DOI:
10.1039/D4CE01214C
(Paper)
CrystEngComm, 2025,
27, 931-938
Structural analysis and water adsorption properties of chloranilate anion–terpyridine metal complexes forming hydrogen-bonded frameworks†
Received
3rd December 2024
, Accepted 14th January 2025
First published on 20th January 2025
Abstract
Emerging microporous hydrogen-bonded organic frameworks (HOFs) are expected to overcome water shortages owing to their potential in harvesting and releasing water at low energies. To investigate their water adsorption properties, two distinct types of HOFs are synthesized. The frameworks are composed of chloranilic acid (H2CA), terpy complexes, and crystal water. The complexes are denoted as [M(terpy)2](H2CA)0.5(HCA)(CA)0.5·H2O (M–A) and [M(terpy)2](CA)·6H2O (M–B), where M represents Fe2+, Co2+, or Ni2+. Structural characterization results reveal that M–A contains H2CA, chloranilate monoanion (HCA−), and chloranilate dianion (CA2−). M–A complexes comprise one-dimensional chains of H2CA–CA2− and HCA−–water, forming a three-dimensional framework via hydrogen bonding with [M(terpy)2]2+. M–B complexes contain [M(terpy)2]2+, CA2−, and six water molecules. The CA2− and water molecules form a two-dimensional layered arrangement via hydrogen bonding, and the water molecules form tetramers within the layers. N2 adsorption measurements indicate that both the M–A and M–B complexes are non-porous. In water adsorption–desorption experiments, M–A adsorbs one water molecule per unit with minimal structural changes, whereas M–B adsorbs six water molecules per unit and undergoes a multi-step isothermal adsorption process, indicating significant structural changes. Furthermore, the adsorption properties were observed to vary with the central metal ion. The differing adsorption behaviors of M–B may be attributed to the hydrogen bonding distances within the crystalline water tetramers forming the hydrogen bonded network. The HOFs explored in this study may be utilized for selectively adsorbing water molecules in low-humidity environments, such as arid regions.
Introduction
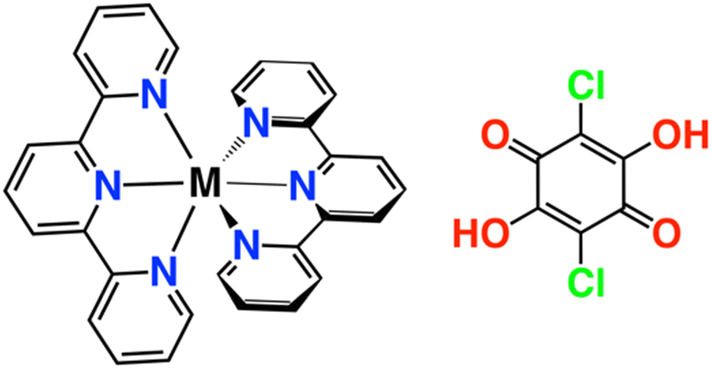
Hydrogen-bonded organic frameworks (HOFs) have attracted considerable attention as a new category of porous crystalline solids.1–5 They are formed by the self-assembly of organic molecules and metal complexes through a variety of weak bonding forces, including hydrogen bonding, π–π, CH–π, van der Waals, dipole–dipole, halogen bonding, and cation–π interactions. These interactions are weaker than covalent and coordination bonds, resulting in increased structural flexibility, reduced directionality, and reversible bonding. The high solubility of HOFs facilitates recrystallization, while their exceptional structural recoverability distinguishes them from other porous crystalline materials, such as metal–organic frameworks6–8 and covalent–organic frameworks,9–11 which rely on coordination bonds and covalent bonds, respectively. HOFs enable the systematic modification of material properties, such as porosity, crystallinity, and structural flexibility, by strategic selection of constituent components, providing a flexible framework for designing tailored functional materials. The discovery of the porous properties of HOFs has expanded their use in various applications, such as gas adsorption12–14 and separation,15–17 proton conductivity,18–20 luminescence,21–23 and catalysis.24–26 Particularly, the gate-opening phenomenon, which governs the influence of structural flexibility on the adsorption properties, plays a critical role in enabling functions such as the storage and separation of guest molecules.27–29 HOFs form supramolecular structures via hydrogen bonding interactions mediated by crystal water molecules.30,31 Significant structural modifications of HOFs have been reported owing to the adsorption and desorption of crystal water.32,33 Porous materials that exhibit large conformational changes show drastic increase or decrease in adsorption in a narrow pressure region and are therefore expected to have considerable potential in harvesting and releasing water easily.34,35 Recently, research on the recovery of wastewater to overcome global water shortages has attracted considerable attention.36–40 Thus, materials that can selectively adsorb water molecules in low-humidity environments, such as in arid regions, are increasingly becoming popular. To utilize the physicochemical properties of HOFs and prepare materials with selective water adsorption ability, this study focused on HOFs composed of chloranilic acid (H2CA) and metal complexes. H2CA is an excellent component for the assembly of HOFs because of its strong hydrogen-bond donor and acceptor properties, as well as its ability to facilitate strong stacking interactions.41–44 The OH groups in H2CA are readily deprotonated because of the inductive effect of the electronegative chlorine substituents, which stabilizes the anion. The pKa values of 0.73 and 3.08 indicate that H2CA readily forms various states of deprotonation and protonation under mild conditions.45 H2CA has attracted considerable attention in the study of complexes with redox properties.46–48 Herein, we focused on the hydrogen-bonded framework of H2CA in which the hydroxyl groups act as proton donors and acceptors. H2CA is expected to form a variety of hydrogen bonds, depending on its charge state. Furthermore, it is expected to form frameworks mediated by water molecules, in addition to hydrogen bonds. The charge state of H2CA can be predicted to considerably affect its water adsorption properties. To controllably modify the hydrogen bonded framework of H2CA and to develop its water adsorption properties, we synthesized two types of HOFs (M–A = [M(terpy)2](H2CA)0.5(HCA)(CA)0.5·H2O and M–B = [M(terpy)2](CA)2·6H2O, where M = Fe2+, Co2+ and Ni2+ and terpy = 2,2′;6′,2′′-terpyridine) comprising three types of terpyridine complexes with different central metal ions and chloranil acids with different charge states (Scheme 1). We evaluated the dependence of water adsorption properties on the crystal structures and central metal ion species of the complexes, as well as the charge states of H2CA. The results of this study provide new insights into the factors controlling the water adsorption behavior of HOFs.
