 Å; Te: 3.06 Å) and the binding energy increases. At −14.2 kcal mol−1, the bonding energy for the tellurium-containing dimer 12c·12c is almost twice as high as the dimerisation energy for the sulphur-containing system (12a·12a: −7.9 kcal mol−1), supporting the missing dimerization in the solid-state structure for sulphur compound 12a. As no stabilising interaction between the aromatic moieties can occur due to the assumed Ci symmetry, the interaction between the monomer building blocks is limited to the two CO⋯E bonds and the two hydrogen bonds (Fig. 5B).
In order to investigate these stabilising interactions in dimers 12·12, bonding energy analyses and IQA calculations were performed using B3LYP-D3BJ/TZ2P (Fig. 5C). Comparison of the data from the energy decomposition analysis (EDA)32 for the calculated structures shows the expected trend: with increasing size of the chalcogen atom (Te > Se > S), the total bonding energy (ΔEtotal) as well as the total orbital interactions (ΔEoi) and the dispersion energy (ΔEDisp) increase. As already observed for other chalcogen bonds,14 ΔEoi increases significantly more than ΔEDisp. For example, ΔEoi for the tellurium dimer 12c·12c is 2.2 times as large as ΔEoi for the sulphur analogue 12a·12a. For ΔEDisp, there is only a 1.6-fold increase. It is also interesting to analyse the sum of the covalent parts of the interaction energies between the atoms of the E⋯O and H⋯O bonds of the dimers 12·12 calculated by means of IQA (Fig. 5C). In the sulphur-containing dimer 12a·12a, the hydrogen bonds dominate over the chalcogen bonds, in the tellurium-containing dimer 12c·12c, it is reversed. A comparison of the analyses for the calculated structure of dimer 12c·12c with the structure obtained in solid-state and evaluated via single point calculations (Fig. 5C) reveals a similar trend, only the absolute values are lower for the solid-state structure. This can be explained by the fact that in the solid-state, the dimerisation via chalcogen bonds is in competition with a large number of other dimerisations.
It should be noted that frequency analysis of the Ci-symmetric structures of 12·12 shows an imaginary frequency for all systems. If the Ci restriction is cancelled in a subsequent geometry optimisation calculation, a minimum containing chalcogen bonds is only obtained for the tellurium compound 12c·12c, verified by the observed dimerization in the solid-state structure (see Fig. 3). In the case of the sulphur and selenium compounds 12a·12a and 12b·12b, dimerization occurs via π⋯π stacking of the aromatic units (see Fig. S48†).
Conclusions
A series of new DBE derivatives 4a–c and 12a–c were synthesized, characterized and investigated regarding their chalcogen–carbonyl interaction in the crystal lattice.To investigate the interactions in relation to the size of the included chalcogen, single crystals of all final compounds were grown and analysed. Sulphur and selenium compounds (4a–b, 12a–b) showed isomorphic crystal structures, with the main interaction driven by their carbonyl oxygen forming hydrogen bonds accompanied by weak E⋯H contacts and π⋯π stacking. Compounds 4a–c only showed conformation 4-II in the crystalline state, revealing the energetic benefit of intramolecular chalcogen–carbonyl bond, supported by quantum chemical calculations. For the tellurium congener 4c, a construction of strand-like patterns by E⋯H and O⋯H contacts was observed, forming almost planar layers connected by hydrogen bonding with tellurium and oxygen donors, leading to a different packing pattern compared to the lighter congeners. This difference aligns with the contribution of interactions evaluated by Hirshfeld analysis. No other interaction of the chalcogen is directly found in the packing, mostly due to a less significant electron-deficient σ-hole as indicated in the ESP.
The crystal structure of the tellurium regioisomer 12c reveals the expected dimer formation based on its self-complementary chalcogen–carbonyl binding motif. The accompanying hydrogen bond was found to be the secondary interaction determining the binding motif, as proven by IQA analysis. In summary, we were able to confirm the formation of dimer-like structures driven by carbonyl–chalcogen bonds in heavy DBEs introducing a benzoyl group in 3-position, which may be suitable for a novel self-complementary binding motif for future applications in supramolecular chemistry or materials science.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Lea Höfmann: methodology, formal analysis, writing, investigation. Christoph Wölper: data curation, formal analysis. Alexander Huber: data curation, investigation. Hannah Siera: data curation, formal analysis. Constantin G. Daniliuc: data curation. Gebhard Haberhauer: data curation, formal analysis, writing. Jens Voskuhl: conceptualization, funding acquisition, writing, supervision, project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Lea Höfmann wants to thank the Professor Werdelmann Stiftung for a PhD fellowship. Dr. Hannah Siera is grateful for a Postdoctoral fellowship from the University of Duisburg-Essen. The authors acknowledge Petra Schneider for her support during the HPLC measurements. We thank reviewer 3 for the insight on the residual electron density in 12c.Notes and references
- (a) S. Chatterjee and R. P. Hausinger, Crit. Rev. Biochem. Mol. Biol., 2022, 57, 461–476, DOI:10.1080/10409238.2022.2141678; (b) A. Raab and J. Feldmann, Anal. Chim. Acta, 2019, 1079, 20–29, DOI:10.1016/j.aca.2019.05.064.
