
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructures and band gaps of lead-free dabconium-containing hybrid alkali-metal halide perovskites†
Hendrik J.
van der Poll
 a,
Rudolph
Erasmus
b and
Melanie
Rademeyer
*a
a,
Rudolph
Erasmus
b and
Melanie
Rademeyer
*a
aDepartment of Chemistry, University of Pretoria, Pretoria, 0002, South Africa. E-mail: melanie.rademeyer@up.ac.za
bSchool of Physics, University of the Witwatersrand, P.O. Wits, 2050 Johannesburg, South Africa
First published on 21st March 2025
Abstract
Ten hybrid metal halide perovskites containing divalent dabconium cations and alkali-metal ions have been structurally characterised and their band gaps determined, with six new crystal structures reported. Five compounds of the formula (C6H14N2)[BX3], where B = Na+ and X = Cl− or Br−, B = K+ and X = Cl− or Br− and B = Cs+ and X = I−, were found to exhibit one-dimensional (1D) hexagonal perovskite structures containing face-sharing metal halide octahedra that form 1D inorganic polymers. Five compounds of the formula (C6H14N2)[BX3], where B = K+ and X = Br− or I−, and B = Cs+ and X = Cl−, Br− or a mixture of Cl− and I−, display three-dimensional (3D) parent perovskite structures containing corner sharing metal halide octahedra that form a 3D framework. A correlation was found between the formation of a 1D or 3D perovskite structure and the sum of ionic radii of the metal and halide ions. The band gaps of the compounds fall in the range 3.42 eV to 5.33 eV. It was found that a 3D perovskite has a smaller band gap than a 1D perovskite, while the halide ion also affects the band gap.
Introduction
Metal halide perovskites have enjoyed much attention from the scientific community over recent years because of their wide range of applications, specifically as sensitisers in perovskite solar cells (PSCs).1 The three-dimensional (3D) parent perovskite structure has the general formula ABX3, where A is either an organic or inorganic cation, B is usually a metal cation, and X is an anion.2,3 In this structure, the A cation has a 12-fold coordination and the B cation a 6-fold coordination, with BX6 octahedra sharing corners. If A is an organic cation and X is a halide anion, a hybrid metal halide perovskite structure is formed.In addition to the 3D parent hybrid perovskite structure described above, other hybrid perovskite structures may also be formed. Relevant to the current study is one-dimensional (1D) hexagonal perovskites which also exhibit the formula ABX3.4 The 1D hexagonal perovskite structure contains face-sharing octahedra, forming 1D halide-bridged polymers.5
To predict whether the A-site cation will “fit” in the BX-framework and allow for the formation of the parent 3D perovskite structure, several prediction factors may be utilised. These include the Goldschmidt tolerance factor6 (t), the octahedral factor7 (μ), and Bartell's tolerance factor8 (τ). More detail on these factors are given in the Section S1.†
The parent hybrid halide perovskites of lead have shown promise as sensitisers in PSCs. The hybrid halide perovskite MAPbI3, with MA = methylammonium, originally synthesised by Weber in 1978,9 was first utilised in PSCs by Miyasaka and Teshima.10–12 Since its initial application in PSC's, MAPbI3 has been modified using other A-cations such as formamidinium (FA) and other organic cations.13 Furthermore, halide mixtures (X = Cl−, Br− or I−) have also been employed,14 as well as combinations of lead and other divalent metal ions, in an attempt to increase the stability and efficiency of PSCs.15
Due to their potential application in PSCs, 3D parent hybrid perovskites are sought after materials. Since lead is a highly toxic metal, there has been a drive to reduce the lead content of these hybrid perovskites, leading to the investigation of the use of lead-less perovskites, where the lead ions in a perovskite structure are partially substituted by other metal ions, or lead-free perovskites, in PSCs.16
In the parent 3D hybrid halide perovskite structure, the organic A cation typically has a charge of +1, while the B metal cation is divalent. However, in the current study we decided to switch these valences, and use B+ metal ions and A2+ organic cations to form perovskite compounds of the formula ABX3. The non-toxic group 1 alkali metal ions17 Li+, Na+, K+ and Cs+ were selected as B+ metal ions, thus avoiding the use of lead as metal ion. In addition, a divalent organic cation, namely the dabconium cation, which is obtained via protonation of the 1,4-diazabicyclo[2.2.2]octane molecule, was selected as A2+ organic cation.
Compounds, and their corresponding structures, will be referred to according to an abbreviation based on their composition. In the abbreviation D-BXn, D indicates that the organic cation is dabconium, B represents the alkali metal ion, X indicates the halide anion, and n gives the ratio of halide ions to metal ions in the molecular formula. For example, the abbreviation D-NaCl3 indicates the compound or structure resulting from the combination of dabconium cations and anionic species comprised of Na+ and Cl− ions. The addition of a number in brackets at the end of the abbreviation, e.g.D-KBr3(1) and D-KBr3(2), indicates polymorphs of the same compound.
A search of the Cambridge Structural Database (CSD (Version 5.44, update June 2023)18) indicated that a number of structures containing dabconium cations, and metal-halide anions comprised of monovalent alkali metal ions and halide anions, have been reported in the literature.
Two 1D structures have been reported. The 1D hexagonal perovskite structure consisting of dabconium cations and KCl-based anions (D-KCl3) has been reported twice, with the CSD18 reference codes DOTHOK19 and DOTHOK01.20 The two structures were determined at 100 K and 120 K respectively, with the same structure present at both temperatures.
The combination of dabconium and a RbI-based anion (D-RbI3) also results in the formation of a 1D hexagonal perovskite structure, with CSD18 reference code HEJHOW,21 with the structure determined at room temperature (293 K).
Five 3D perovskite structures have been reported in the literature for the combination of the dabconium cation and different alkali metal ions and halide ions. The perovskite structure obtained from the combination of dabconium and a KBr-based anion, D-KBr3(2), with CSD18 reference code FIZYIZ,22 was determined at 300 K. In addition, two different structures have been reported for the combination of dabconium and a RbCl-based anion, with CSD18 reference codes GUYNEU20 (D-RbCl3(1)), GUYNEU0121 (D-RbCl3(1)) and GUYNEU0221 (D-RbCl3(2)), with the structures forming a trigonal phase at 120 K and 293 K and a cubic phase at 458 K. Furthermore, two 3D structures have been reported containing dabconium cations and a RbBr-based anion, with CSD18 reference codes HEJGUB21 (D-RbBr3(1)) and HEJGUB0121 (D-RbBr3(2)), with the structures representing trigonal and cubic phases of the compound determined at 293 K and 353 K, respectively. Additionally, a monoclinic 3D perovskite structure was reported to form for the combination of dabconium and a CsCl-based anion (D-CsCl3), with CSD18 reference code GUYNIY,20 determined at 120 K. Finally, the structure of an orthorhombic 3D perovskite resulting from the combination of dabconium with CsBr-based anions (CSD18 reference code BEFXOD01) has also been determined at 193 K.23
The literature survey indicates a tendency of compounds formed by the combination of the dabconium cation and alkali metal halides to form perovskite structures, either of the 3D parent perovskite type or 1D hexagonal type. However, not all the structures in this family have been reported, and those that have been reported, were determined at different temperatures. This aim of this study is to determine the outstanding structures and to re-determine the reported structures at the same temperature. The study will also focus on determination of the band gaps of these compounds, since their band gaps have not been reported.
The three tolerance factors mentioned previously were calculated for all the compounds of interest, with the results of the calculations given in the Section S1.† The ionic radius of the dabconium cation was taken as 339 pm, as per the determination using density functional theory calculations reported in the literature.24 The Shannon ionic radii25 of the metal ions were used.
The analysis showed that if the dabconium cation is combined with LiCl-, NaCl-, KCl-, CsCl-, LiBr-, or LiI-based anions, the resultant structure is unlikely to be of the 3D perovskite type. This is because either the A-site ion is not large enough (indicated by t) or that octahedral coordination is unlikely (indicated by μ). Bartel's tolerance factor agrees with these predictions on combinations containing Li+. Therefore, it is highly improbable that LiX-based anions with dabconium cations would result in 3D perovskite structures.