 |
| | Scheme 1 [M(terpy)2]2+ complex (left) and chloranilic acid (H2CA, right). | |
Experimental
All chemicals and solvents were procured from commercial suppliers and used without additional purification. Fe–A and Fe–B were synthesized under N2 atmosphere, whereas Co–A, Co–B, Ni–A, and Ni–B were synthesized under atmospheric conditions. To assess the thermal stability, thermogravimetric analysis (TGA) with differential thermal analysis (DTA) was performed using an SII EXSTAR6000 TG/DTA 6300 analyzer under N2 atmosphere at a heating rate of 5 °C min−1. Powder X-ray diffraction (PXRD) measurements were performed on the Rigaku MiniFlex X-ray diffractometer using Cu-Kα radiation (λ = 1.5418 Å). The IR spectra were recorded at 298 K in the 4000–550 cm−1 spectral range using an attenuated total reflectance device (ATR) coupled to an FT-IR spectrophotometer (JASCO FTIR-6100). The X-ray tube was operated at 40 kV and 40 mA, and the temperature was varied from 293 K to 298 K. Gas and vapor adsorption isotherms were obtained using an automatic volumetric adsorption analyzer (BELSORP-max). Prior to the adsorption measurements, all samples were dehydrated in a dynamic vacuum at 393 K for 6 h. Gas and vapor adsorption isotherms were obtained using an automatic volumetric adsorption analyzer (BELSORP-max).
Elemental analysis of C, H, and N was performed in the Service Center of the Elementary Analysis of Organic Compounds, Kyushu University. Single crystal XRD data were recorded in the temperature range of 293 K to 298 K using the Rigaku oxford diffraction XtaLAB mini II instrument equipped with Mo-Kα radiation source (λ = 0.71073 Å). The crystal structures were solved by direct methods and refined using full-matrix least squares on F2 with SHELXL-2015.49 All non-hydrogen atoms were refined using the full-matrix least-squares method on F2 by utilizing SHELXL-2015. All calculations were performed using the OLex2 crystallographic software.50 All hydrogen atoms were calculated at idealized positions and refined with the riding models. A summary of the crystallographic data and structural refinement parameters is provided in Table 1. The Cambridge Crystallographic Data Centre (CCDC) numbers 2374727 (Fe–A), 2374762 (Co–A), 2374763 (Ni–A), 2374764 (Fe–B), 2374765 (Co–B), and 2374766 (Ni–B) contain supplementary crystallographic data for this study.
Table 1 Crystallographic information and structural refinement parameters for M–A and M–B
| Crystal |
Fe–A
|
Co–A
|
Ni–A
|
Fe–B
|
Co–B
|
Ni–B
|
|
R
1 = Σ||Fo| – |Fc||/Σ|Fo| and Rw = (Σw(|Fo| – |Fc|)2/ΣwFo2)1/2.
|
| Chemical formula |
C42H26Cl4FeN6O9 |
C42H26Cl4CoN6O9 |
C42H26Cl4NiN6O9 |
C36H34Cl2FeN6O10 |
C36H34Cl2CoN6O10 |
C36H34Cl2NiN6O10 |
| Formula weight |
956.16 |
959.42 |
959.20 |
837.44 |
840.52 |
840.46 |
|
T, K |
293 |
293 |
293 |
293 |
293 |
293 |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P |
P |
P |
P |
|
a, Å |
8.9753(2) |
8.9669(2) |
8.9581(3) |
9.0180(2) |
9.03554(12) |
9.0426(2) |
|
b, Å |
10.8907(2) |
10.9875(2) |
11.0350(3) |
9.02320(10) |
9.04710(13) |
9.0675(3) |
|
c, Å |
20.9485(4) |
20.9096(4) |
20.9833(5) |
22.2494(3) |
22.3304(3) |
22.4477(6) |
|
a, degrees |
85.702(2) |
85.6270(10) |
85.086(2) |
91.6850(10) |
92.0770(12) |
92.511(2) |
|
b, degrees |
80.124(2) |
80.052(2) |
80.136(2) |
95.6840(10) |
95.9851(11) |
96.200(2) |
|
g, degrees |
85.417(2) |
85.213(2) |
85.180(2) |
90.432(2) |
90.5673(11) |
90.655(2) |
|
V, Å3 |
2006.95(7) |
2018.03(7) |
2030.99(10) |
1800.68(5) |
1814.09(4) |
1827.81(9) |
|
Z
|
2 |
2 |
2 |
2 |
2 |
2 |
|
D
calc, g cm−1 |
1.582 |
1.579 |
1.568 |
1.545 |
1.539 |
1.527 |
| μ, mm−1 |
0.708 |
0.756 |
0.807 |
0.636 |
0.688 |
0.744 |
| Refs. meas. |
35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 122 122 |
38624 |
36938 |
34721 |
35106 |
35522 |
| Indep. refs. |
11624 |
11660 |
11630 |
10443 |
10517 |
10601 |
| Refs. used |
11624 |
11660 |
11630 |
10443 |
10517 |
10601 |
|
R
int
|
0.0484 |
0.0279 |
0.0198 |
0.0287 |
0.0208 |
0.0287 |
|
R
1
|
0.0524 |
0.0516 |
0.0432 |
0.0460 |
0.0421 |
0.0546 |
|
R
all
|
0.0673 |
0.0715 |
0.0557 |
0.0731 |
0.0598 |
0.0847 |
|
R
w(F2)a |
0.1418 |
0.1346 |
0.1071 |
0.1025 |
0.0929 |
0.1185 |
| Max res., e− Å−3 |
0.836 |
1.848 |
0.973 |
0.286 |
0.329 |
0.676 |
| Min res., e− Å−3 |
−0.749 |
−1.502 |
−0.880 |
−0.372 |
−0.356 |
−0.400 |
| GOF |
1.096 |
1.032 |
1.017 |
1.019 |
1.031 |
1.046 |
| CCDC |
2374727
|
2374762
|
2374763
|
2374764
|
2374765
|
2374766
|