- Z. He, W. Zhao, J. W. Y. Lam, Q. Peng, H. Ma, G. Liang, Z. Shuai and B. Z. Tang, Nat. Commun., 2017, 8, 416, DOI:10.1038/s41467-017-00362-5.
- (a) J. Korang, W. R. Grither and R. D. McCulla, J. Am. Chem. Soc., 2010, 132, 4466–4476, DOI:10.1021/ja100147b; (b) M. Schmiedtchen, I. Maisuls, H. Siera, J. Balszuweit, C. Wölper, M. Giese, G. Haberhauer, C. A. Strassert and J. Voskuhl, Angew. Chem., Int. Ed., 2025, 64, e202414326, DOI:10.1002/anie.202414326; (c) K. H. Choi, J. M. Kim, W. J. Chung and J. Y. Lee, Molecules, 2021, 26, 2804, DOI:10.3390/molecules26092804; (d) S. Bhandary, R. van Deun, A. M. Kaczmarek and K. van Hecke, Chem. Sci., 2022, 13, 10308–10314, 10.1039/D2SC03729G.
- J. Chen, T. Yu, E. Ubba, Z. Xie, Z. Yang, Y. Zhang, S. Liu, J. Xu, M. P. Aldred and Z. Chi, Adv. Opt. Mater., 2019, 7, 1801593, DOI:10.1002/adom.201801593.
- B. Roy, I. Maisuls, J. Zhang, F. C. Niemeyer, F. Rizzo, C. Wölper, C. G. Daniliuc, B. Z. Tang, C. A. Strassert and J. Voskuhl, Angew. Chem., Int. Ed., 2022, 61, e202111805, DOI:10.1002/anie.202111805.
- M. Zander and G. Kirsch, Z. Naturforsch., A: Phys. Sci., 1989, 44, 205–209, DOI:10.1515/zna-1989-0306.
- M. Jiang, J. Guo, B. Liu, Q. Tan and B. Xu, Org. Lett., 2019, 21, 8328–8333, DOI:10.1021/acs.orglett.9b03106.
- Y. Chen, B. Jing, J. Li and J. Gong, Mater. Chem. Front., 2022, 6, 1874–1881, 10.1039/D2QM00175F.
- (a) N. Biot and D. Bonifazi, Chem. – Eur. J., 2020, 26, 2904–2913, DOI:10.1002/chem.201904762; (b) D. Romito and D. Bonifazi, Helv. Chim. Acta, 2023, 106, e202200159, DOI:10.1002/hlca.202200159.
- A. Kremer, A. Fermi, N. Biot, J. Wouters and D. Bonifazi, Chem. – Eur. J., 2016, 22, 5665–5675, DOI:10.1002/chem.201504328.
- S. Mehrparvar, C. Wölper and G. Haberhauer, Angew. Chem., Int. Ed., 2023, 62, e202304202, DOI:10.1002/anie.202304202.
- A. F. Cozzolino, I. Vargas-Baca, S. Mansour and A. H. Mahmoudkhani, J. Am. Chem. Soc., 2005, 127, 3184–3190, DOI:10.1021/ja044005y.
- (a) C. Bleiholder, R. Gleiter, D. B. Werz and H. Köppel, Inorg. Chem., 2007, 46, 2249–2260, DOI:10.1021/ic062110y; (b) L. Camilli, C. Hogan, D. Romito, L. Persichetti, A. Caporale, M. Palummo, M. Di Giovannantonio and D. Bonifazi, JACS Au, 2024, 4, 2115–2121, DOI:10.1021/jacsau.4c00325; (c) T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76, DOI:10.1002/1521-3773(20020104)41:1%3C48::AID-ANIE48%3E3.0.CO;2-U.
- G. Haberhauer and R. Gleiter, Angew. Chem., Int. Ed., 2020, 59, 21236–21243, DOI:10.1002/anie.202010309.
