
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAqueous solution as a playground of {MoO4}, {Mo4O12}, {Mo8O26}, {Mo8V5O40} and {V7Mo2O27} species in the presence of carboxylic acids and [Co(C2O4)(NH3)4]+ or [Co(en)3]3+ cations†
D.
Kuzman
*a,
V.
Damjanović
 b,
M.
Cindrić
*a and
V.
Vrdoljak
a
b,
M.
Cindrić
*a and
V.
Vrdoljak
a
aDepartment of Chemistry, University of Zagreb Faculty of Science, Horvatovac 102a, Zagreb, Croatia. E-mail: marina.chem@pmf.hr
bDepartment of Chemistry and Biochemistry, University of Zagreb School of Medicine, Šalata 3, Zagreb, Croatia
First published on 23rd January 2025
Abstract
Nine tetraammineoxalatocobalt(III) salts built of different oxomolybdate anions, i.e. {β-Mo8O26}4− (in 1, 2, and 6), {Na2Mo8O29(H2O)4}n4m− (in 3), {[β-Mo8O26(H2O)2][γ-Mo8O26]}8− (in 4), {γ-Mo8O26(H2O)2}4− (in 5, 7), {Mo4O12(C4H4O4)}n4m− (in 8), and {γ-Mo8O26}n4m− (in 9) were isolated as the product(s) of the reaction of sodium molybdate and [Co(C2O4)(NH3)4]NO3·H2O in the presence of acetic or succinic acid. Octamolybdate isomers, namely distinct β- and γ-octamolybdates along with complex cobalt(III) cations and water molecules are the building blocks of three-dimensional supramolecular frameworks (in 1, 2, and 4–7). In some cases, octamolybdate subunits are interconnected by sodium cations or a pair of shared oxygen atoms to form infinite anionic chains (in 3 and 9), or by coordinated succinate anions to assemble into a two-dimensional MOF (in 8). When [Co(CO3)(NH3)4]NO3·H2O was used as a precursor, neutral compounds [Mo5Co2O17(HCO3)(H4C4O4H)(NH3)7] (10) and [Mo7Co2O18(NH3)7] (11) were obtained. Furthermore, two molybdovanadates of proposed formula [Co(C2O4)(NH3)4]2[H2Mo8V5O40Na2(H2O)8] (12) and unique Na3[Co(en)3[V7Mo2O27] (13) were obtained in the reactions of sodium molybdate, ammonium vanadate and [Co(C2O4)(NH3)4]NO3·H2O or [Co(en)3]Cl3 in the presence of acetic or succinic acid, respectively. The products 1–9 were characterized in the solid state via single-crystal X-ray diffraction, infrared spectroscopy, and thermogravimetric and elemental analyses.
Introduction
Polyoxomolybdates represent compounds of highly versatile structures and have potential applications as functional materials in many scientific fields.1–3 The great majority of polyoxomolybdates are discrete molecules, generated via condensation of molybdate anions, MoO42−. However, sometimes the condensation process may continue yielding infinite molecular self-assemblies organized in 1D chains, 2D layers or 3D networks. The design of polyoxomolybdate architectures is non-trivial because their formation is highly dependent on the reaction conditions. Although the pH of the reaction mixture and temperature are considered the most important factors affecting speciation, other parameters such as nature of the used counter ion, ionic strength of the solution or the presence of chelating or reducing agents must also be considered. The ability to control the aggregation process or linkage of the building blocks to pre-establish structure-type as well as engineer the physical properties is an immeasurable challenge.4 One-pot synthesis is a common method used for the preparation of most polyoxomolybdates but despite its apparent simplicity, the above-mentioned conditions under which synthesis is performed act as crucial structure defining parameters. A slight change in one of these parameters can lead to a completely different polyoxomolybdate architecture that prevents the ability for reliable control or design of the framework. Polyoxomolybdates are generally anionic and associated with charge-balancing cations, such as alkali or alkaline earth cations. These cations play a crucial role in controlling the dissolution, precipitation, and purification of POMs. More complex cations, such as [MLn]x+ (where M is a d or an f block element; L is a mono or polydentate ligand), exhibit various interaction modes, including hydrogen bonding, ion–dipole interactions, partial covalency, and van-der-Waals interactions.5,6 These divergent interaction modes facilitate templating and stabilization from both within and outside the POMs, contribute to framework construction and supramolecular assembly, and enhance reactivity and speciation.7 The unique structure-directing properties of different organic, inorganic or inorganic–organic cations have been exploited to design new organic–inorganic hybrid materials using octamolybdate isomers as versatile building blocks.8–10 So far, eight isomers of octamolybdates are known: α, β, γ, δ, ε, ζ, η and θ,11 with the α- and β-isomers being the best-known and frequently encountered.12 In order to explain the inter-conversion of α- and β-forms, Xi and Ozawa13 proposed the γ-isomer as an intermediate in the isomerization process between these two forms. The α-isomer is built up from a ring of six MoO6 edge-shared octahedra bicapped by MoO4 tetrahedra,14 β-[Mo8O26]4− consists of three distinct types of edge-shared MoO6 groups15 unlike the γ-isomer7,16 which is built from six MoO6 octahedra and two MoO5 square pyramid subunits.The various structural isomers of octamolybdates are present and play a significant role in developing many organic–inorganic hybrid materials, which are of great interest in the field. For instance, the δ-isomer can be found in the compounds [(η-C5Me5Rh)2(μ-SMe)3]4[(Mo8O26)]·2CH3CN and [{Cu(4,4′-bipy)}4Mo8O26]. The ε-isomer is present in the compound [{Ni(H2O)2(4,4′-bipy)2}2Mo8O26], while the ξ-isomer is present in the compound [{M(phen)2}(ξ-Mo8O26)], where M can be either Ni or Co and phen refers to 1,10-phenantroline. Such isomeric forms of octamolybdates serve as versatile structural blocks, facilitating the formation of organo–inorganic hybrid materials with tailored properties.17
As part of our efforts to systematize18–20 the factors that direct the polyoxomolybdate composition, in this study we explored the influence of different Co(III) ammine complex cations, along with the pH value of reaction medium and the temperature at which the reaction is conducted. The selected cations exhibit varying stability when dissolved in solution, which can impact how they interact with other species. Moreover, they can act as either donors or acceptors of hydrogen bonds. This ability influences reactions, affects solubility, and contributes to the formation of different hybrids.
Furthermore, the similarity in chemistry of polyoxomolybdates and polyoxovanadates21 prompted us to investigate whether the addition of ammonium vanadate will influence the composition and structure of polyoxomolybdate. By applying solution-based methods we were able to isolate thirteen products of different nuclearity and composition (Scheme 1).
 | ||
| Scheme 1 Schematic representation of the diverse polyoxomolybdates formed utilizing solution-based synthesis methods. | ||
The structures of the seven compounds 3–9 are reported here for the first time. Compound 4 represents the first example of a salt containing both β- and γ-forms of octamolybdate anion in its structure.
