
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Versatile copper(II) discrete and polymeric coordination compounds with (pyridine-2-yl)methylenenicotinohydrazide and azelaic acid†
Mónica
Benito
 a,
Ghodrat
Mahmoudi
*bc,
Elies
Molins
a,
Ennio
Zangrando
d,
Masoumeh
Servati Gargari
e,
Rosa M.
Gomila
f,
Antonio
Frontera
f and
Damir A.
Safin
gh
a,
Ghodrat
Mahmoudi
*bc,
Elies
Molins
a,
Ennio
Zangrando
d,
Masoumeh
Servati Gargari
e,
Rosa M.
Gomila
f,
Antonio
Frontera
f and
Damir A.
Safin
gh
aInstitut de Ciència de Materials de Barcelona (ICMAB-CSIC), Campus de la Universitat Autònoma de Barcelona, 08193 Bellaterra, Spain
bDepartment of Chemistry, Faculty of Science, University of Maragheh, P.O. Box 55136-83111, Maragheh, Iran. E-mail: ghodratmahmoudi@gmail.com
cChemistry Department, Faculty of Engineering and Natural Sciences, Istinye University, Sarıyer, Istanbul 34396, Turkey
dDepartment of Chemical and Pharmaceutical Sciences, University of Trieste, Via L. Giorgieri 1, 34127 Trieste, Italy
eWestern Caspian University, Baku, Azerbaijan
fDepartment of Chemistry, Universitat de les Illes Balears, Crta de Valldemossa km 7.5, 07122 Palma de Mallorca, Baleares, Spain
gScientific and Educational and Innovation Center for Chemical and Pharmaceutical Technologies, Ural Federal University named after the First President of Russia B.N. Yeltsin, Ekaterinburg, 620002, Russian Federation
hDepartment of Technical Sciences, Western Caspian University, Baku, Azerbaijan
First published on 19th November 2024
Abstract
In this contribution, a series of copper(II) discrete and polymeric coordination compounds, namely [Cu3L2(aze)2]n·nH2O (1·nH2O), [CuL2]·H2O (2·H2O) and [CuL(H2O)(Haze)]·H2O (3·H2O) were fabricated from (pyridin-2-yl)methylenenicotinohydrazide (HL) and azelaic acid (H2aze), using evaporative crystallization, grinding and slurry synthesis methods. The solvent crystallization method produced a crystalline precipitate of complex 1·nH2O. The filtrate allowed the production of crystals of complex 3·H2O. The grinding synthetic method gave rise to a mixture of complexes 1·nH2O, 2·H2O and an undetermined phase. Results of the slurry synthetic approach highly depend on the stirring time. The obtained coordination compounds have been fully characterized by single crystal X-ray diffraction revealing different structures of complexes, viz. from mononuclear homo- and heteroleptic complexes (2 and 3) to a 1D heteroleptic coordination polymer (1). DFT calculations on non-polymeric complexes were conducted to evaluate the hydrogen-bonding and π-stacking interactions that play a critical role in the solid-state structures of complexes 2 and 3. Additionally, the impact of co-crystallized water molecules, which enhance the π-stacking interactions, was analyzed using energy decomposition analysis.
1. Introduction
Coordination polymers are adaptable materials with essential properties that make them desirable for a wide range of applications. These polymers are made of bridging ligands and metal ions or entities containing a metal. Additionally, auxiliary ligands of different kinds can be used to adjust the resulting product's characteristics and final structure. Thus, there are potential applications for molecule adsorption and separation, catalysis, and magnetic research due to the large structural variety of coordination polymers, ranging from 1D (chain) to 2D and 3D (framework) networks. That being said, the IUPAC recommends defining a coordination polymer as “a coordination compound with repeating coordination entities extending in 1, 2, or 3 dimensions”.1 Historically, inorganic complexes produced from cyanides (e.g., Hofmann clathrates, Prussian blue) are, likely, the first coordination polymers to be identified and emphasized.2 It was not until much later that the first coordination polymer obtained using an organic linker was revealed.3 Since the discovery of the so-called metal–organic frameworks in 1995,4,5 coordination polymers have been actively developed and produced with exploding interest.Of coordination polymers, those exhibiting a 1D structure are thought to have greater advantages over 2D and 3D frameworks because of their simpler topology, a large and varied library of precursors, and comparatively easier synthesis methods.6–11 In this regard, using organic linkers with two or more N-donor functions seems to be one of the most effective synthetic methods for creating coordination polymers. As such, pyridine is probably the most commonly used N-donor functionality.11 In addition, the so-called Schiff base entity is a promising structural fragment for coordination polymers.12 Consequently, the incorporation of N-donor rings into the Schiff base platform appears to be a potent instrument for designing and manufacturing coordination polymers.
In light of the aforementioned, we have focused our attention on (pyridin-2-yl)methylenenicotinohydrazide (HL). This compound was purposefully created to function as a potentially tridentate chelating ligand with an extended π-system, which additionally contains the 3-pyridine N-donor fragment that has the ability to bridge structural units, thereby producing an extended architecture. The Cu2+ cation was administered as Cu(OAc)2 as a complexation agent. This cation has extensive coordination chemistry because it exhibits a variety of coordination numbers and coordination polyhedra. Furthermore, the Cu2+-containing hydrazide-based coordination compounds also comprise a variety of auxiliary linkers.13 In this regard, we were curious to use two different organic linkers by taking advantage of a multidentate hydrazide ligand with a flexible and long-chain dicarboxylic acid, namely azelaic acid (H2aze), for a potential formation of novel coordination polymers. Notably, a thorough search in the Cambridge Structural Database (CSD) revealed only six crystal structures with doubly deprotonated azelaic acid (aze)14–19 and one crystal structure with monodeprotonated azelaic acid (Haze).20 Thus, the coordination chemistry of H2aze as well as its anionic forms has been extremely poorly studied.
With all this in mind, as well as in continuation of our interest in copper coordination compounds,21–45 in the present work we focused on the preparation of new copper(II) heteroleptic coordination compounds, namely [Cu3L2(aze)2]n·nH2O (1·nH2O), [CuL2]·H2O (2·H2O) and [CuL(H2O)(Haze)]·H2O (3·H2O), fabricated from HL and H2aze, to probe the influence of the applied synthetic approaches, viz. the evaporative crystallization, grinding and slurry synthesis methods. The isolated products were investigated to determine their crystal structures using single crystal and powder X-ray diffraction, and IR spectroscopy. Furthermore, the propensity of non-polymeric complexes to form π-stacking interactions, as well as the influence of lattice water molecules on the strength of these interactions, has been investigated using DFT calculations combined with molecular electrostatic potential (MEP) surface, the quantum theory of atoms-in-molecules (QTAIM), non-covalent interaction plot (NCIplot) and energy decomposition analysis (EDA) computational tools.
2. Results and discussion
The syntheses of complexes were carried out following solvent crystallization, grinding (Fig. 1) and slurry methods by direct reaction of Cu(AcO)2 with a mixture of HL·H2O and H2aze (Scheme 1). | ||
| Fig. 1 Photographs of the grinding synthesis. | ||
 | ||
| Scheme 1 Synthesis of the discussed complexes. | ||
The solvent crystallization method allowed the production of a crystalline precipitate of complex 1·nH2O almost immediately after the addition of a mixture of HL and H2aze (Fig. 2). After the resulting solid material was filtered off, the filtrate was left for slow evaporation under ambient conditions and a few days later green crystals of complex 3·H2O were formed. Thus, this synthetic approach allows the production of both a heteroleptic coordination polymer 1·nH2O with doubly deprotonated azelaic acid aze, and a mononuclear heteroleptic discrete complex 3·H2O with a monodeprotonated Haze without, notably, additional complicated procedures for the separation of these two complexes.
 | ||
| Fig. 2 The calculated powder X-ray diffraction patterns of complexes 1·nH2O (black), 2·H2O (red) and 3·H2O (blue), and the experimental powder X-ray diffraction patterns of complex 1·nH2O (green) and the product obtained using the grinding synthetic method (magenta), and 2·H2O (violet). | ||
The grinding synthetic method, although fully satisfies the principles of a green chemistry procedure, gave rise to a mixture of complexes 1·nH2O, 2·H2O and an undetermined phase (Fig. 2). Thus, this synthetic procedure also allows the production of a mononuclear homoleptic complex 2·H2O. Furthermore, the formation of an undetermined phase also looks intriguing for further detailed studies of this method as well as to an elucidation of new structures.