The single crystal of Fe–A was synthesized in a glass tube under N2 atmosphere, varying the temperature from 293 K to 298 K. An aqueous solution (1 mL) of H2CA (5 mM) was poured in a glass tube. Next, a mixture solution (1 mL) of H2O and EtOH containing Fe(SO4)2·7H2O, terpy (5 mM) was also poured into the same glass tube without mixing the two solutions. Purple platelet-like crystals began to form after one week. A microcrystalline powder was obtained by mixing the two solutions under N2 atmosphere for overnight. Upon stirring, a purple powder was obtained. The suspension was filtered, washed with water and ethanol, and dried in air. The single crystals and microcrystalline powders of Co–A and Ni–A were synthesized following the same procedure as that used for the synthesis of Fe–A. However, Co–A and Ni–A were synthesized using CoCl2·6H2O and NiCl2·6H2O, respectively, under atmospheric conditions. Single-crystal XRD analysis was performed at 293 K. Elemental analysis (%) of Fe–A. Calcd. for C42H26Cl4FeN6O9: C, 52.75; H, 2.74; N, 8.79. Found: C, 52.45; H, 2.81; N, 8.68. Elemental analysis (%) of Co–A. Calcd. for C42H26Cl4CoN6O9: C, 52.58; H, 2.73; N, 8.76. Found: C, 52.51; H, 2.78; N, 8.77. Elemental analysis (%) of Ni–A. Calcd. for C42H26Cl4NiN6O9: C, 52.59; H, 2.73; N, 8.76. Found: C, 52.54; H, 2.77; N, 8.77. The IR spectra of M–A are shown in Fig. S18.†
The single crystal of Fe–B was synthesized in a glass tube under a N2 atmosphere at range of 293 K to 298 K. An aqueous solution (1 mL) of chlorinic acid (5 mM) and NaOH aq. (10 mM) was poured in a glass tube. Next, a mixture solution of H2O and EtOH containing FeCl2·4H2O, terpy (5 mM) was poured into the same glass tube without mixing the two solutions. Purple platelet-like crystals began to form after one week. A microcrystalline powder was obtained by mixing the two solutions for overnight. Upon stirring, a purple powder was obtained. The single crystals and microcrystalline powders of Co–B and Ni–B were synthesized following the same procedure as that used for the synthesis of Fe–B. However, Co–B and Ni–B were synthesized under atmospheric conditions. It is thought that the addition of NaOH during the synthesis of M–B caused the release of all chloranilic acid protons and CA2− became the counter anion. Therefore, the crystal structure of M–B is considered to be different from that of M–A. Elemental analysis (%) of Fe–B. Calcd. for C36H34Cl2FeN6O10: C, 51.63; H, 4.09; N, 10.04. Found: C, 51.37; H, 3.99; N, 9.97. Elemental analysis (%) of Co–B. Calcd. for C36H34Cl2CoN6O10: C, 51.44; H, 4.08; N, 10.00. Found: C, 51.19; H, 3.96; N, 10.00. Elemental analysis (%) of Ni–B. Calcd. for C36H34Cl2NiN6O10: C, 51.46; H, 4.08; N, 10.00. Found: C, 51.26; H, 3.95; N, 10.00. The IR spectra of M–B are shown in Fig. S19.†
Results and discussion
Structure of M–A (M = Fe, Co, Ni)
The crystallographic information and structural refinement parameters for M–A, obtained from single crystal structural analyses, are given in Table 1. M–A crystallizes in the triclinic space group P and has two formula units (Z = 2) per unit cell. In M–A, the asymmetric unit of the structure is composed of a crystallographically independent unit of the [M(terpy)2]2+ cation, half of the neutral H2CA, a chloranilate monoanion (HCA−), half of the chloranilate dianion (CA2−), and one crystal water molecule. The IR spectra of M–A, Fe–A, Co–A, and Ni–A exhibit absorption bands at 3187, 3190, and 3184 cm−1, respectively, which are indicative of hydroxyl (OH) stretching vibrations associated with HCA− and H2CA. Additionally, the bands that appear at 1380, 1386, and 1381 cm−1 correspond to OH bending vibrations. These findings imply the coexistence of HCA− and H2CA within the crystal structure.51Fig. 1a shows the crystal structure of Co–A. Both Fe–A and Ni–A exhibit crystal packing structures identical to those of Co–A, in which the metal ions are hexacoordinated to the nitrogen atoms of the two terpy ligands. The distances and angles between the metal ions and nitrogen atoms in M–A are presented in Table S1.† The crystal packing structure of M–A is characterized by a supramolecular framework stabilized due to the hydrogen bonds formed by H2CA, HCA−, CA2−, crystal water molecules, and [M(terpy)2]2+ cations. The crystal packing structure of M–A consists of an [M(terpy)2]2+ complex, HCA−–crystal water, and H2CA–CA2− layers oriented along the a-axis (Fig. 1b). The [M(terpy)2]2+ complexes are organized into one-dimensional chains through weak π–π interaction of the terpy ligand moieties (Fig. 1c). The π–π interaction distances increase with respect to the central metal ion, with Fe–A showing the shortest distance, followed by Co–A and Ni–A (Table 2). Furthermore, the [M(terpy)2]2+ complex forms weak hydrogen bonds with H2CA, HCA−, CA2−, and a crystal water molecule, as shown by the adjacent distances listed in Table 2. HCA− is dimerized by hydrogen bond interaction via O–H⋯O (Fig. 1f). Owing to the interaction between the two hydrogen bonds and two crystal water molecules, the dimers align in the form of a linear chain that resembles a step pattern (Fig. 1d). Additionally, H2CA and CA2− are formed as one-dimensional chains in an alternating arrangement via hydrogen bonds (Fig. 1e). In the c-axis orientation, chains of HCA− dimer and crystal water molecules are positioned perpendicular to the chains of H2CA and CA2− (Fig. 1g). Crystallographic analyses reveal that [Fe(terpy)2]2+ and [Co(terpy)2]2+ exhibit low-spin states, as indicated by the bond distance between the central metal ion and nitrogen atoms (Table S1†).