Furthermore, when dabconium cations are combined with RbCl-, NaBr-, KBr-, RbBr-, CsBr-, NaI-, KI-, RbI- or CsI-based anions, this could result in 3D parent perovskites. This is also strongly supported by the literature survey, which shows that the combinations that contain KBr-, RbCl- or RbBr-based anions all lead to 3D parent perovskite structures. Only the combination of the dabconium cation with RbI-based anions is predicted incorrectly by all three factors as a 3D perovskite structure, however, a 1D hexagonal perovskite structure was reported in the literature (HEJHOW21).
It can be deduced that the Goldschmidt tolerance factor,6 the octahedral factor,7 as well as Bartel's tolerance factor,8 when used in combination with each other, are often successful in predicting the formation of a 3D parent perovskite structure when considering the structures reported in the literature.
In the current investigation dabconium cations were combined with the alkali metal halides NaCl, NaBr, NaI, KCl, KBr, KI, CsCl, CsBr or CsI in an organic![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :inorganic molar ratio of 1:1, to form hybrid materials. In addition to the structural investigation, the band gaps of the compound were determined using diffuse reflectance spectroscopy (DRS).
:inorganic molar ratio of 1:1, to form hybrid materials. In addition to the structural investigation, the band gaps of the compound were determined using diffuse reflectance spectroscopy (DRS).
Due to the tolerance factors' prediction that the combination of dabconium with Li-halide salts is not expected to yield 3D perovskite structures, the Li+-members of the series were not investigated. Furthermore, Rb-halide salts are prohibitively expensive, and these members of the series were not included in this study, however, some of these structures have been reported in the literature.20,21
Experimental section
Materials
All reagents and solvents used (except distilled water) were reagent grade purchased from Sigma Aldrich and were used without further purification or modification. Distilled water was obtained from the in-house distillation facility. Specific information: NaCl (≥99%), NaBr (≥99%), NaI (≥99%), KCl (≥99%), KBr (≥99%), KI (≥99%), CsCl (≥99%), CsBr (≥99%), CsI (≥99%), 1,4-diazabicyclo[2.2.2]octane (99%), HCl (37% solution), HBr (48% weight) and HI (57% weight, stabilised solution).Synthesis
No attempts were made to optimise the yields of the reactions. All relevant synthetic information is summarised in Table 1.| Combination (BX + HX) | Organic mass (g) | BX mass (g) | HX volume (ml) | Resultant structure | Product mass (g) | Yield (%) | Elemental analysis |
|---|---|---|---|---|---|---|---|
| NaCl + HCl | 1.0079 (8.99 mmol) | 0.5050 (8.64 mmol) | 3.10 | D-NaCl 3 | 0.8437 | 40.0 | Found: C, 29.52%; H, 6.02%; N, 11.50% |
| Calc.: C, 29.59%; H, 5.79%; N, 11.50% | |||||||
| NaBr + HBr | 1.0153 (9.05 mmol) | 0.8933 (8.68 mmol) | 8.90 | D-NaBr 3 | 1.8461 | 56.4 | Found: C, 18.98%; H, 3.60%; N, 7.79% |
| Calc.: C, 19.12%; H, 3.74%; N, 7.43% | |||||||
| KCl + HCl | 1.0060 (8.97 mmol) | 0.6451 (8.65 mmol) | 3.20 | D-KCl 3 | 1.9175 | 85.4 | Found: C, 27.78%; H, 5.24%; N, 10.83% |
| Calc.: C, 27.76%; H, 5.44%; N 10.79% | |||||||
| KBr + HBr | 2.0826 (18.57 mmol) | 2.1467 (18.04 mmol) | 9.00 | D-KBr 3 (1) | 5.8023 | 81.8 | Found: C, 18.41%; H, 3.35%; N, 7.05% |
| Calc.: C, 18.34%; H, 3.59%; N 7.13% | |||||||
| KBr + HBr | 2.0007 (17.84 mmol) | 2.1225 (17.84 mmol) | 9.00 | D-KBr 3 (2) | 6.6743 | 95.3 | Found: C, 18.49%; H, 3.31%; N, 7.07% |
| Calc.: C, 18.34%; H, 3.59%; N 7.13% | |||||||
| KI + HI | 1.5075 (13.44 mmol) | 1.3442 (8.10 mmol) | 5.00 | D-KI 3 | 4.7263 | 68.7 | Found: C, 13.55%; H, 2.20%; N, 5.01% |
| Calc.: C, 13.5%; H, 2.64%; N 5.25% | |||||||
| CsCl + HCl | 1.5086 (13.45 mmol) | 2.2630 (13.44 mmol) | 10.00 | D-CsCl 3 | 2.8467 | 15.0 | Found: C, 20.13%; H, 3.90%; N, 8.19% |
| Calc.: C, 20.39%; H, 3.99%; N 7.93% | |||||||
| CsBr + HBr | 2.0071 (17.89 mmol) | 3.8058 (17.88 mmol) | 4.00 | D-CsBr 3 | 3.8058 | 52.1 | Found: C, 14.57%; H, 2.70%; N, 5.61% |
| Calc.: C, 14.93%; H, 2.70%; N 5.77% | |||||||
| CsI + HI | 0.2178 (1.94 mmol) | 0.4640 (1.79 mmol) | 0.50 | D-CsI 3 | 0.7753 | 69.1 | Found: C, 11.29%; H, 2.22%; N, 4.55% |
| Calc.: C, 11.48%; H, 2.25%; N 4.46% | |||||||
| CsI + HCl | 0.2171 (1.94 mmol) | 0.4660 (1.79 mmol) | 1.20 | D-CsCl 2 I | 0.6597 | 82.7 | Found: C, 15.89%; H, 2.86%; N, 6.33% |
| Calc.: C, 16.20%; H, 3.17%; N 6.30% |
Crystal growth
The general method employed to grow crystals was a slow evaporation method, and was carried out as follows: in a 100 ml beaker, a given mass of 1,4-diazabicyclo[2.2.2]octane was dissolved in 20 ml distilled water. To this solution, a molar excess of HX (X = Cl−, Br−, or I−) was added. Thereafter, an equimolar amount of BX (B = Na+, K+, Cs+ and X = Cl−, Br− or I−) was dissolved in a minimal amount of distilled water and added to the acidic solution. The resultant solution was left at room temperature, open to the atmosphere, for the solvent to evaporate.The crystals were harvested once all the solvent had evaporated and were washed with reagent grade acetone. All resultant crystals were colourless. The slow evaporation method was successful in delivering crystals for the following materials: D-NaCl3, D-NaBr3, D-KCl3, D-KBr3(1), D-KI3, D-CsCl3, D-CsBr3, D-CsI3 and D-CsCl3I.
Bulk powders
The heated evaporation method was employed to obtain bulk powders of the materials. The bulk powders were characterised using powder X-ray diffraction (see Section S2†).This method was performed as follows: a solution of 1,4-diazabicyclo[2.2.2]octane along with an equimolar mass of BX, and excess HX, was prepared in a 100 ml glass beaker with enough distilled water to form a dilute solution. The solution was placed on a hot plate and heated to between 80 °C and 110 °C until all the solvent had evaporated. The resultant product was a white powder in each case.
This method generally resulted in the same materials as obtained from the slow evaporation method, however, with one exception. For the combination of 1,4-diazabicyclo[2.2.2]octane with KBr and HBr, the heated evaporation method delivered a different polymorph, named D-KBr(2) (see Table 1 for synthetic details), from the polymorph obtained by slow evaporation, named D-KBr(1). To distinguish between these materials the suffix (1) or (2) is employed. (1) refers to the slow evaporation method product, whereas (2) refers to the heated evaporation method product.