Finally, the results of the slurry synthetic approach highly depend on the stirring time. In particular, stirring up to about 6 h led to the formation of complex 1·nH2O, while longer stirring (1–2 days) resulted in the formation of a mixture of complexes 1·nH2O and 3·H2O.
The FTIR spectrum of the parent ligand HL·H2O exhibits bands for the C–N, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and CO groups at 1293, 1563 and 1670 cm−1, while broad bands for the NH functionality and the crystal water molecule were found at about 3150 and 3470 cm−1, respectively (Fig. 3). The water molecule in the spectrum of HL·H2O was also shown as a broad band at about 580 cm−1 (Fig. 3). The FTIR spectrum of complex 2·H2O is the simplest among the spectra of the reported complexes with respect to the number of bands. In particular, the CO band was shifted to low wavenumbers in the spectrum of 2·H2O and was found at 1584 cm−1, while the C–N vibration was shifted about 50 cm−1 to higher wavenumbers in the spectrum of this complex in comparison to that in the spectrum of HL·H2O (Fig. 3). The FTIR spectra of 1·nH2O and 3·H2O are more complicated due to the presence of bands from the additional aze and Haze ligands, respectively (Fig. 3). Furthermore, the spectrum of 3·H2O is additionally complicated by the presence of both the carboxylate and carboxylic functionalities of the auxiliary ligand. In particular, the FTIR spectra of 1·nH2O and 3·H2O revealed bands at 1590 and 1587 cm−1, respectively, for Δνasym(OCO) and at about 1365 cm−1 for Δνsym(OCO) (Fig. 3) of the carboxylate fragments. The spectrum of 3·H2O also exhibits a band at 1704 cm−1, which corresponds to the CO vibration of the carboxylic group of Haze. Finally, a broad band at about 3400 cm−1 in the spectra of complexes was attributed to the water molecules (Fig. 3). Thus, the FTIR spectra suggest the formation of the title complexes as well as support their compositions. The FTIR spectra of complexes 1·nH2O and 3·H2O indicate the presence of the additional auxiliary ligand aze and Haze, respectively. Finally, the spectra of all three discussed complexes clearly testify to the exclusive presence of the deprotonated form of the parent hydrazide ligand in their structures.
N and CO groups at 1293, 1563 and 1670 cm−1, while broad bands for the NH functionality and the crystal water molecule were found at about 3150 and 3470 cm−1, respectively (Fig. 3). The water molecule in the spectrum of HL·H2O was also shown as a broad band at about 580 cm−1 (Fig. 3). The FTIR spectrum of complex 2·H2O is the simplest among the spectra of the reported complexes with respect to the number of bands. In particular, the CO band was shifted to low wavenumbers in the spectrum of 2·H2O and was found at 1584 cm−1, while the C–N vibration was shifted about 50 cm−1 to higher wavenumbers in the spectrum of this complex in comparison to that in the spectrum of HL·H2O (Fig. 3). The FTIR spectra of 1·nH2O and 3·H2O are more complicated due to the presence of bands from the additional aze and Haze ligands, respectively (Fig. 3). Furthermore, the spectrum of 3·H2O is additionally complicated by the presence of both the carboxylate and carboxylic functionalities of the auxiliary ligand. In particular, the FTIR spectra of 1·nH2O and 3·H2O revealed bands at 1590 and 1587 cm−1, respectively, for Δνasym(OCO) and at about 1365 cm−1 for Δνsym(OCO) (Fig. 3) of the carboxylate fragments. The spectrum of 3·H2O also exhibits a band at 1704 cm−1, which corresponds to the CO vibration of the carboxylic group of Haze. Finally, a broad band at about 3400 cm−1 in the spectra of complexes was attributed to the water molecules (Fig. 3). Thus, the FTIR spectra suggest the formation of the title complexes as well as support their compositions. The FTIR spectra of complexes 1·nH2O and 3·H2O indicate the presence of the additional auxiliary ligand aze and Haze, respectively. Finally, the spectra of all three discussed complexes clearly testify to the exclusive presence of the deprotonated form of the parent hydrazide ligand in their structures.
 | ||
| Fig. 3 The FTIR spectra of HL·H2O (black), 1·nH2O (red), 2·H2O (blue) and 3·H2O (green). | ||
The thermal behavior of the obtained complexes was revealed under a nitrogen atmosphere from ambient temperature to 500 °C. Complexes 1·nH2O and 3·H2O both started to lose the crystal water at about 100 °C and further become stable up to about 200 °C with the final weight loss of 66.13% and 82.29% (Fig. 4). Complex 2·H2O is stable up to about 270 °C and decomposes without a separate step for losing the crystal water molecule with the final weight loss of 71.34% (Fig. 4).
 | ||
| Fig. 4 The TGA curves of 1·nH2O (black), 2·H2O (red) and 3·H2O (blue). | ||
All the discussed complexes crystallize in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with the hydrazide ligand L being N,N′,O-chelated to the metal ion (Fig. 5). In complex 2, the Cu2+ cation is chelated by two ligands L with the formation of a N4O2 coordination environment of the distorted octahedral structure (Fig. 5). In complex 3, besides the N,N′,O-chelated ligand L, the metal cation is additionally coordinated by one carboxylate oxygen atom of the Haze ligand, and the coordination sphere is completed by the oxygen atom of the coordinated water molecule, yielding a N2O3 coordination environment of the distorted square-pyramidal structure (Fig. 5).
with the hydrazide ligand L being N,N′,O-chelated to the metal ion (Fig. 5). In complex 2, the Cu2+ cation is chelated by two ligands L with the formation of a N4O2 coordination environment of the distorted octahedral structure (Fig. 5). In complex 3, besides the N,N′,O-chelated ligand L, the metal cation is additionally coordinated by one carboxylate oxygen atom of the Haze ligand, and the coordination sphere is completed by the oxygen atom of the coordinated water molecule, yielding a N2O3 coordination environment of the distorted square-pyramidal structure (Fig. 5).
 | ||
| Fig. 5 Molecular structures of 1 (top), 2 (bottom left) and 3 (bottom right). Color code: H = black, C = gold, N = blue, O = red, Cu = green. | ||
The coordination arrangement of complex 1 is more complicated. In particular, two [Cu1L]+ cations are combined in a binuclear centrosymmetric dimer through two carboxylate oxygen atoms of the two aze ligands, and the coordination sphere of the metal cation is completed by the second carboxylate oxygen atom of one of the same aze ligands.
Thus, the Cu2+ cations in this dimer are in a N2O4 coordination environment of the distorted octahedral structure with the Cu1⋯Cu1 separation of about 3.3 Å. These binuclear dimers are combined through the 3-pyridine nitrogen atoms and oxygen atoms of the second carboxylate fragments of the aze ligands being coordinated to the other Cu2 cations, yielding a N2O4 coordination environment of the distorted octahedral structure (Fig. 5). Thus, complex 1 is a 1D heteroleptic coordination polymer with the Cu1⋯Cu2 separations between the mono- and binuclear nodes of about 7.4 and 8.1 Å.