 |
| | Fig. 1 Crystal structure of Co–A. a) Asymmetric unit of Co–A. b) Crystal packing structure along the a-axis. c) [M(terpy)2]2+ complexes layer. d) HCA− – crystal water layer. e) H2CA–CA2− layer. f) Hydrogen bonding of HCA− and crystal water. g) Hydrogen bonding of H2CA and CA2−. | |
Table 2 Hydrogen bond and π–π interaction distances in M–A
| Crystal |
Fe–A
|
Co–A
|
Ni–A
|
| #1 (1 − x, −y, 2 − z), #2 (+x, −1 + y, +z), #3 (−x, −y, 2 − z), #4 (2 − x, −y, 1 − z), #5 (1 + x, +y, +z), #6 (1 − x, 2 − y, 1 − z), #7 (−x, −y, 2 − z), #8 (−x, 2 − y, 2 − z), #9 (−1 + x, +y, +z). |
| HCA−⋯HCA− (Å) |
O(3)⋯O(6)#1 |
2.692(3) |
O(3)⋯O(6)#1 |
2.695(3) |
O(3)⋯O(6)#6 |
2.702(2) |
| HCA−⋯water (Å) |
O(5)⋯O(9)#2 |
2.913(3) |
O(5)⋯O(9)#2 |
2.917(3) |
O(5)⋯O(9)#6 |
2.932(2) |
|
|
O(5)⋯O(9)#3 |
2.929(3) |
O(5)⋯O(9)#3 |
2.935(3) |
O(5)⋯O(9)#7 |
2.903(3) |
| H2CA⋯CA2− (Å) |
O(1)⋯O(7)#4 |
3.007(5) |
O(1)⋯O(7) |
2.700(3) |
O(1)⋯O(7)#8 |
3.161(3) |
|
|
O(1)⋯O(8)#5 |
2.663(7) |
O(1)⋯O(8)#6 |
2.971(3) |
O(1)⋯O(8)#9 |
3.396(4) |
| π–π interaction (Å) |
|
4.0194(17) |
|
3.9187(17) |
|
3.9028(14) |
Structure of M–B (M = Fe, Co, Ni)
Previously, Kawata et al. reported the crystal structure of Fe–B.52 The crystallographic information and structural refinement parameters for M–B, obtained from single crystal structural analyses, are given in Table 1. M–B crystallizes in the triclinic space group P and has two formula units (Z = 2) per unit cell. In M–B, the asymmetric unit of the structure is composed of a crystallographically independent unit of [M(terpy)2]2+ cation, two types of CA2− anions, and six crystal water molecules. The IR spectra of M–B did not reveal any bands associated with the OH stretching and bending modes that were identified in M–A. This finding suggests that HCA− and H2CA are not present in the crystal structure of M–B; rather, it indicates the presence of the CA2− dianion. Fig. 2a shows the crystal structure of Co–B. Similar to the structures of Fe–A and Co–A, the bond distances between the central metal ion and nitrogen atoms suggest that the terpy complexes in Fe–B and Co–B are in a low-spin state (Table S2†). The crystal packing of M–B comprises a hydrogen bond-supported supramolecular framework constructed from CA2−, crystal water molecules, and [M(terpy)2]2+ cations. As shown in Fig. 2b, along the b-axis of M–B, the crystal packing structure consists of an accumulated [M(terpy)2]2+ complex layer and a CA2− dianion–crystal water layer. The [M(terpy)2]2+ complexes are arranged in a two-dimensional layer by weak π–π interaction of the terpy ligand moieties (Fig. 2c). The distances between the centroids of the terpy ligand moieties at π–π interaction sites are listed in Table 3. The CA2− dianions and crystal water molecules form two isomorphic types (types I and II) of layers, via hydrogen bond interactions, with slightly different hydrogen bond distances (Fig. 2d). The CA2− dianions form a one-dimensional chain with adjacent CA2− dianions via hydrogen bonding mediated by two crystal water molecules. Further, the one-dimensional chains interact with neighboring one-dimensional chains via hydrogen bonding, mediated by four clusters of crystal water molecules, to form a two-dimensional layer (Fig. 2e). The evaluated hydrogen bond distances and angles are listed in Table 3. Despite M–A and M–B sharing identical constituents, significant differences are evident from their crystal structures and the quantity of crystalline water that are attributable to the different state states of charge of H2CA. Although the structural framework of the [M(terpy)2]2+ is consistent across both of these materials, the compositions of counter anions differ from each other: M–A contains HCA− and CA2−, whereas M–B exclusively features CA−. Additionally, the crystalline water content varies, with M–A including one molecule and M–B containing six. In M–A, hydrogen bonding networks are established among H2CA and CA2−, and additionally among HCA− and the water molecules. Conversely, in M–B, alongside the hydrogen bonding networks involving CA2− and water molecules, a water tetramer structure is also observed.