Instrumental techniques
Crystallographic discussion of structures
Ten compounds were prepared and the single crystal structures of nine were determined, six of which are new and have not been reported in the literature. Single crystal X-ray diffraction data for all compounds, were collected at 150 K, to allow for the identification of structural trends. Tables 2 and 3 list crystallographic parameters, and Tables S2 and S3 (see Section S3†) list additional parameters. All geometrical parameters for the structures are listed in Tables S4 and S5 in Section S3.†Fig. 1 illustrates the asymmetric units of representative structures.| Structure abbreviation | D-NaCl 3 | D-NaBr 3 | D-KCl 3 | D-KBr 3 (1) | D-CsI 3 |
|---|---|---|---|---|---|
| Empirical formula | 3n(C6H14N2)2+ | 3n(C6H14N2)2+ | 3n(C6H14N2)2+ | 3n(C6H14N2)2+ | n(C6H14N2)2+ |
| n[Na3Cl9]2− | n[Na3Br9]2− | n[K3Cl9]2− | n[K3Br9]2− | n[CsI3]2− | |
| Formula weight (g mol−1) | 243.53 | 376.88 | 259.64 | 392.99 | 627.80 |
| Crystal system | Trigonal | Trigonal | Trigonal | Trigonal | Hexagonal |
| Space group |
R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c c |
Rc |
Rc |
Rc |
P63/mmc |
| Temperature (K) | 150(2) | 150(2) | 150(2) | 150(2) | 150(2) |
| a (Å) | 15.87470(10) | 16.3508(2) | 16.0388(2) | 16.5223(3) | 10.0382(2) |
| b (Å) | 15.87470(10) | 16.3508(2) | 16.0388(2) | 16.5223(3) | 10.0382(2) |
| c (Å) | 21.1921(2) | 21.9979(2) | 22.2094(4) | 23.0126(6) | 7.9849(2) |
| α (°) | 90 | 90 | 90 | 90 | 90 |
| β (°) | 90 | 90 | 90 | 90 | 90 |
| γ (°) | 120 | 120 | 120 | 120 | 120 |
| Z | 18 | 18 | 18 | 18 | 2 |
| Density calculated (g cm−3) | 1.574 | 2.212 | 1.569 | 2.159 | 2.992 |
| Absorption coefficient (mm−1) | 8.077 | 13.230 | 1.164 | 10.312 | 9.267 |
| F(000) | 2268 | 3240 | 2412 | 3384 | 556 |
| Reflections collected | 29780 |
10907 |
23980 |
15695 |
7018 |
| Unique reflections/Rint | 1061/0.0452 | 1207/ 0.0251 | 1613/0.0162 | 1716/0.0711 | 424/0.0554 |
| Goodness-of-fit F2 | 1.034 | 1.146 | 1.157 | 1.064 | 1.293 |
| Final R indices (R1/wR2) | 0.0226/0.0777 | 0.0159/0.0375 | 0.0159/0.0422 | 0.0198/0.0222 | 0.0233/0.0522 |
| R indices (all data) (R1/wR2) | 0.0230/0.0781 | 0.0160/0.0375 | 0.0164/0.0424 | 0.0495/0.0500 | 0.0244/0.0524 |
| Structure abbreviation | D-KI 3 | D-CsCl 3 | D-CsBr 3 | D-CsICl 2 |
|---|---|---|---|---|
| a Since the twinning involves inversion, no Flack parameter could be determined. | ||||
| Empirical formula | n(C6H14N2)2+ | 4n(C6H14N2)2+ | n(C6H14N2)2+ | n(C6H14N2)2+ |
| n[KI3]2− | n[Cs4Cl12]8− | n[CsBr3]2− | n[CsCl2I]2− | |
| Formula weight (g mol−1) | 533.99 | 1413.81 | 486.80 | 444.90 |
| Crystal system | Trigonal | Monoclinic | Orthorhombic | Trigonal |
| Space group | R32 | C2/c | Pbcn | P3121 |
| Temperature (K) | 150(2) | 150(2) | 150(2) | 150(2) |
| a (Å) | 9.6698(7) | 41.1638(16) | 9.8044(2) | 9.77000(10) |
| b (Å) | 9.6698(7) | 9.48990(10) | 9.8273(3) | 9.77000(10) |
| c (Å) | 12.2899(9) | 32.6647(13) | 27.3662(7) | 11.9653(2) |
| α (°) | 90 | 90.00 | 90 | 90 |
| β (°) | 90 | 131.638(7) | 90 | 90 |
| γ (°) | 120 | 90.00 | 90 | 120 |
| Z | 18 | 8 | 8 | 3 |
| Density calculated (g cm−3) | 2.673 | 1.969 | 2.453 | 2.241 |
| Absorption coefficient (mm−1) | 7.342 | 3.735 | 11.862 | 5.515 |
| F(000) | 726 | 5440 | 1792 | 618 |
| Reflections collected | 4000 | 135520 |
20554 |
31157 |
| Unique reflections/Rint | 608/0.0871 | 13501/ 0.0822 |
3387/ 0.0314 | 2095/0.0611 |
| Flack parameter | N/Aa | — | — | −0.004(13) |
| Goodness-of-fit F2 | 1.296 | 1.027 | 1.052 | 1.037 |
| Final R indices (R1/wR2) | 0.0667/ 0.1542 | 0.0295/ 0.0703 | 0.0344/ 0.0802 | 0.0173/0.0354 |
| R indices (all data) (R1/wR2) | 0.0693/0.1553 | 0.0374/0.0735 | 0.0429/0.0832 | 0.0186/0.0357 |
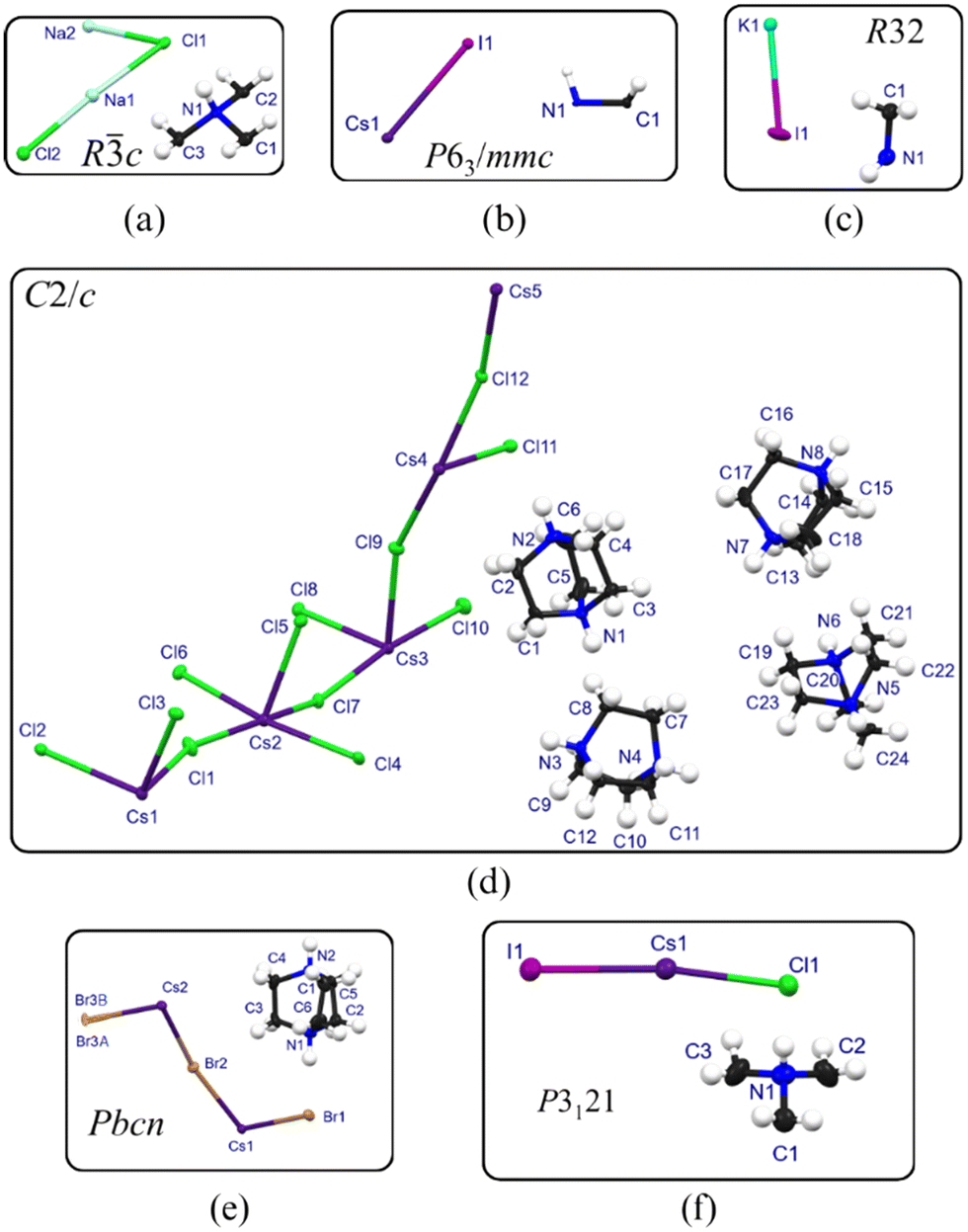 | ||
| Fig. 1 Asymmetric unit of structure (a) D-NaCl3, which is representative of the asymmetric units of D-NaBr3, D-KCl3, and D-KBr3(1), (b) D-CsI3, (c) D-KI3, (d) D-CsCl3, (e) D-CsBr3, and (f) D-CsCl2I. | ||
1D hexagonal perovskite structures
Five structures were found to exhibit the 1D hexagonal perovskite structure, namely D-NaCl3, D-NaBr3, D-KCl3, D-KBr3(1) and D-CsI3, with the first four structures isostructural, and crystallising in the space group Rc, while structure D-CsI3 crystallises in the space group P63/mmc. The crystal structure of D-KCl3 has been reported previously (CSD18 reference code DOTHOK19 (determined at 100 K) and DOTHOK0120 (determined at 120 K)). In this study the structure has been re-determined at 150 K and the compound was found to have the same structure at this temperature as DOTHOK19 and DOTHOK01.20 In addition, the 1D hexagonal perovskite structure of D-RbI3 (HEJHOW) has been reported in the literature,21 crystallising in the space group P![[6 with combining macron]](https://www.rsc.org/images/entities/char_0036_0304.gif) 2c, and will be included for comparison purposes.