The Cu1–N1, Cu1–N2 and Cu1–O1 bond distances within the chelate [CuL]+ cation in the structures of the reported complexes, and the Cu2–N4 bond distances in the structure of complex 1 are 2.002(9)–2.168(5), 1.925(10)–1.962(5), 1.988(7)–2.196(5) and 2.019(10) Å, respectively (Table 1). The Cu–O2 distances in the structures of complexes 1 and 3 vary from 1.923(10) Å to 2.738(10) Å (Table 1). Finally, the Cu1–O6 bond length in the structure of complex 3 is 2.2464(15) Å. Notably, as evidenced from the corresponding dihedral angles between the 2- and 3-pyridine rings (Table 1), the ligand L is almost planar in the structures of complexes 1 and 3, while one of the ligands L in the structure of complex 2 is also almost planar, the second one is remarkably deviated from planarity.
| 1 | 2 | 3 | |
|---|---|---|---|
| Bond length | |||
| Cu1–N1 | 2.002(9) | 2.166(6), 2.168(5) | 2.0529(15) |
| Cu1–N2 | 1.925(10) | 1.955(5), 1.962(5) | 1.9285(14) |
| Cu1–O1 | 1.988(7) | 2.130(4), 2.196(5) | 2.0122(12) |
| Cu1–O2 | 1.923(10), 2.300(8) | — | 2.0122(12) |
| Cu1–O3 | 2.738(10) | — | — |
| Cu1–O6 | — | — | 2.2464(15) |
| Cu2–N4 | 2.019(10) | — | — |
| Cu2–O4 | 1.937(10) | — | — |
| Cu2–O5 | 2.504(12) | — | — |
| Dihedral angle | |||
|---|---|---|---|
| 2-Py⋯3-Py | 6.6(6) | 1.1(3), 30.7(4) | 4.01(9) |
1D polymeric molecules in the structure of 1·nH2O are linked through π⋯π interactions between the 2-pyridine rings, yielding a supramolecular 2D sheet (Fig. 6 and Table 2). Molecules of 2 are linked into a 1D supramolecular ribbon through the π⋯π interactions between the 2- and 3-pyridine rings formed exclusively by the planar ligands L (Fig. 6 and Table 2). The molecular structure of 3 is stabilized by an intramolecular O–H⋯O hydrogen bond formed between one of the hydrogen atoms of the coordinated water molecule and the free carboxylate oxygen atom of the Haze ligand (Fig. 6 and Table 2).
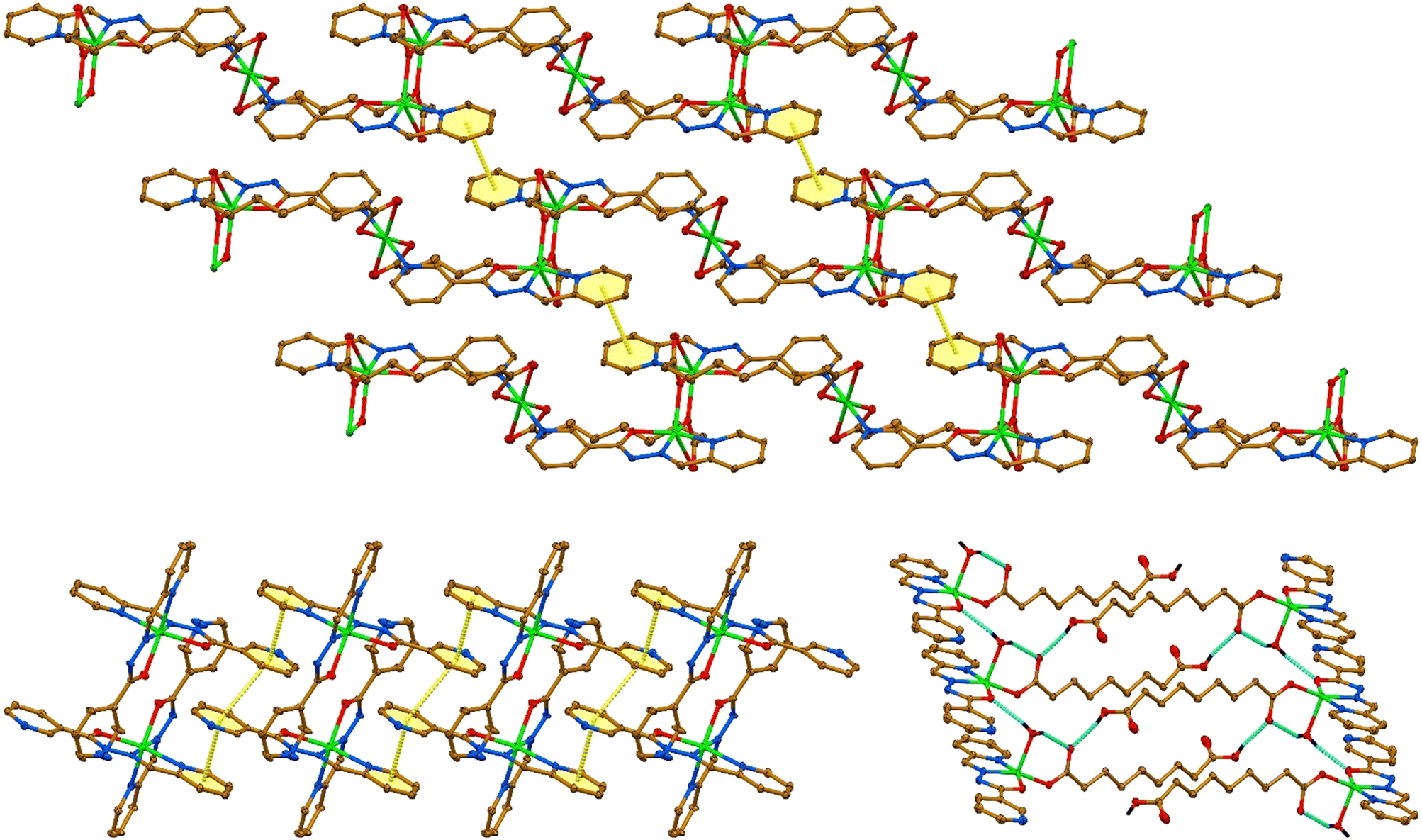 | ||
| Fig. 6 Supramolecular frameworks in the crystal structure of 1·nH2O (top) and 2·H2O (bottom left), constructed from π⋯π interactions, and 3·H2O (bottom right), constructed through O–H⋯N hydrogen bonds (hydrogen atoms, except for the coordinated H2O and carboxylic OH groups, are omitted for clarity). Color code: H = black, C = gold, N = blue, O = red, Cu = green; O–H⋯O hydrogen bonds = cyan dashed line, π⋯π interaction = yellow dashed line. | ||
| D–X⋯A | d(D–X) | d(X⋯A) | d(D⋯A) | ∠(DXA) |
|---|---|---|---|---|
| 1·nH2O | ||||
| Symmetry code: i) x, y, z; ii) −1 − x, −y, 2 − z; iii) 1 − x, −y, 1 − z; iv) 1 + x, y, z; v) −1 + x, −1 + y, z; vi) 1 − x, 2 − y, 1 − z; vii) −1 + x, y, z; viii) −x, 1 − y, 2 − z. | ||||
| O1W–H2W⋯O5i | 0.97(17) | 2.0(2) | 2.89(2) | 151(14) |
| 2·H2O | ||||
|---|---|---|---|---|
| O1W–H1W⋯N4i | 0.96(2) | 1.95(3) | 2.826(8) | 152(4) |
| O1W–H2W⋯O1Wii | 0.96(8) | 1.97(8) | 2.877(10) | 158(6) |
| 3·H2O | ||||
|---|---|---|---|---|
| O5–H3⋯O2iii | 0.76(2) | 1.94(2) | 2.673(2) | 165(3) |
| O6–H2⋯O1iv | 0.73(3) | 2.12(2) | 2.820(2) | 162(3) |
| O6–H1⋯O3 | 0.71(3) | 2.05(3) | 2.705(2) | 154(3) |
| O7–H71⋯N4i | 0.77(3) | 2.13(3) | 2.878(2) | 163(3) |
| O7–H72⋯N3v | 0.86(4) | 2.17(4) | 2.994(3) | 161(3) |
| Cg1⋯Cg2 | d(Cg1⋯Cg2) | α | β | γ |
|---|---|---|---|---|
| 1·nH2O | ||||
| 2-Py⋯2-Pyvi | 3.720(8) | 0.0(6) | 19.9 | 19.9 |
| 2·H2O | ||||
|---|---|---|---|---|
| 2-Py⋯3-Pyiv | 3.626(4) | 1.1(3) | 20.3 | 19.5 |
| 3-Py⋯2-Pyvii | 3.625(4) | 1.1(3) | 19.5 | 20.3 |
| 3-Py⋯3-Pyviii | 3.624(4) | 0.0(3) | 22.1 | 22.1 |
Furthermore, molecules of complex 3 are linked into a 1D supramolecular ribbon through O–H⋯O hydrogen bonds formed between the second hydrogen atom of the coordinated water molecule and the carbonyl oxygen atom of the ligand L from an adjacent molecule, and between the carboxyl hydrogen atom and the free carboxylate oxygen atom of the other neighbouring molecule. The crystalline water molecule in the structure of complex 1·nH2O is linked through the O–H⋯O hydrogen bond to one of the oxygen atoms of the carboxylate group of the aze ligand coordinated to the Cu2 cation (Table 2). Thus, the solvent molecule does not play a structure-forming role in the crystal packing of complex 1·nH2O. Contrarily, in the crystal structures of complexes 2·H2O and 3·H2O, the crystalline water molecules link the above-mentioned supramolecular aggregates through the O–H⋯N hydrogen bonds formed with the 3-pyridine nitrogen atom of the planar ligand L in complex 2·H2O, and with the 3-pyridine and amide nitrogen atoms of the ligands L in complex 3·H2O (Table 2).