Results and discussion
Synthesis
As a continuation of our previous research,14–16 herein we have investigated reaction systems containing [MoO4]2− anions, [Co(C2O4)(NH3)4]+ or [Co(CO3)(NH3)4]+ cations, and acetic or succinic acid at different pH values or temperatures (Scheme 1, Table 1). The pH value of the reaction mixtures was between 2 and 5 (Tables S1–S5†). Depending on the acid concentration, along with the reaction temperature we were able to isolate ten crystalline products of different nuclearity. The crystal structures of compounds 1, 2, and 11 are described in the literature14 but crystals were obtained under the different reaction conditions.| Method | [Co]a + MoO42− | |||||
|---|---|---|---|---|---|---|
| Acetic acid | Succinic acid | |||||
| R.T. | Reflux | Hydrothermal 110 °C | R.T. | Reflux | Hydrothermal 110 °C | |
| Compound | pH range | pH range | ||||
| a Compounds 1–9 were obtained by the solution based methods using [Co(C2O4)(NH3)4]NO3·H2O, while 10 and 11 were obtained using [Co(CO3)(NH3)4]NO3 as a [Co] precursor. b Compound 3 was also obtained by mechanochemical synthesis. | ||||||
| [Co(C2O4)(NH3)4]4[β-Mo8O26]·12H2O (1) | 2.0–5.0 | 3.5–5.0 | 3.5–5.0 | 3.5 | 3.5–5.0 | 3.5–5.0 |
| [Co(C2O4)(NH3)4]4[β-Mo8O26]·10H2O (2) | 2.0–5.0 | 3.5–5.0 | ||||
| [Co(C2O4)(NH3)4]4n[Na2Mo8O29(H2O)4]n·6nH2O (3)b | 3.5–5.0 | 4.5 | 3.5–5.0 | 3.5–5.0 | ||
| [Co(C2O4)(NH3)4]8[β-Mo8O26(H2O)2][γ-Mo8O26]·12H2O (4) | >3.5 | >3.5 | ||||
| [Co(C2O4)(NH3)4]4[γ-Mo8O26(H2O)2]·6H2O (5) | >3.5 | >3.5 | ||||
| [Co(C2O4)(NH3)4]4[β-Mo8O26]·4H2O·C4H6O4 (6) | 2.0–3.5 | 2.5–5.0 | 3.5–5.0 | |||
| [Co(C2O4)(NH3)4]4[γ-Mo8O26(H2O)2]·12H2O (7) | 3.5–5.0 | 3.5–5.0 | ||||
| [Co(C2O4)(NH3)4]2n[Mo4O12(H4C4O4)]n·3nH2O (8) | 3.5–5.0 | 3.5–5.0 | ||||
| [Co(C2O4)(NH3)4]4n[γ-Mo8O26]n·9nH2O (9) | 2.0 | |||||
| [Mo5Co2O17(HCO3)(C4H4O4H)(NH3)7]·5H2O (10) | 3.0–4.0 | 3.0–4.0 | 3.0–4.0 | 3.0–4.0 | 3.0–4.0 | 3.0–4.0 |
| [Mo5Co2O18(NH3)7]·5H2O (11) | 3.0–4.0 | 3.0–4.0 | 3.0–4.0 | 3.0–4.0 | 3.0–4.0 | 3.0–4.0 |
Solution based reactions between molybdate anions, tetraammineoxalatocobalt(III) cations and acid were performed in aqueous medium. When the reactions were performed at room temperature with the addition of acetic acid, they resulted in the formation of a mixture of unstable dark pink rods of [Co(C2O4)(NH3)4]4[β-Mo8O26]·12H2O (1) and stable pink plates of [Co(C2O4)(NH3)4]4[β-Mo8O26]·10H2O (2), regardless of the applied pH value. In the reaction with succinic acid (pH around 4.5), a pink powder precipitated immediately after mixing the reactants, and its transformation into polymer [Co(C2O4)(NH3)4]4n[Na2Mo8O29(H2O)4]n·6nH2O (3) was completed within four days. At higher concentration of succinic acid (pH around 3.5) pink powder transformation resulted in unstable dark pink rods of 1 after several days, while at lower pH values (pH range between 2.0 and 3.5), the final product of transformation was cocrystal [Co(C2O4)(NH3)4]4[β-Mo8O26]·4H2O·C4H6O4 (6).
When the reactions were carried out at higher temperatures in the presence of acetic acid, and within the pH range between 3.5 and 5.0, a mixture of pink plates of polymer 3 and unstable dark pink rods of 1 (under reflux) or a mixture of 1 and 2 (solvothermal) was obtained. At pH above 3.5 and conducting the reaction under reflux or solvothermally yielded pink prisms of [Co(C2O4)(NH3)4]8[β-Mo8O26(H2O)2][γ-Mo8O26]·12H2O (4) that were completely transformed into pink prisms of [Co(C2O4)(NH3)4]4[γ-Mo8O26(H2O)2]·6H2O (5) within 24 hours. It seems that acetic acid as a poor chelating ligand allowed the formation of the corresponding discrete octamolybdates or an octamolybdate-based polymer, as the dominant species.
Most of the reactions carried out at higher temperatures and/or under higher pressure with the addition of succinic acid resulted in the formation of the β- and γ-octamolybdate mixture, or polymeric species {Mo4O12}n, {Na2Mo8O29(H2O)4}n and {γ-Mo8O26}n as the main products. Different reaction outcomes can be attributed to better chelating properties of succinic acid when compared to acetic acid.
Reactions carried out at higher temperatures (under reflux or solvothermally) in the presence of succinic acid (in the pH range 3.5 to 5.0) yielded initially a pink powder which upon standing under room temperature transformed into a mixture of oxomolybdates: hybrid octamolybdates 1 (unstable dark pink rods) and [Co(C2O4)(NH3)4]4[Mo8O26(H2O)2]·12H2O (7) (light pink plates), octamolybdate-based cocrystal 6 (pink prisms), more complex polymer 3 (pink plates), and 2D metal–organic framework [Co(C2O4)(NH3)4]2n[Mo4O12(C4H4O4)]n·3nH2O (8) (light pink needles). Reactions performed under reflux at pH value below 2.5 resulted in the formation of cocrystal 6, while the solvothermally conducted reaction (pH around 2.0) additionally enabled isolation of a few red rods of polymer [Co(C2O4)(NH3)4]4n[γ-Mo8O26]n·9nH2O (9) before the formation of the final cocrystal 6.
By applying the mechanochemical synthesis, all the reactions (independent of the type and the amount of carboxylic acid) yielded polymer 3 after the solid mixture, obtained by milling together sodium molybdate, tetraammineoxalatocobalt(III) nitrate and acid, was exposed to water vapor. Its exclusive formation can be attributed to the increased concentration of building units in the reaction mixture.
The isolation of polyoxomolybdates also depends on the properties of the applied counter ion, its electrostatic potential or its ability to stabilize the present polyoxo species through hydrogen bonding. This could be documented with results when performing the reactions with [Co(CO3)(NH3)4]+ (Table 1). A violet powder of [Mo5Co2O17(HCO3)(C4H4O4H)(NH3)7]·5H2O (10) was observed as the only product of the reactions between tetraamminecarbonatocobalt(III) nitrate and sodium molybdate at pH below 4.0 (independent of the used synthetic approach and used carboxylic acid). If the violet powder was left to stand at room temperature in mother liquor for two months, its transformation resulted in [Mo5Co2O18(NH3)7]·5H2O (11). The transformation of the amorphous precipitate of 10 to the crystalline product 11 is influenced by the stability of intermediates, [Co(NH3)3]3+ and [Co(NH3)4]3+ cations, formed in the reaction of [Co(CO3)(NH3)4]+ cations with carboxylic acids, as known from the literature.14 The same result was obtained at higher temperatures (solvothermal synthesis or under reflux) and lower pH values.