 |
| | Fig. 2 Crystal structure of Co–B. a) Asymmetric unit of Co–B. b) Crystal packing structure along the b-axis c) [M(terpy)2]2+ complexes layer d) CA2−–crystal water layer e) hydrogen bonding of CA2− and crystal water in type I layer. | |
Table 3 Hydrogen bond and π–π interaction distances in M–B
| Crystal |
Fe–B
|
Co–B
|
Ni–B
|
| #1 (1 − x, 1 − y, 1 − z), #2 (+x, 1 − +y, 1 − z), #3 (2 − x, 1 − y, −z), #4 (1 + x, +y, +z), #5 (1 − x, 1 − y, −z), #6 (+x, 1 + y, +z), #7 (−x, 1 − y, 1 − z), #8 (−1 + x, +y, +z), #9 (1 − x, −y, 1 − z), #10 (2 − x, −y, −z). |
| CA2−⋯water type I (Å) |
O1⋯O5 |
2.982(3) |
O1⋯O5 |
2.988(2) |
O1⋯O5 |
2.996(4) |
| O1⋯O5#1 |
2.969(2) |
O1⋯O5#5 |
3.002(2) |
O1⋯O5#9 |
3.020(3) |
| O1⋯O7#2 |
2.828(2) |
O1⋯O7#6 |
2.854(2) |
O1⋯O7#2 |
2.856(3) |
| O5#1⋯O8#2 |
2.733(3) |
O5#5⋯O8#6 |
2.730(2) |
O5#9⋯O8#2 |
2.733(3) |
| O7#2⋯O8#2 |
2.889(2) |
O7#6⋯O8#6 |
2.867(2) |
O7#2⋯O8#2 |
2.864(3) |
| O7#2⋯O8#1 |
2.825(2) |
O7#6⋯O8#5 |
2.846(2) |
O7#2⋯O8#9 |
2.864(4) |
| CA2−⋯water type II (Å) |
O3⋯O6 |
2.958(3) |
O3⋯O6 |
2.972(3) |
O3⋯O6 |
2.997(4) |
| O3⋯O6#3 |
2.998(2) |
O3⋯O6#7 |
3.015(2) |
O3⋯O6#10 |
3.012(4) |
| O3⋯O9#4 |
2.828(2) |
O3⋯O9#8 |
2.845(2) |
O3⋯O9#4 |
2.854(3) |
| O6#3⋯O10#4 |
2.713(3) |
O6#7⋯O10#8 |
2.707(2) |
O6#10⋯O10#4 |
2.705(4) |
| O9#4⋯O10#4 |
2.915(3) |
O9#8⋯O10#8 |
2.889(2) |
O9#4⋯O10#4 |
2.881(3) |
| O9#4⋯O10#3 |
2.822(3) |
O9#8⋯O10#7 |
2.853(2) |
O9#4⋯O10#10 |
2.870(4) |
| π–π interaction |
|
4.0996(13) |
|
4.0165(12) |
|
4.1159(16) |
Characterization results of PXRD and TGA
PXRD data of bulk M–A and M–B, along with the simulated XRD patterns derived from the single-crystal structure data, are presented in Fig. S1 and S2.† The obtained PXRD patterns for both M–A and M–B exhibit sharp peaks that match well with the simulated XRD patterns, indicating that the purities of the synthesized M–A and M–B are high.
The TGA and DTA curves of M–A are shown in Fig. S3–S5.† The TGA and DTA curves of Fe–A, Co–A, and Ni–A indicate weight losses of 2.1%, 1.8%, and 1.7%, respectively, in the temperature range 30–120 °C. This observation can be attributed to the removal of crystal water molecules (calculated at 1.9% for Fe–A, Co–A, and Ni–A). Above 230 °C, the M–A crystal starts to decompose. This is indicated by an abrupt weight loss, which is caused by the decomposition of the [M(terpy)2]2+ complex, H2CA, HCA−, and CA2−. The TGA and DTA curves of M–B are shown in Fig. S6–S8.† The TGA profiles of Fe–B, Co–B, and Ni–B show a two-step weight loss of 12.8%, 12.1%, and 12.3%, respectively, in the temperature range of 30–120 °C. This observation can be attributed to the loss of four crystal water molecules (calculated at 8.6% for Fe–B, Co–B, and Ni–B) and two crystal water molecules (calculated at 4.3% for Fe–B, Co–B, and Ni–B). Above 160 °C, the M–B crystal starts to decompose. This is indicated by an abrupt weight loss, which is the result of decomposition of the [M(terpy)2]2+ complex and CA2−.
Water adsorption properties
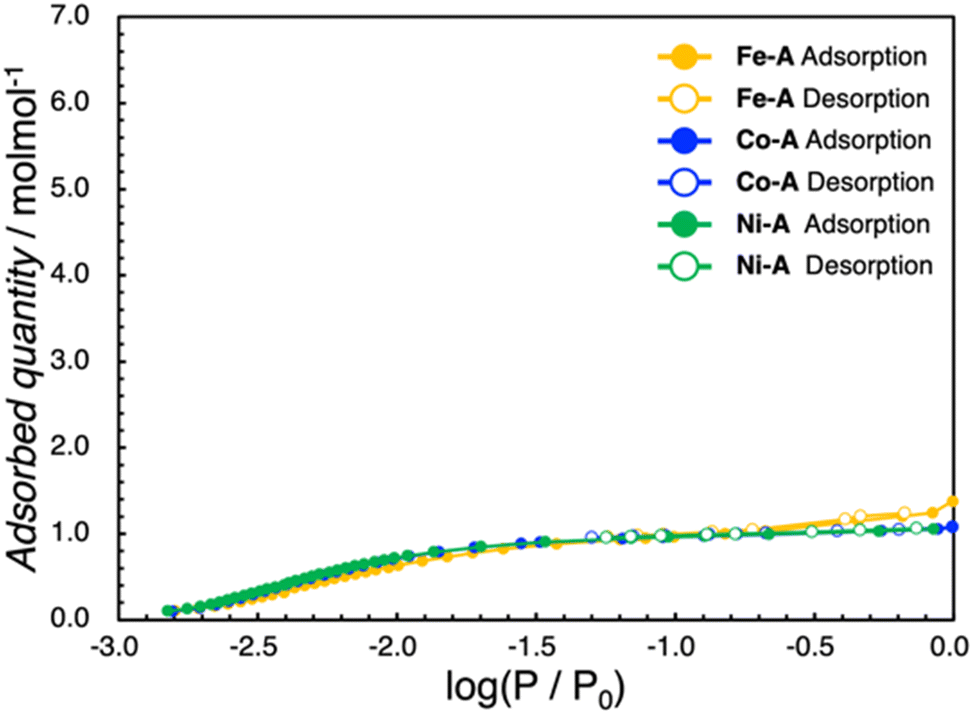
As hydrogen-bonded frameworks involve water molecules, we considered investigating the water adsorption properties of M–A and M–B. The as-synthesized M–A and M–B contain one and six crystal water molecules per asymmetric unit, respectively. To investigate the porosity of the samples, N2 adsorption isotherms of dehydrated Co–A and Co–B were recorded at liquid N2 temperature; however, no adsorption capacity was observed in any of the samples during the adsorption process (Fig. S15†). In conclusion, both the dehydrated samples are non-porous to N2. Other investigated complexes were also observed to be non-porous to N2. The water adsorption–desorption profiles of M–A and M–B are presented in Fig. 3 and 4, S16 and S17.† The dehydrated samples were then subjected to water adsorption and desorption measurements at 298 K. In all the M–A complexes, the quantity of adsorbed water increases in the low-pressure region (log(P/P0) = −3.0 to −2.5) and reaches a plateau at approximately log(P/P0) = −0.08. The quantity of adsorbed water further increases at high relative pressures. The quantity of water adsorbed at log(P/P0) = −0.08 coincides with the quantity of crystal water molecules (1.0 molecule) in the asymmetric unit of M–A. According to the International Union of Pure and Applied Chemistry classifications, the shape of the adsorption isotherm can be categorized as type-I, indicating a rigid framework. The Co–A complex shows no significant structural changes after the desolvation test (Fig. S9–S11†). Conversely, the water adsorption isotherms of the M–B complexes indicate a multi-step adsorption process below log(P/P0) = −0.5. The M–B complexes absorb 0.5 molecules of water per asymmetric M–B unit at around log(P/P0) = −1.9. As the value of log(P/P0) increases, the quantity of water adsorbed increases by an additional 0.5 and 1.0 molecules in discrete steps. Thereafter, four molecules