DFT calculations were carried out to compare the energetics of the π-stacking and hydrogen bonding interactions (Fig. 6) and to examine the role of crystalline water molecules in complexes 2 and 3. Initially, we computed the MEP surfaces of both complexes to identify the most electrophilic and nucleophilic regions (Fig. 7). For 2, the MEP maximum is located at the aromatic hydrogen atoms of the Cu-coordinated pyridine ring (30.1 kcal mol−1), while the minimum is found at the oxygen atoms of the hydrazide group (−44.9 kcal mol−1), closely followed by the non-coordinated pyridine nitrogen atoms (−44.6 kcal mol−1). The nitrogen atom of the hydrazide group also shows a significantly negative MEP value (−30.7 kcal mol−1). Notably, the MEP over the coordinated pyridine ring is positive, whereas it is negative over the non-coordinated ring, suggesting that the π⋯π stacking interactions are electrostatically favourable. For 3, the MEP maximum is located at the hydrogen atom of the coordinated water molecule not involved in the intramolecular O–H⋯O interaction with the non-coordinated carboxylate oxygen atom of the Haze ligand. High positive MEP values are also found at the carboxylic hydrogen (45.5 kcal mol−1) and the aromatic hydrogens of the coordinated pyridine ring (40.8 kcal mol−1). The MEP minimum is at the carboxylate group of the Haze ligand (−45.3 kcal mol−1), followed by the oxygen of the hydrazide group (−43.9 kcal mol−1) and the nitrogen of the non-coordinated pyridine ring (−40.8 kcal mol−1). Additional negative MEP values are observed at the oxygen atom of the carboxylate group (−36.4 kcal mol−1) and the nitrogen atom of the hydrazide group (−18.2 kcal mol−1). This analysis reveals that complex 3 contains several atoms capable of acting as strong hydrogen bond donors and acceptors. As with complex 2, the MEP values over the π-systems of the coordinated and non-coordinated pyridine rings exhibit opposite signs, further supporting the possibility of electrostatically favoured π-stacking interactions.
 | ||
| Fig. 7 MEP surfaces of 2 (top) and 3 (bottom). Isovalue 0.001 a.u. | ||
Next, we analysed both types of antiparallel π-stacking modes observed in complex 2 (Fig. 6). The combined QTAIM/NCIplot analysis and the corresponding energetic features for two dimers (A and B), which serve as models for both stacking modes, were collected (Fig. 8). In dimer A, the π-stacking is characterized by two bond critical points (BCPs) and bond paths interconnecting carbon atoms of the pyridine rings. The π-nature of the interaction is clearly visualized with the NCIplot, which reveals a green reduced density gradient (RDG) isosurface enveloping the entire π-system of the pyridines.
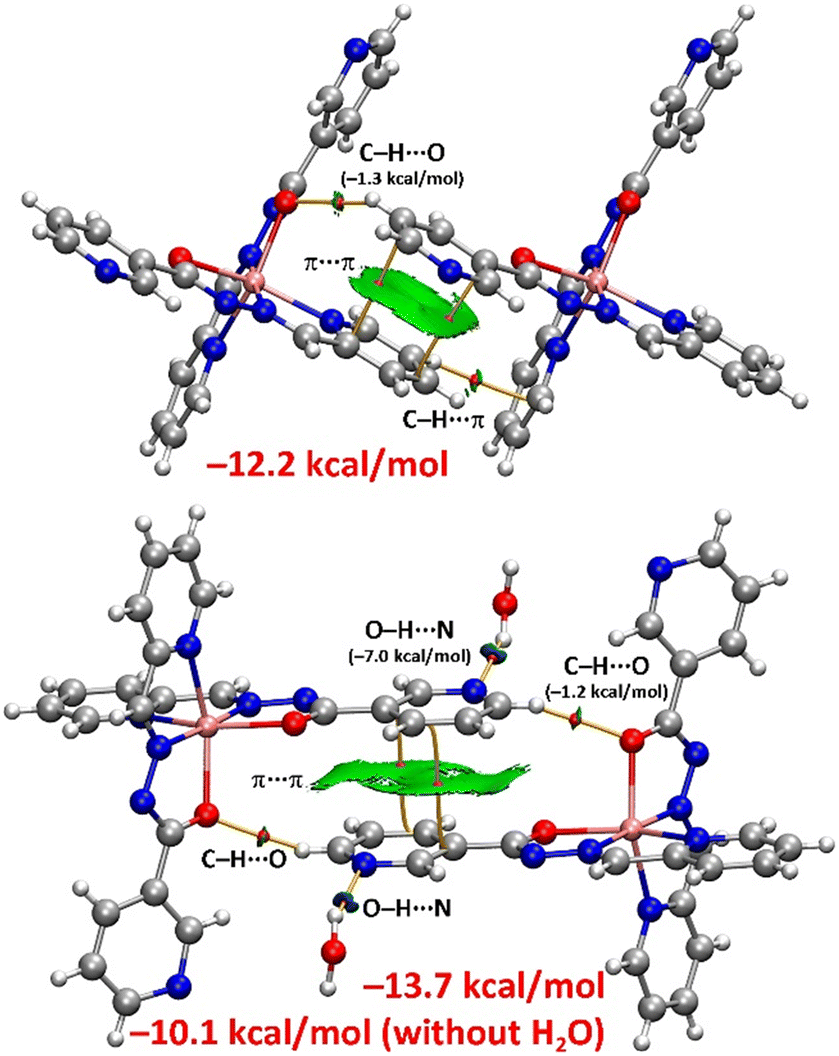 | ||
| Fig. 8 QTAIM/NCIplot analysis of dimers A (top) and B (bottom) of complex 2. BCPs and bond paths are given as small red spheres and solid orange lines, respectively. Only intermolecular BCPs are shown for clarity. Parameters for the NCIplot representation: s = 0.5, ρ cut-off 0.04 a.u., colour scale −0.035 a.u. ≤ (signλ2) ρ ≤ 0.035 a.u. | ||
The combined QTAIM/NCIplot analysis also revealed two ancillary interactions. One involves an aromatic hydrogen atom connected to the coordinated oxygen atom of the hydrazide group via a BCP, bond path and green RDG isosurface, forming a C–H⋯O hydrogen bond. Additionally, on the opposite side, an aromatic hydrogen atom is connected to a carbon atom of the pyridine ring, establishing a C–H⋯π interaction. The dimerization energy is significant (−12.2 kcal mol−1), reflecting the strength of the electrostatically enhanced π⋯π interactions, which play a key role in dictating the solid-state architecture of complex 2. The contribution of the hydrogen bond was estimated using QTAIM descriptors and a method from the literature, yielding an interaction energy of −1.3 kcal mol−1, thereby confirming the dominance of the π-stacking interaction.