The presence of hydrogen succinate and hydrogen carbonate anions in 10 was corroborated by 13C NMR and IR spectroscopy (Table S7, Fig. S9 and S10†). The 13C NMR spectrum of 10 contained three signals at 35.92 ppm, 167.13 and 179.21 ppm arising from C4H4O4H− and CO32−/HCO3− anions. The signals observed at 35.92 and 179.21 belong to H4C4O4H− while the signal at 167.13 to HCO3− anions, respectively. This is in accordance with the literature data.22 Compound 11 was described in the literature but obtained by a different synthetic route.
Inspired by our previous investigation,23 we aimed to explore the correlation of the hydrogen bonding ability of macrocations and the pH of the solution on the molybdovanadate formation. The solution-based synthesis of tetraammineoxalatocobalt(III) performed with the addition of ammonium vanadate led to the isolation of rose sticks of molybdovanadate [Co(C2O4)(NH3)4]2[H2Mo8V5O40Na2(H2O)8]·xH2O (12) only when the reaction was conducted under reflux or solvothermally at pH value around 2.0. In all other solution-based reactions we were not able to isolate a product of defined composition. When [Co(en)3]3+ was used as a counterion, all the solution-based reactions resulted in the formation of a mixture of insoluble powder products. However, by applying a liquid-assisted ball milling method and exposing the solid mixture to water vapor we isolated yellow crystalline product Na3[Co(en)3][HV7Mo2O27]·xH2O (13).
Structural studies
The oxomolybdates and molybdovanadates described in this paper were obtained through various reactions resulting in twelve crystalline compounds. Among these compounds, 1, 2 and 11 have been previously described in the literature.14 Compounds 3–9 are reported here for the first time (Table S9†). Compounds 12 and 13 were characterized in the solid state based on preliminary findings from single-crystal X-ray diffraction, along with data gathered from infrared spectroscopy and elemental analyses (see ESI†). Unfortunately, the crystals lacked the necessary quality for a satisfactory structural determination.Polymer [Co(C2O4)(NH3)4]4n[Na2Mo8O29(H2O)4]n·6nH2O (3) consists of octamolybdate chains interconnected by sodium cations. Each sodium cation is coordinated by seven oxygen atoms forming a distorted capped octahedron. Two vertices of the octahedron are occupied by oxygen atoms of the octamolybdate anion, with the remaining vertices being occupied by four water molecules, two of which are bridging water molecules between two sodium cations making the two oxygen polyhedra around sodium cations have (in 3 and 9) a shared edge. An additional oxygen atom from the molybdate anion also coordinates the sodium cation with a longer bond length (d(Na1–O9) of 2.949(5) Å, as opposed to d(Na1–O4) of 2.325(4) Å or d(Na1–O5) of 2.632(5) Å) capping the octahedron on the face defined by the oxygen atom belonging to the first anion, a bridging water molecule and a terminal water molecule (Fig. 1a).
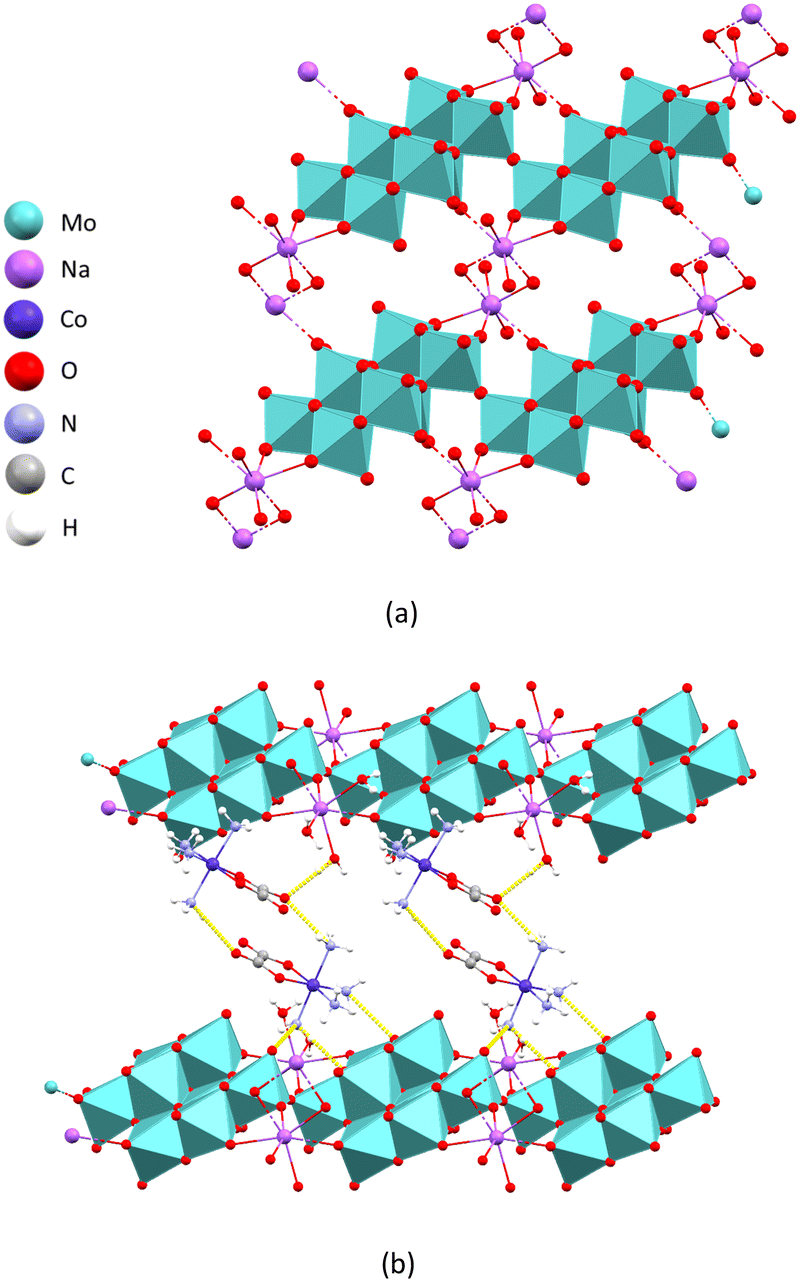 | ||
| Fig. 1 Crystal structure of 3: (a) a fragment of the two-dimensional coordination polymer layer perpendicular to the crystallographic c axis; and (b) crystal packing viewed along the crystallographic b axis of the coordination polymer layers (blue) bridged by hydrogen-bonded cobalt(III) cations and water molecules. | ||
The structure of the polyoxomolybdate anion is in accordance with the ones previously reported.24 There is an extensive network of hydrogen bonds between the coordination polymer layer, the ammine and oxalate ligands of the complex cation and water molecules interconnecting the former two. The ammine ligands of the cobalt(III) cation are oriented towards the negative oxygen atoms of the anion forming long hydrogen bonds (N4–H4B⋯O1a of 2.953(5) Å, N4–H4C⋯O10b of 3.033(6) Å, N4–H4A⋯O13a of 3.029(6) Å, N7–H7A⋯O14a of 2.992(5) Å and N8–H8A⋯O6b of 3.042(5) Å; a = −x, 1 − y, 1 − z; b = x, −1 + y, z).