of water are rapidly adsorbed by Fe–B and Co–B before reaching log(P/P0) = −0.5. The final quantity of adsorbed molecules of water is six, which corresponds to the number of crystal water molecules in the crystal structure of M–B. In contrast to Fe–B and Co–B, Ni–B shows a one-step increase in adsorption around log(P/P0) = −1.0. For Ni–B, the quantity of water adsorbed is 0.5, 0.5, 1.0, 2.0 and 2.0 molecules at approximately log(P/P0) = −2.1, −1.8, −1.4, −0.8 and −0.5, respectively. Unlike Fe–B and Co–B, a two-step adsorption process is observed between log(P/P0) values of −1.0 and −0.5 for Ni–B. Hysteresis is observed in all the M–B complexes during the desorption process, and all the absorbed water molecules are not removed as the pressure reduces at 298 K. M–B is non-porous; however, the multi-step adsorption process suggests structural changes accompanied by adsorption at each step (Fig. S12–S14†). In all the M–B complexes, the three-step adsorption process observed between log(P/P0) values of −2.5 and −1.0, can be attributed to stepwise adsorption in the CA2− layer. The sudden adsorption of four water molecules by Fe–B and Co–B may be due to the formation of water tetramers in the crystal structure. Among all the M–B complexes, the Ni–B complex exhibits the highest hydrogen bond distance for water tetramers. This suggests that the water molecules are gradually adsorbed in a stepwise manner by the Ni–B complex, and water tetramers are formed eventually. The cyclic water adsorption experiments corroborated the adsorption characteristics observed in the initial measurement. This finding indicates that the amorphous state induced by dehydration reverts to the crystalline state through the process of water reabsorption (Fig. S20 and S21†). These phenomena are further supported by the results of the PXRD measurements (Fig. S12–S14†).
 |
| | Fig. 3 Water adsorption isotherms at 298 K for Fe–A (yellow), Co–A (blue), and Ni–A (green). Filled circles indicate adsorption and open circles indicate desorption. | |
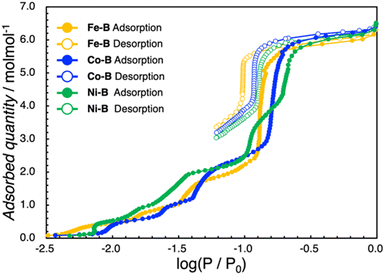 |
| | Fig. 4 Water adsorption isotherms at 298 K for Fe–B (yellow), Co–B (blue), and Ni–B (green). Filled circles indicate adsorption and open circles indicate desorption. | |
Conclusions
In this study, two types of HOFs, denoted as M–A and M–B, were synthesized. Each type of HOF exhibited distinct water adsorption properties, owing to variations in the hydrogen-bonded frameworks resulting from the manipulation of the proton count of H2CA. M–A comprised an assembly of one-dimensional chains of H2CA–CA2− and HCA−–water along with [M(terpy)2]2+ units, forming a three-dimensional framework via hydrogen bonding mediated by one crystal water molecule. M–B consisted of a two-dimensional layered configuration resulting from the hydrogen bonding interactions between CA2− and crystal water mediated by six crystal water molecules. Notably, the crystal water molecules within the layers of M–B complexes formed tetramers. Although both M–A and M–B were observed to be non-porous in the desolvation test, water adsorption measurements revealed an opposite behavior. In the water adsorption experiment, M–A indicated no structural changes, whereas M–B exhibited the capacity to adsorb six water molecules per unit and indicated a multi-step isothermal adsorption process, suggesting a substantial structural transformation during the water adsorption process. Moreover, water adsorption was found to be dependent on the type of the central metal ion. In the Fe–B and Co–B complexes, the adsorbed quantity of water increased in steps of 0.5, 0.5, 1, and 4 molecules in the low-pressure region (log(P/P0) = −2.5 to −0.5). In contrast, in the Ni–B complexes, the adsorption quantity of water increased in steps of 0.5, 0.5, 1, 2 and 2 molecules. The distinct adsorption process observed in M–B was attributed to the hydrogen bond distances of the crystal water tetramers within the hydrogen-bonded framework. These findings suggest that the water adsorption capacity can be modulated by modifying the hydrogen-bonded framework, and that even slight modifications in the metal ion species can influence the adsorption process. The materials studied in this work may be utilized for controlling humidity levels in confined spaces and enhancing water retention in arid areas.
Data availability
The data supporting this article have been included as part of ESI.†
Author contributions
A. Mishima: conceptualization, methodology, resources, investigation, writing – original draft. R. Mori: investigation. K. Kanazashi: investigation. T. Hamaguchi: resources. R. Ishikawa: resources. S. Kawata: conceptualization, resources, writing – review & editing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by JSPS KAKENHI Grant Number JP24K08457 and was partially supported by JSPS KAKENHI Grant Number JP24K08459 and Fukuoka University (Grant No. GW2405-000).