Dimer B in complex 2 is centrosymmetric and has been computed both with and without the presence of lattice water molecule (Fig. 8). These water molecules form hydrogen bonds with the nitrogen atom of the pyridine ring, which, as indicated by the MEP results, is an excellent hydrogen bond acceptor. This is further confirmed by the QTAIM, which estimates the strength of each O–H⋯N hydrogen bond to be −7.0 kcal mol−1. The dimerization energy, in the presence of these water molecules, is −13.7 kcal mol−1, compared to −10.1 kcal mol−1 without them. This suggests that the water molecules enhance the π-stacking interaction. Similar to dimer A, the π-stacking in dimer B is characterized by two BCPs, bond paths and an extended RDG isosurface, confirming the large overlap of the π-clouds. The QTAIM/NCIplot analysis also revealed the existence of two symmetrically equivalent hydrogen bonds between aromatic hydrogen atoms and the oxygen atoms of the hydrazido groups. The strength of these hydrogen bonds, contributing to the dimer formation, is −2.4 kcal mol−1 (−1.2 kcal mol−1 each), indicating that the π-stacking interaction remains dominant, likely due to strong dipole⋯dipole attraction arising from the antiparallel orientation of the pyridine rings.
For complex 3, two dimers were extracted from the 1D supramolecular ribbon (Fig. 6), which dictates its crystal packing. Dimer A corresponds to a centrosymmetric dimer where two strong (blue RDG) O–H⋯O hydrogen bonds and two weak (green RDG) hydrogen bonds are formed, as revealed by the QTAIM/NCIplot analysis (Fig. 9). In addition, hydrophobic C–H⋯H–C interactions are established involving the long aliphatic chains of the Haze ligands. This is confirmed by the presence of two BCPs, bond paths and several small and green RDG isosurfaces connecting the chains. The dimerization energy is calculated to be −18.0 kcal mol−1, which is stronger than the π-stacking observed in complex 2. The contribution of the hydrogen bonds is dominant (−14.0 kcal mol−1), with van der Waals contacts contributing −4.0 kcal mol−1.
 | ||
| Fig. 9 QTAIM/NCIplot analysis of dimers A (top) and B (bottom) of complex 3. BCPs and bond paths are given as small red spheres and solid orange lines, respectively. Only intermolecular BCPs are shown for clarity. Parameters for the NCIplot representation: s = 0.5, ρ cut-off 0.04 a.u., colour scale −0.035 a.u. ≤ (signλ2) ρ ≤ 0.035 a.u. | ||
Dimer B features a complex combination of interactions (Fig. 9). Similar to complex 2, we calculated the dimerization energy with and without the presence of lattice water molecules. In this complex, the O–H⋯N hydrogen bonds between the water molecules and pyridines are estimated to be −3.9 kcal mol−1, weaker than those in complex 2. This aligns with the MEP results, which showed a less negative MEP at the nitrogen atom for complex 3 and a longer O–H⋯N experimental distance (Table 2). The dimerization energy with the lattice water molecules is −25.0 kcal mol−1, reduced to −23.8 kcal mol−1 without them, indicating a positive cooperativity effect. The Cu-coordinated water molecule forms a bifurcated O–H⋯O hydrogen bond with the coordinated oxygen atoms of both co-ligands. Additionally, this water molecule acts as a hydrogen bond acceptor with an aromatic hydrogen atom of the pyridine ring. Another notable interaction is the cation⋯π interaction formed between the copper atom of one monomer and the π-system of the adjacent monomer. The QTAIM and NCIplot analyses also reveal hydrophobic C–H⋯H–C contacts. This intricate combination of interactions explains the strong dimerization energy (−23.8 kcal mol−1), even greater than the hydrogen bonded dimer A. In fact, the contribution of the hydrogen bonds in the dimer (without lattice water molecules) is −6.3 kcal mol−1, confirming that other interactions play a more significant role.
Finally, the EDA was performed to examine the key contributions to the dimerization energies and assess the influence of lattice water molecules on the different interaction terms. The total dimerization energies (Etot) were partitioned into exchange repulsion (Eexc), electrostatic (Eelst), orbital (Eorb), correlation (Ecor) and dispersion (Edisp) components (Fig. 10). For both π-stacking modes in complex 2, dispersion is the most significant attractive contribution, followed by correlation, which is typical of π⋯π stacking interactions. Notably, for dimer B of complex 2, the presence of lattice water molecules enhances the contributions of Eelst, Eorb and Edisp (Fig. 10), with Eelst experiencing the greatest enhancement. This is consistent with the increased antiparallel dipole⋯dipole attraction due to the O–H⋯N hydrogen bonds.
 | ||
| Fig. 10 The total, Etot (black); exchange repulsion, Eexc (red); electrostatic, Eelst (blue); orbital, Eorb (green); correlation, Ecor (violet) and dispersion, Edisp (magenta) energies for the three dimers of complexes 2 (top) and 3 (bottom). | ||
In complex 3, for dimer A, the electrostatic term dominates, reflecting the significance of hydrogen bonds in this dimer. The other contributions are similar, ranging from −5.9 to −7.3 kcal mol−1. In contrast, for dimer B, dispersion dominates due to the prevalence of π-based interactions (Cu⋯π, π-stacking, O⋯π; Fig. 9). In this case, the enhancement of the dimerization energy by lattice water molecules is attributed to an increase in the dispersion component rather than the electrostatic term, which remains constant. This is likely due to the formation of an additional O⋯π interaction involving one of the lattice water molecules (Fig. 9).
Conclusions
In summary, we report three novel copper(II) discrete and polymeric coordination compounds, namely [Cu3L2(aze)2]n·nH2O (1·nH2O), [CuL2]·H2O (2·H2O) and [CuL(H2O)(Haze)]·H2O (3·H2O), which were readily obtained from (pyridin-2-yl)methylenenicotinohydrazide (HL) and azelaic acid (H2aze), using evaporative crystallization, grinding and slurry synthetic approaches. The crystal structures of the obtained coordination compounds have been elucidated by single crystal X-ray diffraction revealing different types of complexes, viz. mononuclear homo- and heteroleptic complexes (2 and 3) and a 1D heteroleptic coordination polymer (1). The solvent crystallization method allowed the production of a crystalline precipitate of complex 1·nH2O. The filtrate allowed the production of crystals of complex 3·H2O. The grinding synthetic method produced a mixture of complexes 1·nH2O, 2·H2O and an unknown phase. Results of the slurry synthetic approach highly depend on the stirring time. Stirring up to about 6 h leads to the exclusive formation of complex 1·nH2O, while longer stirring (1–2 days) results in the formation of a mixture of complexes 1·nH2O and 3·H2O. Finally, the present contribution enriches the library of coordination polymers and provides new insights into the effect of the synthetic procedure onto the resulting compounds.A key outcome of the theoretical analysis is the thorough evaluation of π-stacking and hydrogen bonding interactions in these compounds, with particular emphasis on the role of co-crystallized water molecules. DFT calculations, combined with QTAIM, NCIplot and EDA, provided detailed insights into the energetics and nature of these interactions. The results confirmed that π-stacking interactions are the primary contributors to the solid-state stabilization of complex 2. In contrast, in complex 3, hydrogen bonds dominate in dimer A, while other non-covalent forces prevail in dimer B. Notably, dimer B in complex 2 shows cooperativity due to electrostatic forces in the presence of water molecules, whereas in complex 3, this cooperativity arises primarily from dispersion forces. These findings highlight the crucial role of both non-covalent interactions and co-crystallized water molecules in shaping the final structures of these coordination compounds, offering valuable insights for the design of novel materials.