The ammine groups and the oxalate groups of the cobalt(III) cations form a mutual hydrogen bond (N8–N8C⋯O17 of 2.976(6) Å), as well as hydrogen bonds with the surrounding water molecules, thus further interconnecting the layers into a three-dimensional supramolecular framework (Fig. 1b). The water molecules coordinated on the sodium atom also form hydrogen bonds with the oxygen atoms of the molybdate within the same layer (O3W–H3WB⋯O12b of 2.843(6) Å and O1W–H1WB⋯O7c of 2.888(6) Å; b = x, −1 + y, z; c = 1 + x, y, z).
In the crystal lattice of [Co(C2O4)(NH3)4]8[β-Mo8O26(H2O)2][γ-Mo8O26(H2O)2]·12H2O (4) are present two isomers of the octamolybdate anion: β-form and γ-form. This compound is the first example of a hybrid salt containing two isomers of the octamolybdate anion. The structure of both anions is consistent with similar structures described in the literature.25 In compound 4, anions and cations form a three-dimensional supramolecular network. The NH3 molecules coordinated to the cobalt atom form hydrogen bonds with the oxalate ligands of neighboring cations, connecting cations in two-dimensional layers perpendicular to the crystallographic a axis (Fig. 2a). Layers of cations are bridged by anions forming hydrogen bonds with them (Fig. 2b). The β-octamolybdate anion forms eleven and γ-octamolybdate twelve bonds with [Co(C2O4)(NH3)4]+ cations (Table S10†).
 | ||
| Fig. 2 Crystal structure of 4: (a) supramolecular layer of hydrogen-bonded cations perpendicular to the crystallographic c axis; (b) unit cell packing viewed along the crystallographic a axis with layers of cations (blue) bridged by octamolybdate. | ||
The water molecule coordinated to the molybdenum of the γ-octamolybdate anion forms an intramolecular hydrogen bond with the molybdate oxo ligand (O27–H27A⋯O19 of 3.197(12) Å) while the water molecules present in the crystal lattice also participate in the creation of an extensive supramolecular network.
In compound [Co(C2O4)(NH3)4]4[γ-Mo8O26(H2O)2]·6H2O (5), the γ-octamolybdate anion and four [Co(C2O4)(NH3)4]+ cations crystallize as a hexahydrate salt. The crystal structure is an extensive three-dimensional supramolecular network of cations and anions connected by hydrogen bonds. The cations are connected by hydrogen bonds in a three-dimensional network, where the nitrogen atoms of the NH3 group are donors of hydrogen bonds, and the oxygen atoms of oxalate ligands are acceptors. Water molecules also participate in the network. The structure built by complex cations contains voids in which octamolybdate anions are placed (Fig. 3). A water molecule coordinated to molybdenum of the γ-octamolybdate acts as a hydrogen bond donor and interacts with oxalate ligands (O14–H14B⋯O16a of 3.049(6) Å and O14–H14B⋯O18a of 2.800(6) Å; a = 1 − x, 1 − y, 1 − z), and the other oxygen atoms of molybdate anions are acceptors of hydrogen bonds that they create with NH3 groups of cations.
 | ||
| Fig. 3 Three-dimensional supramolecular network of [Co(C2O4)(NH3)4]+ cations with molybdate anions (blue) located in voids in the crystal structure of compound 5. | ||
The unit cell of [Co(C2O4)(NH3)4]4[β-Mo8O26]·C4H6O4·4H2O (6) comprises four complex cobalt(III) cations, a β-octamolybdate anion, and a molecule of succinic acid. One of the two symmetrically independent complex cations (Co1) forms hydrogen bonds with succinic acid (N3–H3C⋯O22a of 3.400(4) Å, N3–H3B⋯O23a of 2.969(5) Å and O22–H22⋯O16b of 2.830(4) Å; a = 2 − x, 1 − y, 1 − z; b = x, y, z) forming two-dimensional layers perpendicular to the crystallographic b axis (Fig. 4a). The layers are further interconnected by a series of hydrogen bonds (formed between the anions and the cations in the layers as well as the other symmetrically independent cation (Co2) and water molecules) resulting in an extensive three-dimensional supramolecular framework (Fig. 4b). Each anion participates in twenty-four hydrogen bonds with ammine ligands of the surrounding cations and water molecules, with the oxygen atoms of the molybdate anion acting as acceptors.
 | ||
| Fig. 4 Crystal structure of 6: (a) fragment of the two-dimensional layer perpendicular to the crystallographic b axis comprising hydrogen-bonded cations bridged by succinic acid molecules; and (b) crystal packing viewed along the crystallographic a axis with layers of cations and succinic acid (gray) interconnected by molybdate anions and complex cobalt cations. | ||
The structure of [Co(C2O4)(NH3)4]4[γ-Mo8O26(H2O)2]·12H2O (7) contains a three-dimensional supramolecular framework consisting of γ-octamolybdate anions26 hydrogen-bonded to [Co(C2O4)(NH3)4]+ cations and water molecules. The NH3 ligands of the cation form hydrogen bonds with oxygens from oxalate ligands of other cations (N1–H1C⋯O22a of 2.966(5) Å, N2–H2A⋯O21b of 3.006(6) Å, N4–H4B⋯O18b of 2.986(6) Å, N5–H5C⋯O20b of 2.986(5) Å, N6–H6A⋯O22b of 3.051(6) Å, N7–H7B⋯O18c of 2.797(5) Å and N8–H8C⋯O18c of 3.092(6) Å; a = x, y, 1 + z; b = 2 − x, −y, 1 − z; c = x, y, z) forming a two-dimensional layer perpendicular to the crystallographic a axis (Fig. 5a). The oxygen atoms of the molybdate anions act as acceptors of hydrogen bonds formed with the cation layers (N1–H1B⋯O11 of 2.946(6) Å, N2–H2B⋯O12 of 2.961(6) Å, N4–H4A⋯O1 of 3.088(6) Å and N5–H5A⋯O9 of 3.098(5) Å) expanding the layers into a 3D network.
 | ||
| Fig. 5 Crystal structure of 7: (a) fragment of the hydrogen-bonded two-dimensional cation layer perpendicular to the crystallographic a axis; and (b) crystal packing viewed along the crystallographic c axis with cations and anions forming channels containing water molecules. | ||
The hydrogen-bonded network of anions and cations is arranged in a way that it leaves channels along the crystallographic c axis that contain water molecules (Fig. 5b). Additionally, two water molecules coordinated on molybdenum atoms also form hydrogen bonds with neighboring water molecules. This is the probable reason for a longer Mo1–O1W bond length (2.160(4) Å) compared to other terminal oxygen atoms with an average bond length of about 1.710 Å.