References
- B. Wang, R.-B. Lin, Z. Zhang, S. Xiang and B. Chen, J. Am. Chem. Soc., 2020, 142, 14399–14416 CrossRef CAS PubMed.
- I. Hisaki, C. Xin, K. Takahashi and T. Nakamura, Angew. Chem., Int. Ed., 2019, 58, 11160–11170 CrossRef CAS PubMed.
- J. Luo, J.-W. Wang, J.-H. Zhang, S. Laia and D.-C. Zhong, CrystEngComm, 2018, 20, 5884–5898 RSC.
- R.-B. Lin, Y. He, P. Li, H. Wang, W. Zhou and B. Chen, Chem. Soc. Rev., 2019, 48, 1362–1389 RSC.
- Z.-H. Zhu, H.-L. Wang, H.-H. Zou and F.-P. Liang, Dalton Trans., 2020, 49, 10708–10723 RSC.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- A. J. Howarth, A. W. Peters, N. A. Vermeulen, T. C. Wang, J. T. Hupp and O. K. Farha, Chem. Mater., 2017, 29, 26–39 CrossRef CAS.
- A. Schneemann, V. Bon, I. Schwedler, I. Senkovska, S. Kaskel and R. A. Fischer, Chem. Soc. Rev., 2014, 43, 6062–6096 RSC.
- C. S. Diercks and O. M. Yaghi, Science, 2017, 355, eaal1585 CrossRef PubMed.
- H. Wang, H. Wang, Z. Wang, L. Tang, G. Zeng, P. Xu, M. Chen, T. Xiong, C. Zhou, X. Li, D. Huang, Y. Zhu, Z. Wanga and J. Tang, Chem. Soc. Rev., 2020, 49, 4135–4165 RSC.
- R. Freund, O. Zaremba, G. Arnauts, R. Ameloot, G. Skorupskii, M. Dincă, A. Bavykina, J. Gascon, A. Ejsmont, J. Goscianska, M. Kalmutzki, U. Lächelt, E. Ploetz, C. S. Diercks and S. Wuttke, Angew. Chem., Int. Ed., 2021, 60, 23975–24001 CrossRef CAS PubMed.
- P. Li, P. Li, M. R. Ryder, Z. Liu, C. L. Stern, O. K. Farha and J. F. Stoddart, Angew. Chem., Int. Ed., 2019, 58, 1664–1669 CrossRef CAS PubMed.
- D. W. Kang, M. Kang, H. Kim, J. H. Choe, D. W. Kim, J. R. Park, W. R. Lee, D. Moon and C. S. Hong, Angew. Chem., Int. Ed., 2019, 58, 16152–16155 CrossRef CAS PubMed.
- J. Gao, Y. Cai, X. Qian, P. Liu, H. Wu, W. Zhou, D.-X. Liu, L. Li, R.-B. Lin and B. Chen, Angew. Chem., Int. Ed., 2021, 60, 20400–20406 CrossRef CAS PubMed.
- M. Zhao, L. Sun, Y. Yang, X.-S. Gu and C.-J.-S. Lai, Coord. Chem. Rev., 2024, 514, 215881 CrossRef CAS.
- Y. Yang, H. Zhang, Z. Yuan, J.-Q. Wang, F. Xiang, L. Chen, F. Wei, S. Xiang, B. Chen and Z. Zhang, Angew. Chem., Int. Ed., 2022, 61, e20220757 Search PubMed.
- P. Li, Y. He, Y. Zhao, L. Weng, H. Wang, R. Krishna, H. Wu, W. Zhou, M. OKeeffe, Y. Han and B. Chen, Angew. Chem., Int. Ed., 2015, 54, 574–577 CrossRef CAS PubMed.
- A. Karmakar, R. Illathvalappil, B. Anothumakkool, A. Sen, P. Samanta, A. V. Desai, S. Kurungot and S. K. Ghosh, Angew. Chem., Int. Ed., 2016, 55, 10667–10671 CrossRef CAS PubMed.
- X. Chen, R.-K. Huang, K. Takahashi, S. Noro, T. Nakamura and I. Hisaki, Angew. Chem., Int. Ed., 2022, 61, e202211686 CrossRef CAS PubMed.
- F.-F. Yang, X.-L. Wang, J. Tian, Y. Yin and L. Liang, Nat. Commun., 2024, 15, 3930 CrossRef CAS PubMed.
- Y. Han, T. Zhang, X. Chen, Q. Chen, J. Hao, W. Song, Y. Zeng and P. Xue, ACS Appl. Mater. Interfaces, 2021, 13, 32270–32277 CrossRef CAS PubMed.
- Y. Shi, S. Wang, W. Tao, J. Guo, S. Xie, Y. Ding, G. Xu, C. Chen, X. Sun, Z. Zhang, Z. He, P. Wei and B. Z. Tang, Nat. Commun., 2022, 13, 1882 CrossRef CAS PubMed.
- C.-C. Wang, C.-C. Yang, W.-C. Chung, G.-H. Lee, M.-L. Ho, Y.-C. Yu, M.-W. Chung, H.-S. Sheu, C.-H. Shih, K.-Y. Cheng, P.-J. Chang and P.-T. Chou, Chem. – Eur. J., 2011, 17, 9232–9241 CrossRef CAS PubMed.
- P. Zhai, C. Wang, Y. Li, D. Jin, B. Shang, Y. Chang, W. Liu, J. Gao and J. Hou, Nano Lett., 2024, 24, 8687–8695 CrossRef CAS PubMed.
- Q. Yin, E. V. Alexandrov, D.-H. Si, Q.-Q. Huang, Z.-B. Fang, Y. Zhang, A.-A. Zhang, W.-K. Qin, Y.-L. Li, T.-Fu Liu and D. M. Proserpio, Angew. Chem., Int. Ed., 2022, 61, e202115854 CrossRef CAS PubMed.
- B. Yu, T. Meng, X. Ding, X. Liu, H. Wang, B. Chen, T. Zheng, W. Li, Q. Zeng and J. Jiang, Angew. Chem., Int. Ed., 2022, 61, e202211482 CrossRef CAS PubMed.