Experimental
Materials
All the reagents and solvents were commercially available and used without further purification. The parent ligand HL was synthesized as previously described.46,47 The isolated solid product was obtained in the form of the corresponding monohydrate HL·H2O as evidenced from the elemental analysis data. Anal. calc. for C12H12N4O2 (244.25): C 59.01, H 4.95 and N 22.94%; found: C 58.88, H 4.89 and N 22.73%. The presence of water is also supported by TGA, which revealed a mass loss of about 7.4% between 50 and 150 °C, corresponding to elimination of one molecule of water per one molecule of HL.Physical measurements
The IR spectra were collected using a Jasco 4700LE spectrometer equipped with an attenuated total reflectance accessory. A NETZSCH-STA 449 F1 Jupiter system was used to perform thermal analysis. Samples (3–8 mg) were placed in an open alumina pan and measured at a scan speed of 10 °C min−1 from ambient temperature to 500 °C under a nitrogen atmosphere as a protective and purge gas with the flow velocities of 20 and 40 mL min−1, respectively. Elemental analysis was performed with a CHNS Thermo Scientific Flash 2000 elemental analyzer.Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mL) was added to the copper solution, followed by the addition of a mixture containing the ligand HL·H2O and H2aze giving a green crystalline precipitate of complex 1·nH2O, which was filtered off. Yield: 73%.
:1 mL) was added to the copper solution, followed by the addition of a mixture containing the ligand HL·H2O and H2aze giving a green crystalline precipitate of complex 1·nH2O, which was filtered off. Yield: 73%.
The resulting filtrate was left for the slow evaporation under ambient conditions. A few days later, several green crystals of complex 3·H2O were formed.
:1 mL) followed by acetone. The solid was allowed to air dry and collected for powder X-ray diffraction and IR analyses, which revealed the formation of a mixture of complexes 1·n(H2O) and 2·H2O accompanied with an undetermined phase.
Crystal data for 1·nH2O. C42H46Cu3N8O10, 2(H2O); Mr = 1049.52 g mol−1, triclinic, space group P
, a = 10.201(4), b = 10.349(4), c = 11.762(5) Å, α = 86.291(10), β = 74.948(8), γ = 78.418(9)°, V = 1174.6(8) Å3, Z = 1, ρ = 1.484 g cm−3, μ(Mo-Kα) = 1.413 mm−1, reflections: 25628 collected, 3970 unique, Rint = 0.216, R1(all) = 0.2009, wR2(all) = 0.2669, S = 1.056.
Crystal data for 2·H2O. C24H18CuN8O2, H2O; Mr = 532.03 g mol−1, triclinic, space group P
, a = 8.7721(12), b = 11.1218(16), c = 13.0204(18) Å, α = 91.724(3), β = 107.466(3), γ = 105.767(3)°, V = 1157.5(3) Å3, Z = 2, ρ = 1.526 g cm−3, μ(Mo-Kα) = 0.989 mm−1, reflections: 28339 collected, 4720 unique, Rint = 0.233, R1(all) = 0.2104, wR2(all) = 0.1699, S = 0.961.
Crystal data for 3·H2O. C21H26CuN4O6, H2O; Mr = 512.01 g mol−1, triclinic, space group P
, a = 5.1202(3), b = 7.4808(4), c = 30.0866(17) Å, α = 94.0670(10), β = 92.6730(10), γ = 95.8620(10)°, V = 1141.81(11) Å3, Z = 2, ρ = 1.489 g cm−3, μ(Mo-Kα) = 1.006 mm−1, reflections: 33716 collected, 5697 unique, Rint = 0.031, R1(all) = 0.0363, wR2(all) = 0.0872, S = 1.045.
Powder X-ray diffraction
The powder X-ray diffraction data were collected at room temperature with a Siemens D5000 powder diffractometer (Cu-Kα radiation, graphite monochromator). The step size of 2θ was 0.02° and the counting time was 1 s per step.DFT calculations
Theoretical calculations were computed at the PBE0-D4/def2-TZVP level of theory,52–54 using the crystallographic coordinates within the Turbomole 7.7 program.55 The “atoms-in-molecules” (AIM)56 and noncovalent interaction plot (NCIplot)57 analyses were performed at the same level of theory using the Multiwfn program.58 The QTAIM/NCIplot analysis was represented using the VMD software.59 The energy decomposition analysis (EDA) was performed using the Turbomole 7.7 program55 using the Kitaura–Morokuma partition scheme.60Data availability
Crystallographic data for compounds 1–3 have been deposited at the CCDC under deposition numbers CCDC 2370358–2370360.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. Benito and E. Molins acknowledge financial support from the State Investigation Agency, through the Severo Ochoa Programme for Centres of Excellence (projects CEX2019-00917-S and CEX2023-001263-S). M. Benito and E. Molins thank the X-ray diffraction, thermal analysis and spectroscopic services from the ICMAB. This research was funded by the MICIU/AEI of Spain (projects PID2021-1245720B-C32, PID2020-115637GB-I00 and PID2023-148453NB-I00, FEDER funds).Notes and references
- S. R. Batten, N. R. Champness, X.-M. Chen, J. Garcia-Martinez, S. Kitagawa, L. Öhrström, M. O'Keeffe, M. P. Suh and J. Reedijk, Terminology of metal–organic frameworks and coordination polymers (IUPAC Recommendations 2013), Pure Appl. Chem., 2013, 85, 1715–1724 CrossRef CAS.
- J. F. Keggin and F. D. Miles, Structures and formulæ of the prussian blues and related compounds, Nature, 1936, 137, 577–578 CrossRef CAS.
- Y. Kinoshita, I. Matsubara, T. Higuchi and Y. Saito, The crystal structure of Bis(adiponitrilo)copper(I) Nitrate, Bull. Chem. Soc. Jpn., 1959, 32, 1221–1226 CrossRef CAS.
- O. M. Yaghi and H. Li, Hydrothermal Synthesis of a Metal-Organic Framework Containing Large Rectangular Channels, J. Am. Chem. Soc., 1995, 117, 10401–10402 CrossRef CAS.
- O. M. Yaghi, G. Li and H. Li, Selective binding and removal of guests in a microporous metal-organic framework, Nature, 1995, 378, 703–706 CrossRef CAS.
- (a) J.-B. Huang, L. Yin, T.-C. Yue, L.-L. Wang and D.-Z. Wang, Assembly of Functional Co(II) Metal–Organic Frameworks through a Mixed Ligand Strategy: Structure and Photocatalytic Degradation Properties, Inorg. Chem., 2024, 63, 6928–6937 CrossRef CAS PubMed; (b) Q.-W. Cao, T.-C. Yue, Q.-W. Dong, Q.-C. Ma, Z.-B. Xie, D.-Z. Wang and L.-L. Wang, Effective detection of Ag+, Hg2+ and dye adsorption properties studies of Ln-MOFs based on a benzimidazole carboxylic acid ligand, Dalton Trans., 2023, 52, 6008–6018 RSC; (c) L. Yin, J.-B. Huang, T.-C. Yue, L.-L. Wang and D.-Z. Wang, Two 2D Metal–Organic Frameworks Based on Purine Carboxylic Acid Ligands for Photocatalytic Oxidation of Sulfides and CO2 Chemical Fixation, Inorg. Chem., 2024, 63, 9109–9118 CrossRef CAS PubMed.
- (a) C.-T. Chen and K. S. Suslick, One-dimensional coordination polymers: Applications to material science, Coord. Chem. Rev., 1993, 128, 293–322 CrossRef CAS; (b) W. L. Leong and J. J. Vittal, One-dimensional coordination polymers: complexity and diversity in structures, properties, and applications, Chem. Rev., 2011, 111, 688–764 CrossRef CAS PubMed.