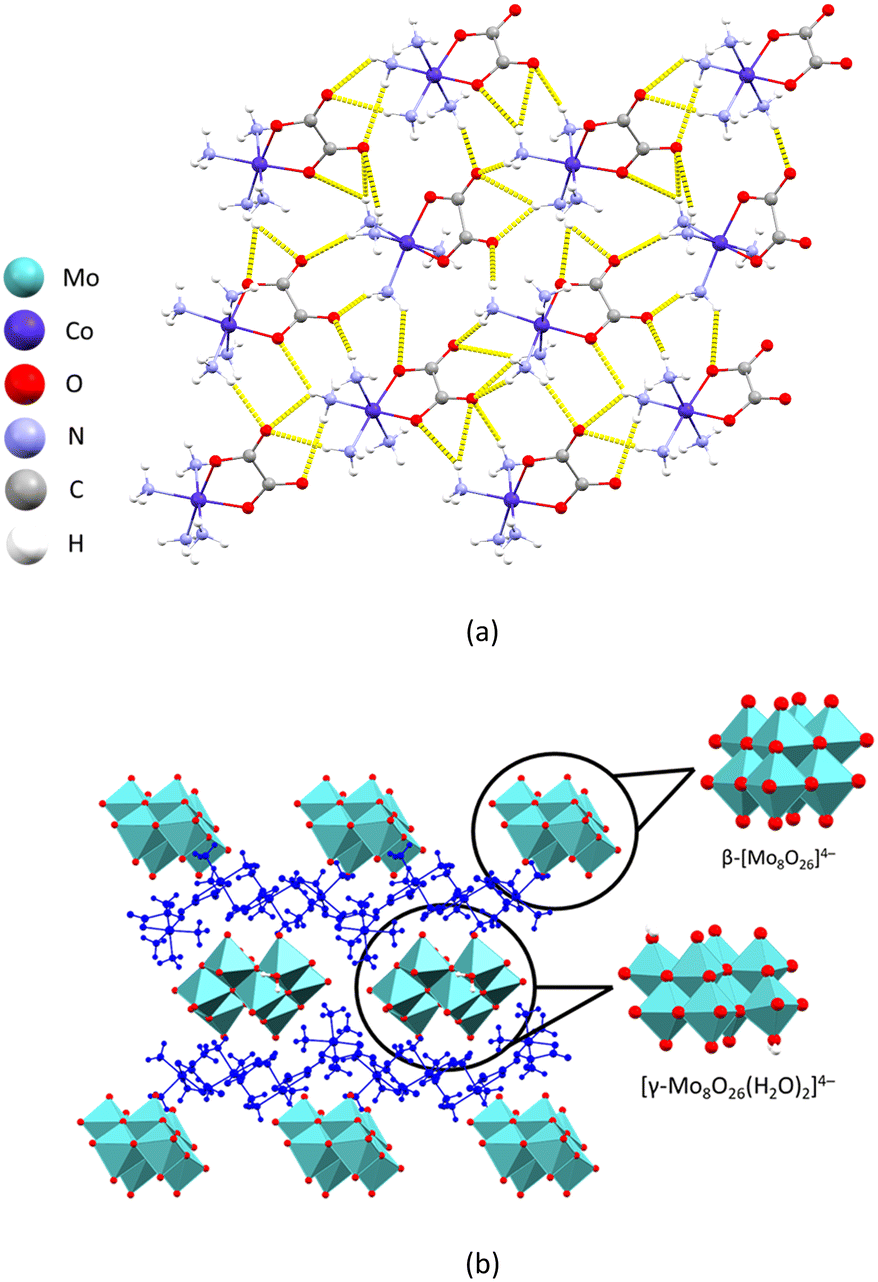
Compound [Co(C2O4)(NH3)4]2n[Mo4O12(C4H4O4)]n·3nH2O (8) is a two-dimensional metal–organic framework with polyoxomolybdate chains interconnected by coordinated succinate ions. The molybdate octahedra form a chain in a way that every octahedron shares an edge with two neighbouring ones making the three octahedra have a shared vertex. The succinate ions act as bridging ligands between four molybdenum ions, bidentate coordinating two neighbouring octahedra of the first chain and the two neighbouring octahedra of the second chain via two carboxyl groups (Fig. 6a). The geometry of the anion chain in 8 is similar to chains formed by MoO6 octahedra in molybdenum(VI) oxide, the layers being separated by the succinate anions (Fig. 6b).27
 | ||
| Fig. 6 Crystal structure of 8: (a) fragment of the two-dimensional metal–organic framework layer perpendicular to the crystallographic a axis; (b) crystal packing viewed along the crystallographic b axis with a network of hydrogen-bonded cations and MOF layers; (c) polyhedral representation of the anionic chain in 8 (left) and layered structure of MoO3 (right). | ||
The layers are interconnected by a series of hydrogen bonds formed between the oxygen atoms of the molybdate anions and the ammine ligands of the complex cobalt cation. Each monomeric unit of the anion participates in the formation of eleven N–H⋯O hydrogen bonds with the surrounding cations (Table S11†). The extensive network of hydrogen bonds connects the cations and anions into a three-dimensional supramolecular network with voids that contain additional water molecules (Fig. 6c).
The crystal structure of [Co(C2O4)(NH3)4]4n[γ-Mo8O26]n·9nH2O (9) consists of complex cobalt(III) cations and infinite anionic molybdate chains built of γ-octamolybdate units connected by a pair of shared vertices (Fig. 7a). The octamolybdate subunits found in 9 are isostructural with those found in the literature.28 The polyanions are interconnected by a series of hydrogen bonds formed between oxygens of the anion and the hydrogens from the ammine ligands of the cations. Each {Mo8O24} monomer participates in the formation of eleven hydrogen bonds with the surrounding cations (Table S12†). The cations are further connected through hydrogen-bonding of oxygen atoms of the oxalate ligands and the hydrogen atoms of the ammine ligands connecting the overall structure into a three-dimensional supramolecular network that leaves channels containing water molecules (Fig. 7).
 | ||
| Fig. 7 Crystal structure of 9: (a) polyhedral representation of polyanionic [γ-Mo8O26]4− chains; (b) crystal packing viewed along the crystallographic a axis showing a three-dimensional supramolecular network of hydrogen-bonded cations and anions with channels containing water molecules. | ||
XPRD, spectroscopic and thermal investigations
To further study the purity of all crystals, the bulk products were crushed into a fine powder suitable for PXRD analysis. The PXRD patterns confirmed that the bulk powder is a single-phase material. As shown in Fig. 8 and Fig. S11–S14 (see ESI†), the diffraction peak positions in the experimental PXRD patterns for samples 1–9 agree well with the simulated patterns. This indicates that the structures of the bulk powders are consistent with those of single crystals, confirming that the phase purity is satisfactory. | ||
| Fig. 8 PXRD patterns of compound 3: red – simulated pattern; blue – experimental pattern. | ||
The IR spectra of the prepared compounds 1–13 were examined in detail (Table S7†) and related to the structural information obtained by X-ray analysis and to the data reported in the literature.29,30 In 1–10, 12 and 13 the strong bands between 993 cm−1 and 822 cm−1 were assigned to the stretching vibrations of the terminal Mo–O bonds. The very strong bands observed in the regions between 836 and 796 cm−1 and 575 and 484 cm−1, respectively, were assigned to Mo–O stretching within the Mo–O–Mo bridge. The broad and medium intensity bands present at 3498–3286 cm−1 confirmed the existence of the O–H⋯O hydrogen bonds. In the IR spectra of all the compounds, the strong bands at 1744–1699 cm−1 and 1560–1289 cm−1 were assigned to the asymmetric and symmetric stretching vibrations of C–O and C–C from the oxalate ligand present in the Co(III) complex cation and succinate ligand (present in 8 and 10) or succinic acid (present in 6).