2c, and will be included for comparison purposes.
The asymmetric units of the isostructural structures D-NaCl3, D-NaBr3, D-KCl3 and D-KBr3(1) consist of half a dabconium cation moiety and a [B2X2]− anion, as illustrated for structure D-NaCl3 in Fig. 1(a), with the anion containing two crystallographically independent metal ions. The crystal structure of D-NaCl3 will be discussed as an example of the structures in the isostructural series. Two crystallographically independent halide ions bridge the metal ions, with repetition of the inorganic part of the asymmetric unit forming a face-shared, halide-tri-bridged polymer extending along the c-direction, as illustrated in Fig. 2(a). Due to the two metal ions in the asymmetric unit, two different BX6 octahedra are present, one for each crystallographically independent metal ion, Na1 and Na2. In the polymer, the metal ion sequence is such that two Na1 metal ions are followed by one Na2 metal ion. Thus, the metal ion sequence displayed is Na1⋯Na1⋯Na2⋯Na1⋯Na1⋯Na2 etc., as illustrated in Fig. 2(a). This sequence results in different B⋯B and B–X distances in the halide-tri-bridged polymer chain.
 | ||
| Fig. 2 (a) 1D halide-bridged polymer, (b) hydrogen bonding interactions, (c) packing diagram and (d) cation orientation in structure D-NaCl3. Note that structure D-NaCl3 is also representative of the rest of the structures in the isostructural series. (e) 1D halide-bridged polymer, (f) hydrogen bonding interactions, (g) packing diagram and (h) cation orientation in structure D-CsI3. | ||
The B⋯B distances increase with increasing metal- and halide-ion size. Specifically, Na⋯Na distances range between 3.4390(17) Å and 3.7028(13) Å, K⋯K distances fall between 3.6753(6) Å and 3.83 90(6) Å and the one Cs⋯Cs distance is 3.99245(10) Å, with individual distances listed in Table S4 in the Section S3.†
In addition, all the metal ions are co-linear, hence the B–B–B angles are 180° in each case. In structure D-NaCl3, the Cl–Na2–Cl angles have values of 180°, 88.925(10)° and 91.074(10)°, which are closer to a perfect octahedral arrangement (angles of 180° and 90°) than the Cl–Na1–Cl angles which exhibit values of 172.248(13)°, 83.51(2)°, 88.04(2)°, 89.59(7)° and 99.256(7)°. This means that the geometry of the octahedron containing metal ion Na2 is closer to the ideal octahedral geometry than that of the octahedron containing metal ion Na1. X–B–X values for the rest of the compounds in the isostructural series are listed in Table S4.† The ideal B–X–B angle for face-sharing octahedra has been reported as 70.5°, with relatively little structural flexibility possible in face-shared systems, compared to corner- and edge-shared polyhedral structures.27 The B–X–B angles of the structures in the isostructural family are listed in Table S4,† and range from 63.742(6)° to 75.761(15)°.
A three-dimensional hydrogen bonding network is formed in structure D-NaCl3 and the other isostructural structures in the family, made possible by the relative orientation of both the dabconium cation and the polymeric chains, with the dabconium cations optimally positioned to form hydrogen bonds to bridging halide ion acceptors on the 1D polymers. The hydrogen bonds are shown as light blue dotted lines in Fig. 2(b) for structure D-NaCl3, and hydrogen bonding parameters are listed in Table S4 (Section S3†). Each dabconium cation forms two strong, charge assisted N–H+⋯−Cl–Na hydrogen bonding interactions to two different halide-tri-bridged polymers, as shown in Fig. 2(b).
The packing diagram for structure D-NaCl3 is illustrated in Fig. 2(c) and is also representative of the packing of the other members of the isostructural family. Each inorganic polymer is surrounded by six cations. The cations, packing in a row in the c-direction, alternate in orientation, as illustrated in Fig. 2(d), with every third cation displaying the same orientation. The cations adopt the specific orientations to position their N–H+ groups to form strong hydrogen bonding interactions to the bridging halide ions of neighbouring halide-tri-bridged polymers.
The asymmetric unit of structure D-CsI3 is shown in Fig. 1(b) and comprises a H–C–N–H portion of the dabconium cation and a CsI moiety. The N–H bond of the H–C–N–H moiety of the cation lies on a three-fold rotation axis, which, in combination with a two-fold rotation axis, three mirror planes and a six-fold roto-inversion axis, forms the full dabconium cation. Repetition of the inorganic portion results in the formation of a face-shared, halide-tri-bridged polymer, along the c-direction, shown in Fig. 2(e).
Only one metal ion is present in the inorganic polymer, namely atom Cs1, and only one Cs–I bond length of 3.7806(3) Å, one Cs⋯Cs distance of 3.99245(10) Å and one type of octahedron are present, unlike what was observed for the 1D hexagonal perovskite structures in the isostructural series. The three I–Cs–I angles are 180.0°, 94.692(4)° and 85.308(4)°, respectively. These values are closer to the ideal octahedral arrangement than any of those observed for the structures in the isostructural series discussed previously. The Cs–I–Cs angle is equal to 63.742(6)°(13). The D-RbI3 structure21 reported in the literature, is similar to the structure D-CsI3 in terms of its structural features, however they are not isostructural.
In structure D-CsI3 each dabconium cation forms two trifurcated hydrogen bonds to six different I− acceptor ions on three different neighbouring polymers, as illustrated in Fig. 2(f) and in (g). Thus, each dabconium cation forms six strong, charge assisted N–H+⋯−I–Cs hydrogen bonding interactions, all with the same D⋯A length of 3.773(4) Å, to three different halide-tri-bridged polymers. This is made possible by the identical orientation of all the dabconium cations present in the structure (Fig. 2(h)). The same trifurcated hydrogen bonds are also present in structure D-RbI3.21
Structural trends in 1D hexagonal perovskites
The fact that several structures containing the same organic cation but a range of metal and halide ions crystallise as 1D hexagonal perovskite structures allow for the identification of structural trends in this family of compounds. Firstly, the distortion of the octahedron in the 1D polymer will be considered. The distortion in an octahedron (BX6) can be calculated using different parameters, including B–X bond lengths, B–X–B angles or B⋯B distances, and then evaluating their range (spread of the values), calculated using eqn (1).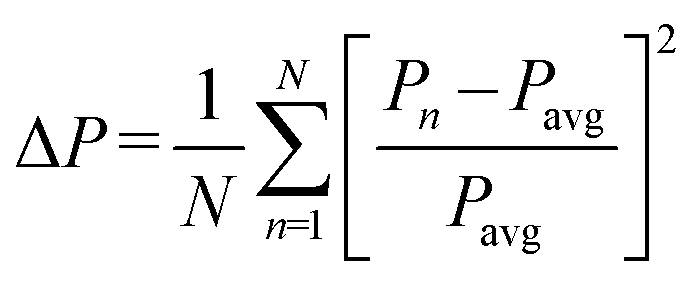 | (1) |
The spread of the structural parameters B–X (Δd), B⋯B (ΔB⋯B) and B–X–B (ΔB–X–B) are listed in Table S7 (Section S4†), with the sum of the ionic radii of the B- and X-constituents of the different 1D hexagonal perovskite structures. In the last two columns of Table S7,† the terms “Equatorial” and “Axial” are used. These terms do not refer to the traditional axial and equatorial positions, instead the term “equatorial angles” refers to angles that are approximately 90° and “axial angles” refers to angles close to 180°, since the assignment of axial and equatorial ligands in the octahedra would be arbitrary. The so-called “Average Spread” parameter is defined as the average value of the five different individual spread values. Considering the data in Table S7,† the following trends can be identified and conclusions drawn.