- (a) D. Romito, H. Kählig, P. Tecilla, G. Sosso and D. Bonifazi, Chem. – Eur. J., 2024, e202401346, DOI:10.1002/chem.202401346; (b) S. Mehrparvar, C. Wölper, R. Gleiter and G. Haberhauer, Org. Mater., 2022, 4, 43–52, DOI:10.1055/a-1883-6076.
- P. C. Ho, J. Rafique, J. Lee, L. M. Lee, H. A. Jenkins, J. F. Britten, A. L. Braga and I. Vargas-Baca, Dalton Trans., 2017, 46, 6570–6579, 10.1039/c7dt00612h.
- (a) P. Wonner, A. Dreger, L. Vogel, E. Engelage and S. M. Huber, Angew. Chem., Int. Ed., 2019, 58, 16923–16927, DOI:10.1002/anie.201910639; (b) S. Mehrparvar, C. Wölper, R. Gleiter and G. Haberhauer, Angew. Chem., Int. Ed., 2020, 132, 17154–17161, DOI:10.1002/ange.202005374.
- J. Liu, M. Zhou, R. Deng, P. Zheng and Y. R. Chi, Nat. Commun., 2022, 13, 4793, DOI:10.1038/s41467-022-32428-4.
- (a) X. Lian, J. Sun and Y. Zhan, J. Lumin., 2023, 263, 120023, DOI:10.1016/j.jlumin.2023.120023; (b) J. Chen, X. Chen, Y. Liu, Y. Li, J. Zhao, Z. Yang, Y. Zhang and Z. Chi, Chem. Sci., 2021, 12, 9201–9206, 10.1039/d1sc02094c.
- X. Miao, Z. Cai, J. Li, L. Liu, J. Wu, B. Li, L. Ying, F. Silly, W. Deng and Y. Cao, ChemPhotoChem, 2021, 5, 626–631, DOI:10.1002/cptc.202100041.
- K. Nishino, Y. Ogiwara and N. Sakai, Chem. – Eur. J., 2018, 24, 10971–10974, DOI:10.1002/chem.201802475.
- (a) C. Chen, Z. Chi, K. C. Chong, A. S. Batsanov, Z. Yang, Z. Mao, Z. Yang and B. Liu, Nat. Mater., 2021, 20, 175–180, DOI:10.1038/s41563-020-0797-2; (b) K. C. Chong, C. Chen, C. Zhou, X. Chen, D. Ma, G. C. Bazan, Z. Chi and B. Liu, Adv. Mater., 2022, 34, 2201569, DOI:10.1002/adma.202201569.
- (a) W. Ding, C. Wang, J. R. Tan, C. C. Ho, F. León, F. García and N. Yoshikai, Chem. Sci., 2020, 11, 7356–7361, 10.1039/d0sc02737e; (b) M. Bielawski, Licentiate Thesis, Stockholm University, 2008 Search PubMed; (c) M. Bielawski, Dissertation thesis, Stockholm University, 2011 Search PubMed.
- M. Wang, Q. Fan and X. Jiang, Org. Lett., 2016, 18, 5756–5759, DOI:10.1021/acs.orglett.6b03078.
- K. Nishino, Y. Ogiwara and N. Sakai, Eur. J. Org. Chem., 2017, 2017, 5892–5895, DOI:10.1002/ejoc.201701155.
- S. Riebe, C. Wölper, J. Balszuweit, M. Hayduk, M. E. Gutierrez Suburu, C. A. Strassert, N. L. Doltsinis and J. Voskuhl, ChemPhotoChem, 2020, 4, 398–406, DOI:10.1002/cptc.202000002.
- R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, Oxford, U.K., 1990 Search PubMed.
- M. A. Blanco, A. Martín Pendás and E. Francisco, J. Chem. Theory Comput., 2005, 1, 1096–1109, DOI:10.1021/ct0501093.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, J. Appl. Crystallogr., 2021, 54, 1006–1011, DOI:10.1107/S1600576721002910.
- (a) A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100, DOI:10.1103/physreva.38.3098; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789, DOI:10.1103/physrevb.37.785; (c) B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200–206, DOI:10.1016/0009-2614(89)87234-3.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465, DOI:10.1002/jcc.21759.
- F. M. Bickelhaupt and E. J. Baerends, in Reviews in Computational Chemistry, ed. K. B. Lipkowitz and D. B. Boyd, Wiley, 1993, vol. 15, pp. 1–86 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2389699, 2389700 and 2403909–2403915. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ce01248h |
| This journal is © The Royal Society of Chemistry 2025 |