Thermogravimetric studies were conducted on all products under an oxygen atmosphere and in the temperature interval of 25–600 °C (Fig. S1–S8,† Table S6). The dehydration and decomposition of the anhydrous part of the examined compounds were observed, and agreed with other investigations, e.g. structural analysis and literature data.31 The decompositions of compounds 1–4, 6, 8, and 10–12 proceeded in three steps, starting with the endothermic process associated with the loss of water molecules of crystallization (in the range of 36–146 °C for 1; 46–155 °C for 2; 37–166 °C for 3; 126–296 °C for 4; 36–103 °C for 6; 33–291 °C for 7; 39–100 °C for 8; 28–155 °C for 10; 31–154 °C for 11; 40–282 °C for 12).
For compound 4, the initial step began at a notably higher temperature of 126 °C, unlike the other compounds examined. This observation can be attributed to the interactions between the two-dimensional layers, specifically to a series of hydrogen bonds involving water molecules, β- and γ-octamolybdate anions, and Co(III) cations. In contrast, compound 7 exhibits solvent loss at a significantly lower temperature of 33 °C. This can likely be attributed to the presence of water molecules within the channels of the structure, which are formed by the hydrogen bonding between the cations and γ-octamolybdate anions. The TG curves for compounds 4 and 7 are presented in Fig. 9. Additional data for compounds 1–3, 5, 6, and 8–12 can be found in the ESI,† Fig. S1–S8.
 | ||
| Fig. 9 The TG-curves for compounds 4 (red) and 7 (blue). | ||
The notable further weight loss can be attributed to the degradation of the unsolvated species in two distinct stages. The first one occurred in the temperature range of 146–316 °C (for 1; 155–316 °C for 2; 166–287 °C for 3; 206–297 °C for 4; 103–331 °C for 6; 291–383 °C for 7; 120–300 °C for 8; 155–298 for 10; 154–298 °C for 11; 282–342 °C for 12) and the second stage in the range of 316–361 °C (for 1; 316–398 °C for 2; 287–353 °C for 3; 297–434 °C for 4; 331–499 °C for 6; 320–385 °C for 8; 298–395 °C for 10; 298–365 °C for 11; 342–389 °C for 12). These two stages represent the decomposition of Co-cation and oxomolybdate cores. The residual solids consisted of mixtures of CoxMoyOz (of 1, 2, 4, 6, 7, 8, 10, and 11) or NaCoxMoyOz (of 3) or CoxMozVy (of 12).
Conclusions
We synthesized and structurally characterized seven polyoxomolybdates and two molybdovanadates with varying nuclearities. These salts include {Na2Mo8O29(H2O)4}n4m−, {[β-Mo8O26(H2O)2][γ-Mo8O26]}8−, {β-Mo8O26(H2O)2}4−, {Mo4O12(C4H4O4)}n4m−, and {γ-Mo8O26}n4m− anions, which represent to the best of our knowledge the first reported structures of that kind. The assembly processes are sensitive to the type of carboxylic acid used, and both succinic and acetic acids promote the formation of hybrid polyoxomolybdates. However, it is important to note that reaction temperature and pressure also play a significant role in influencing the polymerization process. Under reflux or hydrothermal conditions, octamolybdate32 emerges as the primary structural framework in the products obtained. This is largely attributed to the thermal stability of the species involved in the equilibria present in aqueous solution. The less dense three-dimensional supramolecular framework, comprising either β- or γ-octamolybdates, is preferentially formed within the pH range between 2 and 5. In this pH range at room temperature, the dominant species consist of discrete Mo8O264− units or the chains built up from Mo8O264− connected with sodium cations. Lastly, we isolated the molybdovanadate Na3[Co(en)3][HV7Mo2O27] by exposing a solid reaction mixture to water vapor, which opens up new avenues for exploring the chemistry of molybdovanadates.Experimental section
General methods and materials
Starting cobalt(III) complex salts, [Co(CO3)(NH3)4]NO3·H2O, [Co(en)3]Cl3 and [Co(C2O4)(NH3)4]NO3·H2O used as reaction precursors, were prepared according to the literature data.33 Na2MoO4·2H2O, NH4VO3, acetic acid and succinic acid were commercially available reagent-grade chemicals that were employed as received without further purification. Elemental analyses were provided by the Analytical Services Laboratory of the Ruđer Bošković Institute, Zagreb and by the Analytical Services Laboratory of the Faculty of Science and Faculty of Forestry, University of Zagreb. Thermal studies (TGA-SDTA) were performed on a Mettler Toledo TGA/DSC3+ STARe Systems instrument using aluminum oxide crucibles under an O2 atmosphere and in the temperature range from 25 to 600 °C. The heating rate was 10 °C min−1. Infrared spectra were recorded on a PerkinElmer Spectrum RXI FTIR-ATR spectrometer in a 4000–400 cm−1 range. 13C NMR spectra were recorded on a Bruker Avance III HD 400 spectrometer operating at 400 MHz. Compounds were dissolved in a mixture of NaOH and D2O and measured in 5 mm NMR tubes at 298 K with TMS as the internal standard. The sample concentration was 10 mg mL−1. Single-crystal X-ray diffraction data of 1–9 and 11 were collected on an XtaLAB Synergy-S CCD diffractometer with CuKα (λ = 154![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 184 Å) radiation at room temperature or at 170 K. Data reduction was performed using the CrysAlis software package.34 Solution, refinement and analysis of the structures were performed using the programs integrated in the WinGX35 and OLEX236 systems. All structures were solved and refined with the SHELX program suite.37 Structural refinement was performed on F2 using all data. All hydrogen atoms were placed at calculated positions and treated as riding on their parent atoms. Geometrical calculations were performed using PLATON.38 Drawings of the structures were prepared using PLATON and MERCURY programs.39 Powder X-ray diffraction (PXRD) data were collected on a Malvern Panalytical Aeris powder diffractometer in the Bragg–Brentano geometry with a PIXcel1D detector, using CuKα radiation (λ = 1.5406 Å). Samples were contained on a Si sample holder. Powder patterns were collected at room temperature in the range from 5° to 50° (2θ) with a step size of 0.043° and 7.14 s per step. The data were collected and visualized by using the Malvern Panalytical HighScore Software Suite.40
184 Å) radiation at room temperature or at 170 K. Data reduction was performed using the CrysAlis software package.34 Solution, refinement and analysis of the structures were performed using the programs integrated in the WinGX35 and OLEX236 systems. All structures were solved and refined with the SHELX program suite.37 Structural refinement was performed on F2 using all data. All hydrogen atoms were placed at calculated positions and treated as riding on their parent atoms. Geometrical calculations were performed using PLATON.38 Drawings of the structures were prepared using PLATON and MERCURY programs.39 Powder X-ray diffraction (PXRD) data were collected on a Malvern Panalytical Aeris powder diffractometer in the Bragg–Brentano geometry with a PIXcel1D detector, using CuKα radiation (λ = 1.5406 Å). Samples were contained on a Si sample holder. Powder patterns were collected at room temperature in the range from 5° to 50° (2θ) with a step size of 0.043° and 7.14 s per step. The data were collected and visualized by using the Malvern Panalytical HighScore Software Suite.40
All isolated products were filtered off, washed with cold water and dried to constant mass in a desiccator. In the case when the final product was a mixture of crystals, they were separated mechanically, washed with cold water and dried to constant mass in a desiccator.