Two types of octahedra are present in the structures isostructural to D-NaCl3, with the noticeable trend, from consideration of the X–B–X angles of both sets of octahedra, that the octahedra approach the ideal octahedral geometry with an increase in halide ion radius (Cl− to Br−) and increasing metal ion radius (Na+ to K+). This trend can be explained by looking at the B⋯B distances and B–X bond lengths. With an increase in halide ion size or metal ion size, the B⋯B distances and B–X bond lengths increase, allowing for a more symmetrical arrangement of the ions, allowing the octahedra in the 1D face-sharing polymer to tend toward the ideal conformation.
In terms of the B⋯B distances in the structure D-NaCl3 and its isostructural counterparts, the B1⋯B1 distances are shorter than the B1⋯B2 distances, and as the metal ion radius is increased (Na+ to K+), the B⋯B distances start to converge (i.e., the difference between them decreases). This allows for the octahedra of the two different metal ions to become similar in geometry and approach the ideal octahedral geometry.
A similar trend appears in the B–X bond lengths. Three different B–X bond lengths are found in each of the structures in the isostructural series. The B–X bond lengths increase with increasing halide ion radius (from Cl− to Br−) and increasing metal ion radius (Na+ to K+). Also, the difference between the three bond lengths for each structure becomes smaller as halide and metal ion radii are increased.
The values in Table S7† show that with a combination of larger metal and halide ions, i.e. a larger sum of ionic radii, a smaller degree of distortion from the ideal octahedral geometry, or spread in the structural parameters, is observed. This means that the “average spread” listed in column eight is reduced with increasing B+ + X− ionic radii. This agrees with the fact that structure D-CsI3 crystallises in the ideal space group (hexagonal P63/mmc) for 1D hexagonal perovskites since it shows almost no variation in its structural parameters, bar one, and that the calculated average spread is small (1.8 × 10−4).28
Furthermore, considering all the structures crystallising in the space group Rc, the structures D-NaCl3, D-NaBr3, D-KCl3, and D-KBr3(1) show less distortion in their inorganic framework with the combination of larger metal and halide ions. The space group Rc, in which the structures D-NaCl3, D-NaBr3, D-KCl3 and D-KBr3(1) crystallise, is of lower symmetry than space group P2c in which structure D-RbI3 crystallises, which, in turn, is again of lower symmetry than space group P63/mmc, exhibited by structure D-CsI3. Thus, the larger the metal and halide ion (indicated in column 2 of Table S7†), the higher the symmetry of the 1D hexagonal perovskite structure formed. Moreover, the space group P63/mmc is seen as the ideal and, therefore, the most symmetrical space group for 1D hexagonal perovskite structures,28 meaning that structure D-CsI3 represents the ideal 1D hexagonal perovskite structure.
Continuing the comparison between the higher symmetry structures D-RbI3 and D-CsI3, it is evident that only one axial bond angle is present in either structure. This angle has a value of 172.585(19)° in structure D-RbI3 and 180.0° in structure D-CsI3, indicating an ideal geometry in the latter. Additionally, the distortion from the ideal octahedral geometry noted in Table S7† for the equatorial angles is less in the case of the structure D-CsI3 than for the structure D-RbI3. These observations imply that the octahedra in structure D-CsI3 are less distorted and closer to the ideal octahedral arrangement than the octahedra in structure D-RbI3, or in fact, any of the 1D hexagonal perovskite structures reported here. Hence, from this analysis, it may be concluded that the optimal combination of metal and halide with the dabconium cation to form a 1D hexagonal perovskite structure, is Cs+ with I−. This combination leads to the most symmetrical 1D hexagonal perovskite structure possible, since larger ionic radii will prevent the formation of the 1D hexagonal structure.
3D parent perovskite structures
A total of four 3D parent perovskites were structurally characterised at 150 K, namely D-KI3, D-CsCl3,20D-CsBr3 and a mixed halide structure D-CsCl2I. The structure D-CsCl3,20 previously reported in the literature, as determined at 293 K and 120 K respectively, was re-determined at 150 K, for comparison purposes, and the same phase as those reported in the literature was observed at 150 K. In addition, the 3D perovskite structures D-KBr3(2),22D-RbCl3(1)20 and D-RbBr3(1)21 have been reported in the literature, but not investigated in the current study. Their crystallographic parameters and geometric parameters are included in Tables S5 and S6, respectively (see Section S3†). These structures will not be discussed in detail, however, where relevant, they will be included in the structural comparison.The structure of D-KBr3(2)
The compound D-KBr3(2) was only synthesised as a bulk powder in this study, since good quality crystals could not be obtained. It was confirmed by powder X-ray diffraction that the powder pattern calculated from the structure reported in literature matches the experimental powder pattern of the bulk sample prepared in this study. The crystal structure of D-KBr2(2) has been reported in the literature, with CSD18 reference code FIZYIZ.22 FIZYIZ crystallises in the trigonal space group P3121, which is a member of the pair of chiral enantiomorphic space groups P3221 and P3121, with the former containing a left handed three-fold screw axis, and the latter a right handed one.29 The structural features of structure D-KBr3(2) are similar to those of structure D-CsCl2I, which is discussed later.Of specific interest is that structure D-KBr3(2) and the 1D hexagonal perovskite structure D-KBr3(1) discussed previously, are polymorphs.
The 1D polymorph, D-KBr3(1), is formed from crystallisation at room temperature and the 3D polymorph, D-KBr3(2), is formed from crystallisation at a higher temperature of ca. 80 °C, from the same solvent, namely water. Hence, the desired polymorph may be selectively obtained via adjustment of the crystallisation temperature. This suggests that the 1D polymorph is more stable since it is the thermodynamic product. Moreover, both crystallise in a trigonal space group (Rc versus P3121), and with the switch from face-sharing octahedra to corner-sharing octahedra, chirality is introduced in the 3D structure D-KBr3(2).
The structure of D-CsCl3
The structure D-CsCl3 was previously determined at 120 K and reported by Paton et al.,20 with CSD18 reference code GUYNIY.20 The structure determined in the current study, at 150 K, is the same as that reported by Paton et al.,20 and only its main structural features will be highlighted.Structure D-CsCl3 crystallises in the space group C2/c. Of specific interest is large unit cell, with a volume of 9536.4(12) Å3. The asymmetric unit, illustrated in Fig. 1(f), contains five caesium ions, twelve chloride ions, and four dabconium cations, all of which are crystallographically independent. One of the caesium atoms lie on an inversion centre and a mirror plane, while another is located on a mirror plane.
Repetition of the inorganic portion of the asymmetric unit results in a 3D perovskite inorganic framework, with the cations packing in the cavities formed by the inorganic framework, as shown in the packing diagram in Fig. 3(a). Due to the presence of five crystallographically independent caesium ions in the asymmetric unit, five different octahedra comprise the inorganic framework. Four unique inorganic cages are formed in the structure, each containing one of the crystallographically independent cations, as illustrated in Fig. 3(d). Cations pack in a row in the inorganic framework, and neighbouring cations differ in relative orientation, as illustrated in Fig. 3(g).
 | ||
Fig. 3 The packing diagrams of structures (a) D-CsCl3, (b) D-CsBr3, (c) D-CsCl2I. The hydrogen bonding interactions in structures (d) D-CsCl3, (e) D-CsBr3, (f) D-CsCl2I. The relative packing of the dabconium ions down the packing diagrams shown in (a), (b), and (c) of (g) D-CsCl3, (h) D-CsBr3, (i) D-CsCl2I, respectively. (j) The inorganic sheet of structure D-CsCl2I parallel to the (033) Miller plane, viewed perpendicular to the sheet. The same sheet is formed parallel to the (30) and (1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 1) Miller planes. (k) and (l) show the screw axes of the ClCs and ICs substructures found in the chiral structure D-CsCl2I. Note that, for structure D-CsBr3, only the Br atoms with the highest occupancy are shown. 1) Miller planes. (k) and (l) show the screw axes of the ClCs and ICs substructures found in the chiral structure D-CsCl2I. Note that, for structure D-CsBr3, only the Br atoms with the highest occupancy are shown. | ||
Each of the four unique dabconium cations forms two charge assisted hydrogen bonding interactions of the type N–H+⋯−Cl–Cs, thus eight of the twelve chloride ions are involved in hydrogen bonding, as illustrated in Fig. 3(d). The D⋯A values of the hydrogen bonds range from 3.0038(17) Å to 3.0523(16) Å. In each of the four types of cages, the bridging chloride ions accepting hydrogen bonds are displaced towards the N–H+ group of the cation hydrogen bonded to it, resulting in non-linear Cs–Cl–Cs angles, and distortion of the cage and the inorganic framework, with the Cs–Cl–Cs angles ranging from 155.242(14)° to 167.730(16)°.