- N. Nijem, H. Wu, P. Canepa, A. Marti, K. J. Balkus, Jr., T. Thonhauser, J. Li and Y. J. Chabal, J. Am. Chem. Soc., 2012, 134, 15201–15204 CrossRef CAS PubMed.
- D. Fairen-Jimenez, S. A. Moggach, M. T. Wharmby, P. A. Wright, S. Parsons and T. Düren, J. Am. Chem. Soc., 2011, 133, 8900–8902 CrossRef CAS PubMed.
- Q. Dong, X. Zhang, S. Liu, R.-B. Lin, Y. Guo, Y. Ma, A. Yonezu, R. Krishna, G. Liu, J. Duan, R. Matsuda, W. Jin and B. Chen, Angew. Chem., Int. Ed., 2020, 59, 22756–22762 CrossRef CAS PubMed.
- M. Tadokoro, Y. Ohata, Y. Shimazaki, S. Ishimaru, T. Yamada, Y. Nagao, T. Sugaya, K. Isoda, Y. Suzuki, H. Kitagawa and H. Matsui, Chem. – Eur. J., 2014, 20, 13698–13709 CrossRef CAS PubMed.
- S. B. Tayade, S. S. Bhat, R. Illathvalappil, V. M. Dhavale, V. A. Kawade, A. S. Kumbhar, S. Kurungot and C. Näther, CrystEngComm, 2018, 20, 1094–1100 RSC.
- J. Boonmak, M. Nakano, N. Chaichit, C. Pakawatchaid and S. Youngme, Dalton Trans., 2010, 39, 8161–8167 RSC.
- R. Yano, M. Yoshida, T. Tsunenari, A. Sato-Tomita, S. Nozawa, Y. Iida, N. Matsunaga, A. Kobayashi and Masako Kato, Dalton Trans., 2021, 50, 8696–8703 RSC.
- A. M. Kałuża, S. Mukherjee, S.-Q. Wang, D. J. O'Hearn and M. J. Zaworotko, Chem. Commun., 2020, 56, 1940–1943 RSC.
- S.-Q. Wang, S. Darwish, C. R. M. O. Matos, Z. M. Wong, A. Nalaparaju, J. Yifei Luo, X. Zhu, Z. Xu Zhang and M. J. Zaworotko, Inorg. Chem. Front., 2024, 11, 3254–3262 RSC.
- N. Hanikel, X. Pei, S. Chheda, H. Lyu, W. Jeong, J. Sauer, L. Gagliardi and O. M. Yaghi, Science, 2021, 374, 454–459 CrossRef CAS PubMed.
- C. Nie, N. Yan, C. Liao, C. Ma, X. Liu, J. Wang, G. Li, P. Guo and Z. Liu, J. Am. Chem. Soc., 2024, 146, 10257–10262 CrossRef CAS PubMed.
- W. Gong, X. Chen, M. Wahiduzzaman, H. Xie, K. O. Kirlikovali, J. Dong, G. Maurin, O. K. Farha and Y. Cui, J. Am. Chem. Soc., 2024, 146, 2141–2150 CrossRef CAS PubMed.
- A. H. Alawadhi, S. Chheda, G. D. Stroscio, Z. Rong, D. Kurandina, H. L. Nguyen, N. Rampal, Z. Zheng, L. Gagliardi and O. M. Yaghi, J. Am. Chem. Soc., 2024, 146, 2160–2166 CrossRef CAS PubMed.
- M. A. van der Veen, S. Canossa, M. Wahiduzzaman, G. Nenert, D. Frohlich, D. Rega, H. Reinsch, L. Shupletsov, K. Markey, D. E. De Vos, M. Bonn, N. Stock, G. Maurin and E. H. G. Backus, Adv. Mater., 2024, 36, 2210050 CrossRef CAS PubMed.
- S. Yagishita, A. Himegi, K. Kanazashi, T. Ohishi, R. Ishikawa, T. Hamaguchia and S. Kawata, Dalton Trans., 2017, 46, 2966–2973 RSC.
- S. Kawata, S. Kitagawa, H. Kumagai, T. Ishiyama, K. Honda, H. Tobita, K. Adachi and M. Katada, Chem. Mater., 1998, 10, 3902–3912 CrossRef CAS.
- S. Kawata, S. Kitagawa, H. Kumagai, C. Kudo, H. Kamesaki, T. Ishiyama, R. Suzuki, M. Kondo and M. Katada, Inorg. Chem., 1996, 35, 4449–4461 CrossRef CAS PubMed.
- K. Nagayoshi, Md. K. Kabir, H. Tobita, K. Honda, M. Kawahara, M. Katada, K. Adachi, H. Nishikawa, I. Ikemoto, H. Kumagai, Y. Hosokoshi, K. Inoue, S. Kitagawa and S. Kawata, J. Am. Chem. Soc., 2003, 125, 221–232 CrossRef CAS PubMed.
- K. Wallenfels and K. Friedrich, Chem. Ber., 1960, 93, 3070–3082 CrossRef CAS.
- J. A. DeGayner, I.-R. Jeon, L. Sun, M. Dincă and T. D. Harris, J. Am. Chem. Soc., 2017, 139, 4175–4184 CrossRef CAS PubMed.
- K. A. Collins, R. J. Saballos, M. S. Fataftah, D. Puggioni, J. M. Rondinelli and D. E. Freedman, Chem. Sci., 2020, 11, 5922–5928 RSC.
- K. Mou, F. Meng, Z. Zhang, X. Li, M. Li, Y. Jiao, Z. Wang, X. Bai and F. Zhang, Angew. Chem., Int. Ed., 2024, e202402446 CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A:Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- A. Pawlukojć, G. Bator, L. Sobczyk, E. Grech and J. Nowicka-Scheibe, J. Phys. Org. Chem., 2003, 16, 709–714 CrossRef.
- Md. K. Kabir, H. Tobita, H. Matsuo, K. Nagayoshi, K. Yamada, K. Adachi, Y. Sugiyama, S. Kitagawa and S. Kawata, Cryst. Growth Des., 2003, 3, 791–798 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *,
Runa
Mori
,
Ken
Kanazashi
,
Tomohiko
Hamaguchi
*,
Runa
Mori
,
Ken
Kanazashi
,
Tomohiko
Hamaguchi