General procedure for liquid-assisted ball milling followed by vapour-assisted aging
a) Na2MoO4·2H2O (5 mmol), Co(III) salt (2 mmol), acetic or succinic acid (1.22, 2.44, 7.53, 15.90 mmol) and acetone (25 μL) were placed within a 10 mL stainless steel jar. The reactants were milled for 1 hour at 25 Hz frequency. The produced pink solid reaction mixture was exposed to 100% humidity at room temperature. In all cases, the transformation resulted in the crystalline product 3 (yield of 3: 14.00 mg 11.78% or 11.67 mg 9.82% or 14.89 mg 12.52%), regardless of the added acid or its amount.b) Na2MoO4·2H2O (5 mmol), NH4VO3 (5 mmol), Co(III) salt (2 mmol), acetic or succinic acid (1.22, 2.44, 7.53, 15.90 mmol) and acetone (25 μL) were placed in a 10 mL stainless steel jar. The reactants were milled for 1 hour at 25 Hz frequency. The produced rose solid reaction mixture was exposed to 100% humidity at room temperature. From all the solid reaction mixtures, after their exposure to water vapour, crystalline product 13 (yield of 13: 4.21 mg) was obtained, regardless if the reaction was conducted with or without carboxylic acid.
For compounds 5, 9 and 13 thermogravimetric analysis as well as elemental analysis for 5 and 9 could not be performed since only several crystals were produced. The obtained amount of these compounds was only sufficient for their single-crystal XRD analysis and IR spectroscopic characterization.
All products were filtered off, washed with cold water and dried to constant mass in a desiccator.
Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for 3–9, 12, and 13 have been deposited at the Cambridge Structural Database under deposition numbers CCDC 2404581–2404589.Author contributions
Conceptualization, M. C.; investigation, M. C., D. K., V. D.; writing—original draft preparation, M. C., D. K.; project administration, V. V.; funding acquisition, V. V.; review and editing, V. V. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Croatian Science Foundation under the project number HRZZ-IP-2022-10-7368. We acknowledge the support of project CIuK co-financed by the Croatian Government and the European Union through the European Regional Development Fund—Competitiveness and Cohesion Operational Programme (Grant KK.01.1.1.02.0016).Notes and references
- (a) X. L. Wang, C. Qin, E. B. Wang, Z. M. Su, Y. G. Li and L. Xu, Angew. Chem., Int. Ed., 2006, 45, 7411 CrossRef CAS PubMed; (b) M. T. Pope and A. Müller, Angew. Chem., Int. Ed. Engl., 1991, 103, 56 CrossRef CAS.
- D. L. Long, E. Burkholder and L. Cronin, Chem. Soc. Rev., 2007, 36, 105 RSC.
- (a) D. L. Long, R. Tsunashima and L. Cronin, Angew. Chem., Int. Ed., 2010, 49, 1736 CrossRef; (b) N. Mizuno and M. Misono, Chem. Rev., 1998, 98, 199 CrossRef; (c) Q. S. Yin, J. M. Tan, C. Besson, Y. V. Geletii, D. G. Musaev, A. E. Kuznetsov, Z. Luo, K. I. Hardcastle and C. L. Hill, Science, 2010, 328, 342 CrossRef.
- J. J. Cruywagen, A. G. Draaijer, J. B. B. Heyns and E. A. Rogwer, Inorg. Chim. Acta, 2002, 331, 322 CrossRef.
- J. Pisk, V. Vrdoljak, M. Mandarić, T. Hrenar, D. Agustin and M. Rubčić, RSC Adv., 2024, 14, 19029 RSC.
- N. Bebić, E. Topić, M. Mandarić, T. Hrenar and V. Vrdoljak, CrystEngComm, 2021, 23, 6349 RSC.
- A. Misra, K. Kozma, C. Streb and M. Nyman, Angew. Chem., 2020, 59(2), 596 CrossRef PubMed.
- (a) V. W. Day, M. F. Fredrich, W. G. Klemperer and W. Shum, J. Am. Chem. Soc., 1977, 99, 6146 CrossRef; (b) T. C. Hsieh, S. N. Shaikh and J. Zubieta, Inorg. Chem., 1987, 26(24), 4079 CrossRef; (c) B. Kamenar, B. Korpar-Čolig, M. Penavić and M. Cindrić, J. Chem. Soc., Dalton Trans., 1990, 1125 RSC; (d) M. L. Niven, J. J. Cruywagen and J. B. B. Heyns, J. Chem. Soc., Dalton Trans., 1991, 2007 RSC; (e) M. Inoue and T. Yamase, Bull. Chem. Soc. Jpn., 1995, 68, 3055 CrossRef.
- (a) T. J. R. Weakley, Polyhedron, 1982, 1(1), 17 CrossRef CAS; (b) I. Zebiri, S. Boufas, S. Mosbah, L. Bencharif and M. Bencharif, J. Chem. Sci., 2015, 127(10), 1769 CrossRef CAS; (c) R. Z. Wang, J. Q. Xu, G. Y. Yang, W. M. Bu, Y. H. Xing, D. M. Li, S. Q. Liu, L. Ye and Y. G. Fan, Polyhedron, 1999, 18, 2971 CrossRef CAS; (d) R. Z. Wang, J. Q. Xu, G. Y. Yang, Y. F. Li, Y. H. Xing, D. M. Li, S. Q. Liu, L. W. M. Bu, L. Ye and Y. G. Fan, Solid State Sci., 2000, 2, 705 CrossRef CAS.
- (a) D. Hagrman, C. Zubieta, D. J. Pink, J. Zubieta and R. C. Haushalter, Angew. Chem., Int. Ed. Engl., 1997, 36, 873 CrossRef CAS; (b) J. Q. Xu, R. Z. Wang, G. Y. Yang, Y. H. Xing, D. M. Li, W. M. Bu, L. Ye, Y. G. Fan, G. D. Yang, Y. Xing, Y. H. Lin and H. Q. Jia, Chem. Commun., 1999, 983 RSC.
- A. J. Bridgeman, J. Phys. Chem. A, 2002, 106, 12151 CrossRef CAS.
- M. T. Pope and A. Müller, Polyoxometalate Chemistry From Topology via Self-Assembly to Applications, Kluwer, Dordrecht, 2001, p. 269 Search PubMed.
- R. Xi, B. Wang, K. Isobe, T. Nishioka, K. Toriumi and Y. Ozawa, Inorg. Chem., 1994, 33, 833 CrossRef CAS.
- J. Fuchs and H. Hartl, Angew. Chem., Int. Ed. Engl., 1976, 15, 375 CrossRef.
- L. Atovmyan and O. Krasochka, Struct. Chem., 1972, 13, 319 CrossRef.
- A. Szymańska, W. Nitek, D. Rutkowska-Zbik and W. Łasocha, Monatsh. Chem., 2014, 145(6), 921 CrossRef.
- P. J. Hagrman, D. Hagrman and J. Zubieta, Angew. Chem., Int. Ed., 1999, 38(18), 2638 CrossRef.
- D. Kuzman, V. Damjanović, V. Stilinović, M. Cindrić and V. Vrdoljak, New J. Chem., 2021, 45, 19764 RSC.
- V. Damjanović, D. Kuzman, V. Vrdoljak, S. Muratović, D. Žilić, V. Stilinović and M. Cindrić, Cryst. Growth Des., 2019, 19, 6763 CrossRef.
- D. Kuzman, V. Damjanović, V. Vrdoljak, V. Stilinović and M. Cindrić, Inorg. Chim. Acta, 2020, 510, 119765 CrossRef.