The structure of D-CsBr3
Structure D-CsBr3 crystallises in the orthorhombic space group Pbcn, and has been reported before, with CSD reference code BEFXOD01.23 The structure D-CsBr3 determined in this study at 150 K corresponds to the structure BEFXOD0123 determined at 193 K, except that all three Br atoms are disordered over two positions each at 150 K. The site occupancy of one of the Br atoms is much higher than that of the other, for all three Br atoms, with the occupancy of the higher of the two being 0.976, 0.979 and 0.971 respectively for atoms Br1, Br2 and Br3. The asymmetric unit of structure D-CsBr3, illustrated in Fig. 1(e), comprises a Br3–Cs2–Br2–Cs1–Br1 moiety and a complete dabconium cation, with atoms Br1, Br2 and Br3 disordered over two positions, as described above. Two crystallographically independent caesium ions are present, with one of the caesium ions lying on a mirror plane, and one on an inversion centre. Repetition of the asymmetric unit results in a 3D parent perovskite structure, with dabconium cations filling cavities formed by the inorganic framework, as illustrated in Fig. 3(b). Two different types of octahedra alternate in the BX-framework along the c-direction, forming sheets of either Cs1Br6 or Cs2Br6 octahedra, parallel to the ab-plane, alternating along the c-direction. The Cs1 octahedra consist of only Cs1, Br1 and Br2 ions, whereas the Cs2 octahedra are comprised of Cs2, Br2 and Br3 ions. From consideration of the X–B–X angles listed in Table S5,† it is evident that the Cs2-octahedra are less distorted than the Cs1-octahedra. Two different CsBr-cages, each containing one crystallographically independent dabconium cation, are present, as illustrated in Fig. 3(e). Each cation forms two strong, charge assisted N–H+⋯−Br–Cs hydrogen bonds, however, only the Br atoms with highest occupancy will be considered as hydrogen bonding acceptors. D⋯A distances of 3.222(4) Å (to atom Br1A) and 3.139(4) Å (to atom Br3A) are observed. The Cs–Br–Cs angles of bromide ions involving N–H+⋯−Br1A–Cs1 hydrogen bonds deviate more from linearity than those which participate in N–H+⋯−Br3A–Cs2 hydrogen bonds.The packing of the dabconium cations in structure D-CsBr3, viewed down the c-axis, is illustrated in Fig. 3(h). Along the c-direction, adjacent cations show different orientations, with every fourth cation exhibiting the same orientation.
The structure of D-CsCl2I
Structure D-CsCl2I is a mixed halide structure which has not been reported previously, with the ratio of chloride to iodide ions equal to 2:1. The compound crystallises in the chiral, trigonal space group P3121, which contains a right-handed, three-fold screw axis as symmetry element. The structures D-KBr3(2),22D-RbCl3(1)20 and D-RbBr3(1),21 crystallising in the enantiomorphic space groups P3121 or P3221, are related to structure D-CsCl2I, but not isostructural to it. The asymmetric unit of structure D-CsCl2I consists of half a dabconium cation (an NH(CH2)(CH2)(CH2) moiety) and a Cl–Cs–I moiety, as shown in Fig. 1(f). A two-fold rotation axis generates the rest of the cation, and the chloride ion is located on a two-fold screw axis, while the caesium and iodide ions lie on a two-fold rotation axis. A 3D perovskite inorganic framework, comprised of Cs+-, Cl−- and I−-ions, is formed, with the ratio of Cl−- to I−-ions equal to 2:1. The dabconium cations pack in cavities created by the inorganic framework, as can be seen in Fig. 3(c) and (f). Three inorganic sheets comprised of Cs+-, Cl−- and I−-ions combine to form the inorganic framework. The sheets are all exactly the same, and parallel to the (30), (033) and (11) Miller planes. Fig. 3(j) illustrates the sheet, with three of these sheets packing approximately perpendicular to each other.
The chirality of the structure is evident in both the packing of the cations and the inorganic framework. The cations pack to form a right-handed helix around the three-fold screw axis, illustrated in Fig. 3(i), and portions of the inorganic framework forms right-handed helices, with one type of helix comprised of Cs+-ions and Cl−-ions and the other of Cs+-ions and I−-ions, as illustrated in Fig. 3(k) and (l), respectively.
The crystal structure of D-CsCl2I can be viewed as the repeat of dabconium cations occupying the body centre position of an inorganic cage consisting of Cs+-, Cl−- and I−-ions. The orientation of the cations in neighbouring cages differ, but all cages are the same. Each cage comprises four Cs+-ions, one on each corner, eight bridging chloride ions and four bridging iodide ions. The cation forms two strong, charge assisted N–H+⋯−Cl–Cs hydrogen bonds to two different chloride ligands, diagonally across the cage, which can be seen in Fig. 3(f). Only chloride ions act as hydrogen bond acceptors, with no iodide ions involved in hydrogen bonding. This can be ascribed to the higher electronegativity of the chloride ion compared to the iodide ion, which means that N–H+⋯−Cl–Cs hydrogen bonds are stronger than N–H+⋯−I–Cs hydrogen bonds.
The structure of D-KI3
The chiral structure D-KI3 crystallises in the Sohncke space group R32 and has not been reported previously. The asymmetric unit of structure D-KI3 consists of a H2C–NH organic moiety and an inorganic K–I moiety, as shown in Fig. 1(c). The nitrogen atom and the hydrogen atom bonded to it lie on three-fold rotation axis, which generates the full dabconium cation.In addition, the potassium ion lies on both a two-fold and a three-fold rotation axis. Repetition of the inorganic portion results in a 3D perovskite inorganic framework containing one type of octahedron, with the cations packing in cavities in the inorganic framework, as illustrated in Fig. 4(a). Each dabconium cation is anchored to the inorganic cage via hydrogen bonding interactions, as illustrated in Fig. 4(b), with each cation forming two trifurcated, charge assisted hydrogen bonds of the type N–H+⋯−I–K. The length of these hydrogen bonds exceed that of the definition of classic hydrogen bonds of this type, however, these interactions definitely provide cohesion in the structure. The inorganic framework contains right-handed helices, as illustrated in Fig. 4(c). The cations pack in rows in the organic framework, with all the cations in a row displaying the same orientation, as illustrated in Fig. 4(d). The octahedra in the inorganic framework are slightly distorted, with I–K–I angles of 85.255(19)°, 88.733(2)°, 98.03(2)° and 170.83(3)° and a K–I–K angle of 170.83(3)°.
 | ||
| Fig. 4 (a) Packing diagram of structure D-KI3, viewed down an arbitrary axis. (b) Packing of dabconium cation in inorganic framework and hydrogen bonding interactions. (c) Metal halide helix formed in structure D-KI3. (d) Packing of dabconium cations along the b-direction. | ||
Comparison of 3D structures
The 3D perovskite structures obtained in this study crystallise in a range of phases and space groups, making direct structural comparison difficult. However, the inorganic frameworks of the 3D structures will be compared using the same spread of values, calculated using eqn (1), as for the 1D hexagonal perovskites. The results are summarised in Table S8 in Section S4.† The literature structures D-RbCl3(1),20D-RbBr3(1)21 and D-KBr3(2)22 are also included in this comparison, though determined at different temperatures than the rest of the structures, namely at 120 K, 293 K and 300 K, respectively.The values in Table S8† show an interesting overall trend: all the structures, though not comparable in a direct sense, have a similar average spread of values, all of the magnitude of approximately 10−3. Furthermore, the spread in their parameters does not differ significantly. The structures D-RbBr3(1) and D-CsCl3 have the same summed ionic radii (348 pm) and average spread. However, the range of the sum of the ionic radii is not extensive, with a minimum sum of 322 pm, and a maximum sum of 363 pm. Therefore, since the cation is constant in these structures, and since there is not much difference in the B+ and X− ionic radii, it makes sense that the distortion in these structures should be similar.