- E. Loukopoulos and G. E. Kostakis, Review: Recent advances of one-dimensional coordination polymers as catalysts, J. Coord. Chem., 2018, 71, 371–410 CrossRef CAS.
- K. C. Bentz and S. M. Cohen, Supramolecular metallopolymers: from linear materials to infinite networks, Angew. Chem., Int. Ed., 2018, 57, 14992–15001 CrossRef CAS PubMed.
- X. Meng, W. Shi and P. Cheng, Magnetism in one-dimensional metal-nitronyl nitroxide radical system, Coord. Chem. Rev., 2019, 378, 134–150 CrossRef CAS.
- J. Zhao, J. Yuan, Z. Fang, S. Huang, Z. Chen, F. Qiu, C. Lu, J. Zhu and X. Zhuang, One-dimensional coordination polymers based on metal–nitrogen linkages, Coord. Chem. Rev., 2022, 471, 214735 CrossRef CAS.
- N. Dunski and T. H. Crawford, Coordination polymers of Schiff base ligands and their monomeric analogs, J. Inorg. Nucl. Chem., 1973, 35, 2707–2717 CrossRef CAS.
- N. Ribeiro and I. Correia, A review of hydrazide-hydrazone metal complexes' antitumor potential, Front. Chem. Biol., 2024, 3, 1398873 CrossRef.
- Y.-Q. Zheng, J. Sun and J.-L. Lin, An Azelaato-bridged Dinuclear Copper(II) Complex, [Cu2(phen)2(C9H14O4)2]·6H2O (phen = 1,10-phenanthroline), Z. Anorg. Allg. Chem., 2000, 626, 1271–1273 CrossRef CAS.
- S. M. Krishnan, R. M. Supkowski and R. L. LaDuca, One- and two-dimensional divalent copper coordination polymers based on kinked organodiimine and long flexible aliphatic dicarboxylate ligands, J. Mol. Struct., 2008, 891, 423–428 CrossRef CAS.
- F.-H. Zhao, Y.-X. Che and J.-M. Zheng, Three 3D complexes constructed from azelaic aci and rigid bis(imidazole) ligand: Syntheses, structures, thermal and photoluminescent properties, Inorg. Chem. Commun., 2012, 24, 200–204 CrossRef CAS.
- L.-L. Han, S.-N. Wang, Z. Jagličić, S.-Y. Zeng, J. Zheng, Z.-H. Li, J.-S. Chen and D. Sun, Synthesis, structural versatility and magnetic properties of a series of copper(II) coordination polymers based on bipyrazole and various dicarboxylate ligands, CrystEngComm, 2015, 17, 1405–1415 RSC.
- J. Q. Xu, Y. Q. Zheng and W. Xu, Synthesis, crystal structures, and properties of copper(II) dicarboxylate complexes with [bis(2-pyridylcarbonyl)amido], Russ. J. Coord. Chem., 2017, 43, 63–72 CrossRef CAS.
- G. Liu, J. Zhao, S. Liang, Y. Li, Z. Chang, X. Wang and B. Chen, Ten polytorsional-amide-induced helical-based coordination polymers with difunctional electrochemical activities, CrystEngComm, 2021, 23, 1263–1271 RSC.
- W. Xu, K. Hu, S. Jin, Y. Zhang and D. Wang, Constructions of seven noncovalent-bonded supramolecules from reactions of Cu(II)/Cd(II)/Zn(II) with isonicotinamide and carboxylates, Inorg. Nano-Metal Chem., 2021, 51, 1842–1859 CrossRef CAS.
- D. A. Safin, M. G. Babashkina, F. D. Sokolov, N. G. Zabitov, J. Galezowska and H. Kozlowski, Complexes of a novel N-(diisopropxythiophosphoryl)thiourea derivative of 1,4,8,11-tetraazacyclotetradecane with Na+, K+ and Cu(PPh3)2+ cations, Polyhedron, 2007, 26, 1113–1116 CrossRef CAS.
- F. D. Sokolov, M. G. Babashkina, D. A. Safin, A. I. Rakhmatullin, F. Fayon, N. G. Zabirov, M. Bolte, V. V. Brusko, J. Galezowska and H. Kozlowski, Complexes of N-thiophosphorylthioureas (HL) with copper(I). Crystal structures of [Cu3L3] and [Cu(PPh3)2L] chelates, Dalton Trans., 2007, 4693–4700 RSC.
- D. A. Safin, M. G. Babashkina, T. R. Gimadiev, M. Bolte, M. V. Pinus, D. B. Krivolapov and I. A. Litvinov, Complexes of N-thiophosphorylthiourea (EtO)2P(O)CH2C6H4-4-[NHC(S)NHP(S)(OiPr)2] with Zn(II), Cd(II), Co(II) and Cu(PPh3)(I), Polyhedron, 2008, 27, 2978–2982 CrossRef CAS.
- F. D. Sokolov, M. G. Babashkina, F. Fayon, A. I. Rakhmatullin, D. A. Safin, T. Pape and F. E. Hahn, Novel bicyclic hexanuclear copper(I) aggregate: Structure and solid state 31P CPMAS NMR spectra of [(Cu3L3)2] and [Cu(PPh3)2L] complexes of N-(diisopropoxythiophosphinyl)-N′-phenylthiourea (HL), J. Organomet. Chem., 2009, 694, 167–172 CrossRef CAS.
- M. G. Babashkina, D. A. Safin, Ł. Szyrwiel, M. Kubiak, F. D. Sokolov, Y. V. Starikov and H. Kozlowski, Complexes of N-Thiophosphorylthiourea α-NaphthylNHC(S)NHP(S)(OiPr)2 (HL) with Copper(I). Crystal Structures of HL and Cu(PPh3)2L, Z. Anorg. Allg. Chem., 2009, 635, 554–557 CrossRef CAS.
- R. C. Luckay, X. Sheng, C. E. Strasser, H. G. Raubenheimer, D. A. Safin, M. G. Babashkina and A. Klein, Competitive bulk liquid membrane transport of some metal ions using RC(S)NHP(S)(OiPr)2 as ionophores. Unusual supramolecular “honeycomb” aggregate of the polynuclear copper(I) complex of H2NC(S)NHP(S)(OiPr)2, Dalton Trans., 2009, 4646–4652 RSC.
- D. A. Safin, M. G. Babashkina, M. Bolte, F. D. Sokolov and V. V. Brusko, Complexes of N-thiophosphorylthioureas RNHC(S)NHP(S)(OiPr)2 (HL) (R = pyridin-2-yl, pyridin-3-yl, 6-amino-pyridin-2-yl) with copper(I): Crystal structures of Cu(PPh3)nL (n = 1, 2), Inorg. Chim. Acta, 2009, 362, 1895–1900 CrossRef CAS.
- D. A. Safin, M. G. Babashkina, M. Bolte and A. Klein, Synthesis, characterization and luminescent properties of heteroligand copper(I) complexes with N-thiophosphorylated thioureas RNHC(S)NHP(S)(OiPr)2 (R = iPr, tBu, Ph, 2,6-Me2C6H3, 2,4,6-Me3C6H2) and phosphines (PPh3, Ph2P(C5H4FeC5H4)PPh2), Inorg. Chim. Acta, 2010, 363, 1897–1901 CrossRef CAS.
- M. G. Babashkina, D. A. Safin, F. Fayon, A. I. Rajhmatullin, F. D. Sokolov, A. Klein, D. B. Krivolapov, T. Pape, F. E. Hahn and H. Kozlowski, Copper(I) complexes with N-(diisopropoxythiophosphoryl)thiobenzamide PhC(S)NHP(S)(OiPr)2, Dalton Trans., 2010, 39, 8261–8268 RSC.