- N. I. Gumerova and A. Rompel, Chem. Soc. Rev., 2020, 49, 7568 RSC.
- S. Morete, P. J. Dyson and G. Laurenczy, Dalton Trans., 2013, 42, 4353 RSC.
- V. Damjanović, J. Pisk, D. Kuzman, D. Agustin, V. Vrdoljak, V. Stilinović and M. Cindrić, Dalton Trans., 2019, 48, 9974 RSC.
- (a) I. Böschen, B. Buss and B. Krebs, Acta Crystallogr., Sect. B:Struct. Sci., Cryst. Eng. Mater., 1974, 30, 48 CrossRef; (b) T. Yamase and H. Naruke, J. Chem. Soc., Dalton Trans., 1991, 285 RSC; (c) T. Yamase, T. Ozeki and I. Kawashima, Acta Crystallogr., Sect. C:Struct. Chem., 1995, 51, 545 CrossRef; (d) A. S. Sukhikh, P. Khranenko, T. V. Basova and S. A. Gromilov, J. Struct. Chem., 2022, 63, 310 CrossRef.
- (a) A. Kitamura, T. Ozeki and A. Yagasaki, Inorg. Chem., 1997, 36, 4275 CrossRef; (b) T. J. R. Weakley, Polyhedron, 1982, 1, 17 CrossRef; (c) A. B. Kama, R. Dessapt, H. Serier-Brault, M. Sidibe, D. Cheikh, A. K. Diop and R. Gautier, J. Mol. Struct., 2017, 1141, 698 CrossRef; (d) X. Wang, D. Liu, H. Lin, G. Liu, X. Wang, M. Le and X. Rong, CrystEngComm, 2016, 18, 888 RSC; (e) M. H. Rosnes, C. Y. D.-L. Long and L. Cronin, Dalton Trans., 2012, 41, 10071 RSC.
- (a) E. Cartuyvels, K. Van Hecke, L. Van Meervelt, C. Gröller-Walrand and T. N. Parac-Vogt, J. Inorg. Biochem., 2008, 102, 1589 CrossRef; (b) M. Inoue and T. Yamase, Bull. Chem. Soc. Jpn., 1995, 68, 3055 CrossRef; (c) M. Tahmasebi, M. Mirzaei, H. Eshtiagh-Hosseini, J. T. Mague, A. Bauzá and A. Frontera, Acta Crystallogr., Sect. C:Struct. Chem., 2019, 75, 469 CrossRef; (d) R. Atencio, A. Briceño, P. Silva, J. A. Rodríguez and J. C. Hanson, New J. Chem., 2007, 31, 33 RSC; (e) E. M. McCarron III and R. L. Harlow, J. Am. Chem. Soc., 1983, 105, 6179 CrossRef; (f) T. Ito, K. Mikurube, K. Hasegawa, T. Matsumoto, K. Kosaka, H. Naruke and A. Koguchi, Crystals, 2014, 4, 42 CrossRef; (g) B. Kamenar, M. Penavić and B. Marković, Acta Crystallogr., Sect. C:Struct. Chem., 1988, 44, 1521 CrossRef.
- N. Wooster, Z. Kristallogr. – Cryst. Mater., 1931, 80, 504 CrossRef.
- (a) Q.-G. Zhai, X.-Y. Wu, S.-M. Chen, Z.-G. Zhao and C.-Z. Lu, Inorg. Chem., 2007, 46, 5046 CrossRef; (b) X. Wang, J. Sun, H. Lin, Z. Chemng, X. Wang and G. Liu, Dalton Trans., 2016, 45, 12465 RSC; (c) H.-J. Du, Z.-Z. Shu, Y.-Y. Niu, L.-S. Song and Y. Zhu, J. Solid State Chem., 2012, 190, 296 CrossRef CAS; (d) H. S. Casalongue, S. J. Choyke, A. N. Sarjeant, J. Schrier and A. J. Norquist, J. Solid State Chem., 2009, 182, 1297 CrossRef; (e) K. J. Thorn, A. N. Sarjeant and A. J. Norquist, Acta Crystallogr., Sect. E:Crystallogr. Commun., 2005, 61, m1665 CrossRef; (f) M. Evain, V. Petricek, V. Coué, R. Dessapt, M. Bujoli-Doeuff and S. Jobic, Acta Crystallogr., 2006, 62, 790 CrossRef PubMed.
- M. Cindrić, V. Stilinović, M. Rubčić, G. Medak, D. Šišak Jung and V. Vrdoljak, CrystEngComm, 2018, 20, 1889 RSC.
- (a) Y. Chen, X. Shen, H. Zhang, C. Huang, Y. Cao and R. Sun, Vib. Spectrosc., 2006, 40, 142 CrossRef; (b) G. Mahata and K. Biradha, Inorganica Chim. Acta, 2007, 360, 281 CrossRef; (c) E. McCarron, J. F. Whitney and D. B. Chase, Inorg. Chim. Acta, 1984, 23, 3275 CrossRef; (d) R. P. Sharma, R. Bala, R. Sharma and P. Venugopalan, J. Coord. Chem., 2004, 57, 1563 CrossRef; (e) S. Ikegami and A. Yagasaki, Materials, 2009, 2, 869 CrossRef.
- P. Roman, A. Lique, J. M. Gutiérrez-Zorrilla and S. Garcia-Granada, Polyhedron, 1991, 10, 2057 CrossRef.
- J. Noack, F. Rosowski, R. Schlögl and A. Trunschke, Z. Anorg. Allg. Chem., 2014, 640(14), 2730 CrossRef.
- (a) N. Hur, W. G. Klemperer and R. C. Wang, Inorganic Synthesis, John Wiley & Sons, New York, 1990, vol. 27, p. 77 Search PubMed; (b) M. Mori, M. Shibata, E. Kyuno and E. Hoshiyama, Bull. Chem. Soc. Jpn., 1958, 31, 291 CrossRef; (c) P. C. Junk and J. W. Steed, Polyhedron, 1999, 18, 3593 CrossRef CAS; (d) R. A. Krause and E. A. Megargle, J. Chem. Educ., 1976, 53(10), 667 CrossRef CAS.
- Agilent, CrysAlis PRO, Agilent Technologies Ltd., Yarnton, Oxfordshire, England, 2014 Search PubMed; Oxford Diffraction, Xcalibur CCD system, CrysAlis CCD and CrysAlis RED software, Version 1.170, Oxford Diffraction Ltd., 2003 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A, 2015, 71, 3 CrossRef.
- A. L. Spek, Acta Crystallogr., Sect. D:Biol. Crystallogr., 2009, 65, 148 CrossRef CAS.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van de Streek, J. Appl. Crystallogr., 2006, 39, 453 CrossRef CAS.
- T. Degen, M. Sadki, E. Bron, U. König and G. Nénert, Powder Diffr., 2014, 29(S2), S13 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: (1) Additional experimental data, (2) thermogravimetric data, (3) IR data, (4) analytical data, (5) crystallographic data, (6) tables of selected bond distances and angles and of hydrogen bond parameters, (7) TGA curves, and (8) NMR spectra. Crystallographic data sets for the structures 3–9 are available through the Cambridge Structural Database with deposition numbers CCDC 2404581–2404587. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ce01187b |
| This journal is © The Royal Society of Chemistry 2025 |