Of interest is the fact that a number of 3D perovskite structures, namely structures D-KI3 and D-CsCl2I and literature structures D-RbCl3(1),20D-RbBr3(1)21 and D-KBr3(2)22 are chiral, even though the components comprising the structures are not chiral.
Comparison of 1D and 3D perovskite structures
Comparison of the 1D hexagonal perovskite structures and the 3D parent perovskite structures allows for the identification of structural trends. Note that compound D-KBr3 has two polymorphs, a 1D hexagonal perovskite polymorph, D-KBr3(1) (obtained in this study) and a 3D perovskite polymorph D-KBr3(2) (reported previously22), which may be selectively formed by either allowing crystallisation at room temperature or heating the reaction mixture, respectively.In Fig. 5 the different structures are presented as a function of the sum of the B+ and X− ionic radii, which range from 283 pm to 387 pm. In the figure, the colour red indicates 1D hexagonal perovskite structures, while dark blue indicates 3D perovskite structures. The figure illustrates the change in structural dimensionality as a function of the sum of the ionic radii of the B+ and X− ions. When considering this figure, the structures D-KBr3(1) and KBr3(2) represent a tipping point, with a summed B+ and X− ionic radii of 334 pm, since both a 3D parent perovskite structure and a 1D hexagonal perovskite structure can be formed at this point.
 | ||
| Fig. 5 Schematic illustration of the evolution of the perovskite structure dimensionality between 1D and 3D for dabconium containing structures. The colours in the arrow change as a gradient from red (indicating 1D ABX3-type structures) to dark blue (indicating 3D perovskite structures). | ||
The structure D-KBr3(2) represents the 3D parent perovskite structure, thus containing only corner sharing octahedra, with the lowest combined summed radii suggesting a tipping point in the relation between dimensionality and the sum of the ionic radii at 334 pm since both the 1D and 3D structural dimensionalities are possible. It is possible that the combination of dabconium, Na+ and I−, which has a sum of 322 pm would give further insight into the relationship between these parameters.
Furthermore, structure D-CsBr3 is the 3D perovskite with the highest summed ionic radii of 363 pm, indicating a second tipping point. For larger values of the sum of ionic radii, compounds D-RbI3 (sum of radii equal to 372 pm) and D-CsI3 (sum of radii 387 pm) revert back to the 1D hexagonal perovskite structure.
Below the value of 334 pm for the sum of ionic radii, only 1D hexagonal perovskite structures exhibiting face sharing octahedra are formed. Thereafter, the tipping-point (334 pm) is reached with D-KBr3 which exhibits both a 1D hexagonal perovskite and a 3D perovskite structure, depending on the reaction conditions. When the sum of radii range from 334 pm to 363 pm, several 3D perovskite phases are obtained, but at a sum of radii value of 373 pm, a 1D hexagonal perovskite is again formed. These identified trends allow for crystal engineering since a direct correlation may be drawn between the summed ionic radii of the B+- and X−-ions and the resultant structure dimensionality. Hence, any combination of B+- and X−-ions for which the summed B+ and X− ionic radii lie between approximately 330 pm and 365 pm may be combined with dabconium cations to form a 3D perovskite structure.
Structure D-CsCl2I is a prime example of crystal engineering. It has a summed ionic radii of 361 pm, lying in the range expected to deliver a pure corner-sharing 3D parent perovskite, while D-CsCl3 is a 3D parent perovskite and D-CsI3 is a 1D hexagonal perovskite. The identification of these trends now allows for different mixed halide, mixed metal or mixed halide and metal structures to be predicted for this family of compounds.
Success of tolerance factor predictions
The ability of different tolerance factors to predict the structural dimensionality of the structures of interest was assessed, based on the experimental structural results obtained in this study, and the results are summarised in Section S5.†Only the Goldschmidt tolerance factor6 had a significant degree of success, managing to predict seven out of the nine structure dimensionalities correctly. However, the octahedral factor7 and Bartel's tolerance factor8 had less success, only predicting four and five out of nine dimensionalities correctly, respectively.
Band gap measurements
The optical band gaps of the all the compounds synthesised in this study were measured via diffuse reflectance spectroscopy (DRS), and the DRS spectra were interpreted according to the Kubelka–Munk theory (see Section S6† for a detailed description). The processed spectra are shown in Fig. 6(a) and (b), with the original spectra included in Section S6.† The experimental band gap values are listed in Table 4. The DRS technique possesses an accuracy of ±0.05 eV, and hence the values reported here need to be regarded with this error margin in mind.30 A correlation (R2) factor of 0.98 or higher was used as a guideline to determine the linear range from which the band gap was determined.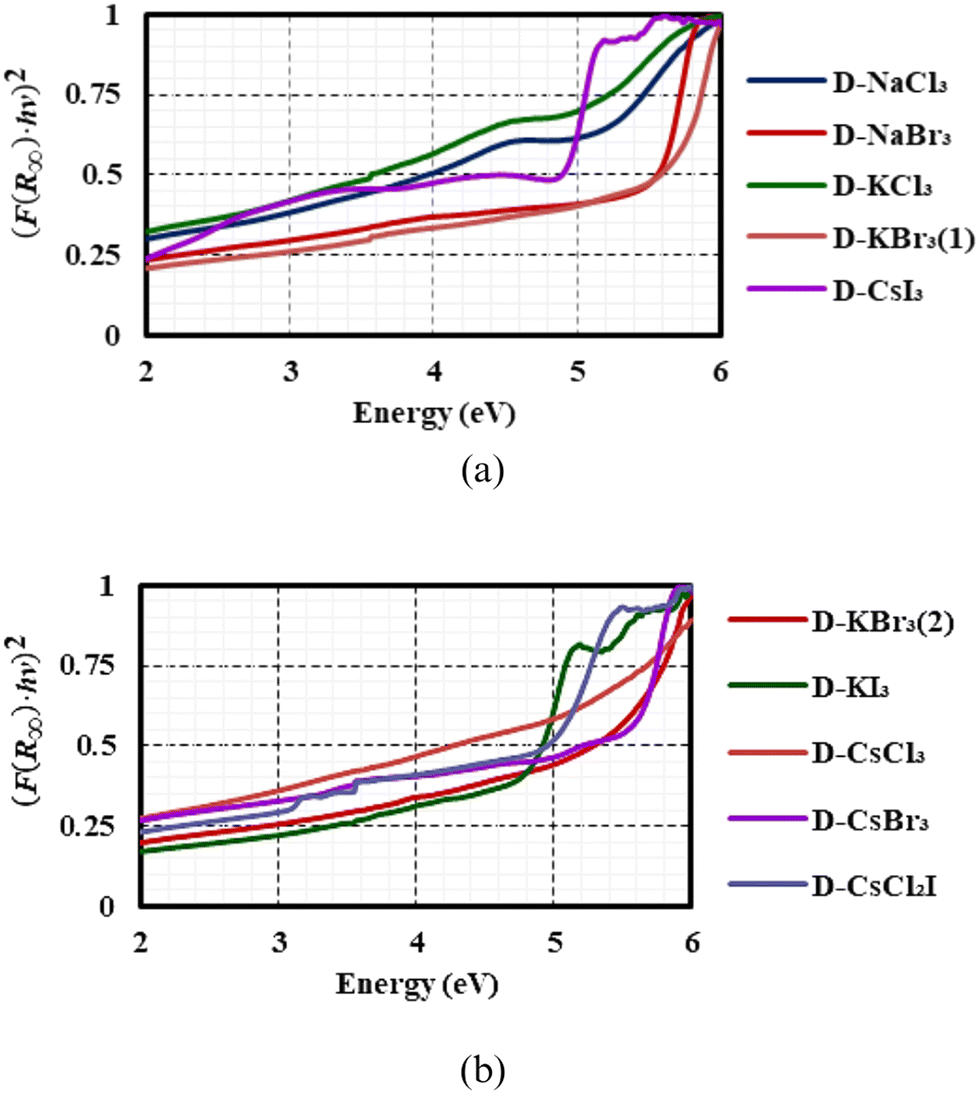 | ||
| Fig. 6 Processed and normalised DRS spectra for (a) the 1D hexagonal perovskite materials (b) the 3D perovskite materials. | ||
| Material | Band gap (eV) | Perovskite structure type |
|---|---|---|
| D-NaCl 3 | 3.99 | 1D hexagonal |
| D-NaBr 3 | 5.33 | 1D hexagonal |
| D-KCl 3 | 3.42 | 1D hexagonal |
| D-KBr 3 (1) | 5.30 | 1D hexagonal |
| D-KBr 3 (2) | 5.00 | 3D parent |
| D-KI 3 | 4.50 | 3D parent |
| D-CsCl 3 | 5.10 | 3D parent |
| D-CsBr 3 | 5.26 | 3D parent |
| D-CsI 3 | 4.63 | 1D hexagonal |
| D-CsCl 2 I | 4.54 | 3D parent |
The band gaps of the dabconium-containing perovskites with alkali metal halide anions lie in the range of 3.42 eV to 5.33 eV. Trend identification from the values in Table 4 is made difficult due to the compounds exhibiting different structural dimensionalities and crystallising in different space groups. That said, a systematic approach of comparing halide and metal changes within a series of D-BX3 is possible.