- D. A. Safin, M. G. Babashkina, M. Bolte, T. Pape, F. E. Hahn, M. L. Verizhnikov, A. R. Bashirov and A. Klein, Polynuclear and mixed-ligand mononuclear CuI complexes with N-thiophosphorylated thioureas and 1,10-phenanthroline or PPh3, Dalton Trans., 2010, 39, 11577–11586 RSC.
- M. G. Babashkina, D. A. Safin, M. Bolte and A. Klein, Copper(I) complexes with N-thiophosphorylated thioureas and phosphines with versatile structures and luminescence, CrystEngComm, 2010, 12, 134–143 RSC.
- R. C. Luckay, X. Sheng, C. E. Strasser, H. G. Raubenheimer, D. A. Safin, M. G. Babashkina and A. Klein, Aerial oxidation of tetrahydrofuran to 2-hydroxotetrahydrofuran in the presence of a trimeric CuI complex [Cu3L3] (HL = tBuNHC(S)NHP(S)(OiPr)2) and trapping of the unstable product at recrystallization, New J. Chem., 2010, 34, 2835–2840 RSC.
- M. G. Babashkina, E. R. Shakirova, D. A. Safin, F. D. Sokolov, A. Klein, Ł. Szyrwiel, M. Kubiak, H. Kozlowski and D. B. Krivolapov, Dinuclear Complexes of Copper(I) with Crown Ether-containing N-Thiophosphorylated Bis-thioureas and 2,2′-Bipyridine or 1,10-Phenanthroline: Synthesis, Characterization, and Picrate Extraction Properties, Z. Anorg. Allg. Chem., 2010, 636, 2626–2632 CrossRef CAS.
- M. G. Babashkina, D. A. Safin, A. Klein and M. Bolte, Synthesis, Characterisation and Luminescent Properties of Mixed-Ligand Copper(I) Complexes Incorporating N-Thiophosphorylated Thioureas and Phosphane Ligands, Eur. J. Inorg. Chem., 2010, 2010, 4018–4026 CrossRef.
- D. A. Safin, M. G. Babashkina, M. Bolte and M. Köckerling, Heteroligand copper(I) complexes of N-thiophosphorylated thioureas and phosphanes: Versatile structures and luminescence, Inorg. Chim. Acta, 2011, 370, 59–64 CrossRef CAS.
- D. A. Safin, M. G. Babashkina, M. Bolte and A. Klein, Versatile structures and photophysical properties of poly- and mononuclear CuI complexes with N-thiophosphorylated thioureas RNHC(S)NHP(S)(OiPr)2 and phosphanes, CrystEngComm, 2011, 13, 568–576 RSC.
- D. A. Safin, M. G. Babashkina, M. Bolte and Y. Garcia, Homoleptic polynuclear CuI and AgI complexes of N-thiophosphorylated thioureas o-RO(O)CC6H4NHC(S)NHP(S)(OiPr)2 (R = Me, Et), CrystEngComm, 2012, 14, 774–778 RSC.
- M. G. Babashkina, D. A. Safin and Y. Garcia, Heteroleptic Cu(I) and Ag(I) complexes of N-thiophosphorylated tris-thioureas and triphenylphosphine, Polyhedron, 2012, 33, 114–118 CrossRef CAS.
- M. M. Ditru, Y. Boland, D. Gillard, B. Tinant, K. Robeyns, D. A. Safin, E. Devlin, Y. Sanakis and Y. Garcia, New Mononuclear Cu(II) Complexes and 1D Chains with 4-Amino-4H-1,2,4-triazole, Int. J. Mol. Sci., 2013, 14, 23597–23613 CrossRef PubMed.
- D. A. Safin, M. P. Mitoraj, K. Robeyns, Y. Filinchuk and C. M. L. Vande Velde, Luminescent mononuclear mixed ligand complexes of copper(I) with 5-phenyl-2,2′-bipyridine and triphenylphosphine, Dalton Trans., 2015, 44, 16824–16832 RSC.
- D. A. Safin, C. M. L. Vande Velde, M. G. Babashkina, K. Robeyns and Y. Filinchuk, Mononuclear heteroleptic complexes of copper(I) with 5-phenyl-2,2′-bipyridine and triphenylphosphine: crystal structures, Hirshfeld surface analysis and luminescence properties, New J. Chem., 2016, 40, 6156–6163 RSC.
- O. Semyonov, K. A. Lyssenko and D. A. Safin, Copper(II) acetate structures with benzimidazole derivatives, Inorg. Chim. Acta, 2019, 488, 238–245 CrossRef CAS.
- A. A. Shiryaev, T. M. Burkhanova, M. P. Mitoraj, M. Kukulka, F. Sagan, G. Mahmoudi, M. G. Babashkina, M. Bolte and D. A. Safin, Supramolecular structures of NiII and CuII with the sterically demanding Schiff base dyes driven by cooperative action of preagostic and other non-covalent interactions, IUCrJ, 2021, 8, 351–361 CrossRef CAS PubMed.
- E. V. Panova, J. K. Voronina and D. A. Safin, Copper(II) Chelates of Schiff Bases Enriched with Aliphatic Fragments: Synthesis, Crystal Structure, In Silico Studies of ADMET Properties and a Potency against a Series of SARS-CoV-2 Proteins, Pharmaceuticals, 2023, 16, 286 CrossRef CAS PubMed.
- E. V. Panova and D. A. Safin, Computational Studies of Copper(II) Complexes Derived from N-Cyclohexyl-3-methoxysalicylideneimine and N-Cyclohexyl-3-ethoxysalicylideneimine, Russ. J. Gen. Chem., 2024, 94, 703–710 CrossRef CAS.
- P. V. Bernhardt, P. Chin, P. C. Sharpe and D. R. Richardson, Hydrazonechelators for the treatment of iron overload disorders: iron coordination chemistry and biological activity, Dalton Trans., 2007, 3232–3244 RSC.
- M. Kuriakose, M. R. P. Kurup and E. Suresh, Six coordinate Ni(II) complexes of ONN donor aroylhydrazone ligands: synthesis, spectral studies, and crystal structures, Struct. Chem., 2007, 18, 579–584 CrossRef CAS.
- Bruker, APEX2 Software, Bruker AXS Inc. v2019.11-0, Madison, Wisconsin, USA, 2019 Search PubMed.
- R. H. Blessing, An empirical correction for absorption anisotropy, Acta Crystallogr., Sect. A: Found. Crystallogr., 1995, 51, 33–38 CrossRef PubMed.
- G. M. Sheldrick, SADABS, Program for Empirical Absorption Correction of Area Detector Data, University of Goettingen, Germany, 1997 Search PubMed.
- G. M. Sheldrick, A short history of SHELX, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- C. Adamo and V. Barone, Toward reliable density functional methods without adjustable parameters: The PBE0 model, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- F. Weigend, Accurate Coulomb-fitting basis sets for H to Rn, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- E. Caldeweyher, S. Ehlert, A. Hansen, H. Neugebauer, S. Spicher, C. Bannwarth and S. Grimme, A generally applicable atomic-charge dependent London dispersion correction, J. Chem. Phys., 2019, 150, 154122 CrossRef PubMed.
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Electronic structure calculations on workstation computers: The program system Turbomole, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- R. F. W. Bader, A quantum theory of molecular structure and its applications, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- J. Contreras-Garcia, E. R. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. N. Beratan and W. Yang, NCIPLOT: A Program for Plotting Noncovalent Interaction Regions, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef CAS PubMed.
- T. Lu and F. Chen, Multiwfn: A multifunctional wavefunction analyzer, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, VMD: visual molecular dynamics, J. Mol. Graph., 1996, 14, 33–38 CrossRef CAS PubMed.
- K. Kitaura and K. Morokuma, A new energy decomposition scheme for molecular interactions within the Hartree-Fock approximation, Int. J. Quantum Chem., 1976, 10, 325–340 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: CCDC 2370358–2370360. For crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ce01021c |
| This journal is © The Royal Society of Chemistry 2025 |