In the isostructural 1D hexagonal perovskite series, D-NaCl3, D-NaBr3, D-KCl319,20 and D-KBr3(1) a direct comparison of their band gaps is possible. The band gap values for D-KX3 structures are generally lower than those of the D-NaX3 structures, for X = Cl−, Br−, even though only slightly in the Br− case. Also, upon substituting Cl− for Br− in D-MX3, with B = Na+ or K+, an increase in the band gap is seen for the 1D hexagonal perovskites. Hence, the optimal, and indeed the most optimal combination is that of D-KCl3 which has a band gap of 3.42 eV, the smallest of all the materials studied.
Continuing with the comparison of the 1D structures, in comparing D-NaX3, D-KX3 and D-CsI3, a direct comparison is challenging because they are not isostructural, however, because of their similar dimensionality some insight may be gathered. D-CsI3 has a band gap of 4.63 eV which falls between that of the others. Its band gap is higher than that of D-NaCl3 or D-KCl3, but lower than that of D-NaBr3 or D-KBr3. It is difficult to ascribe this observation to one effect since two parameters are changing, namely the halide anion and the metal cation. More insight may be gathered when looking at the other materials that contain caesium, however, to do so, one must first understand the inherent dimensionality–band-gap-relationship.
The two polymorphs of D-KBr3 allows for such a comparison. Since two different polymorphs of D-KBr3 were structurally characterised, with D-KBr3(1) exhibiting a 1D hexagonal perovskite structure, and D-KBr3(2)22 a 3D parent perovskite structure, it is the only material that allows for the direct study of the effect of dimensionality of the structure on the band gap. Because the ions comprising the compounds are the same, any difference in band gap should arise purely due to structural differences. The 3D material has a narrower band gap of 5.00 eV compared to the band gap of 5.30 eV of the 1D material. This narrowing of the band gap in the 3D case may be attributed to the presence of face-sharing octahedra in the 1D structure versus corner-sharing octahedra in the 3D structure, resulting in better orbital overlap of B+ and X− ions in the 3D structure.
That said, the effect of the halide ion on the band gap in the case of the 3D parent perovskites must also be considered. When comparing compounds D-KBr3(2)22 and D-KI3, the bromide analogue has a wider band gap (5.30 eV) than the iodide analogue (5.00 eV). In addition, compounds D-CsCl320 and D-CsBr3 may be compared, with the chloride analogue having a wider band gap (>5.30 eV) than the bromide analogue (5.26 eV). Thus, in the 3D case, the chloride analogues have larger band gaps than the bromide-containing compounds, which, in turn have larger band gaps than the iodide analogues.
In addition, the band gap of the material D-CsCl2I is narrower than that of the other Cs-containing materials D-CsCl3, D-CsBr3 or D-CsI3, correlating with all the observations thus far, which are that 3D perovskites of the same constituents have narrower band gaps than that of the 1D hexagonal perovskites (D-KBr3(1)versusD-KBr3(2)), and that an iodide containing perovskite should have a narrower band gap that the corresponding bromide or chloride analogues. This further implies that within a specific combination of dabconium and B+, it should be possible to tune the band gap utilising a combination of iodide and bromide as the anion.
Even though the band gap values of the dabconium-containing alkali halide perovskites are significantly wider than the ideal band gap required for sensitiser materials in PSC devices, which is equal to 1.1 eV, as suggested by the Shockley–Queisser Theory,31 they may be suitable for other applications. Perovskite materials may be used in a range of applications as reported in the literature.32
The wide band gaps of the materials indicate that they are either semi-conductors or insulators. Semi-conductor materials have potential applications in various optoelectronics (LEDs, photovoltaics etc.), and insulators mostly as insulating materials in electronics.33–35 Moreover, the D-CsBr3 material has been proposed for use as a flexible X-ray detector by Cui et al.23
Conclusions
All the compounds prepared by the combination of dabconium cations and alkali metal halides in this study formed perovskite structures of the formula ABX3, with both 1D and 3D structural dimensionalities exhibited. It was found that the type of structure formed could be correlated with the value of the sum of the ionic radii of the B+ and X− ions, where values ranging from 283 pm to 334 pm result in 1D hexagonal perovskite structures. The two polymorphs of D-KBr3 (1D hexagonal and 3D) are formed for a sum of radii of 334 pm, which represent a tipping point, where either a 1D or a 3D structure can form, depending on crystallisation conditions. In the range of 334 pm to 363 pm, 3D perovskite structures are formed, and when the sum of radii falls in the range of 372 pm to 387 pm, a 1D hexagonal perovskite structure is again formed.All the compounds studied possess wide band gaps (>3.0 eV), placing most of them in the category of semi-conductors (<5.0 eV). The band gap values suggest that a 3D structure results in a narrower band gap than a 1D structure. With increasing halide ionic radius, a narrower band gap is obtained in the 3D case, and the opposite is true for the 1D case.
The structural trends identified in this study allows for structural prediction of members of this family based on the sum of the ionic radii of the B+ and X− ions, thereby allowing for design of materials with a specific, desired structural dimensionality, namely 1D or 3D.
Data availability
All crystallographic data reported in the paper entitled “Structures and Band Gaps of Lead-Free Dabconium-Containing Hybrid Alkali-Metal Halide Perovskites” may be sourced using the following information: CCDC 2381380–2381383, 2381385–2381387, 2381389 and 2381390 contain the crystallographic information for the structures reported in this paper.Author contributions
The author contributions were as follows H. J. van der Poll: conceptualisation, data curation, formal analysis, investigation, methodology, project administration, visualisation, writing – original draft, writing – review & editing. R. Erasmus: data curation, resources, validation, writing – review & editing. M. Rademeyer: conceptualisation, formal analysis, funding acquisition, resources, supervision, validation, writing – original draft, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Dr. F. Malan for assistance with crystallographic aspects. MR acknowledges financial support from the National Research Foundation (Grant No.: SRUG210427597644).Notes and references
- G. Kieslich, S. Sun and A. K. Cheetham, Chem. Sci., 2014, 5, 4712–4715 RSC.
- H. Megaw, Nature, 1945, 155, 484–485 CAS.
- C. Møller, Nature, 1957, 180, 981–982 CrossRef.
- C. C. Stoumpos, L. Mao, C. D. Malliakas and M. G. Kanatzidis, Inorg. Chem., 2017, 56, 56–73 CAS.
- L. Katz and R. Ward, Inorg. Chem., 1964, 3, 205–211 CrossRef CAS.
- V. M. Goldschmidt, Naturwissenschaften, 1926, 14, 477–485 CrossRef CAS.
- X. L. C. Li, L. F. W. Ding and Z. G. Y. Gao, Acta Crystallogr., Sect. B:Struct. Sci., 2008, 64, 702–707 CrossRef PubMed.
- C. J. Bartel, C. Sutton, B. R. Goldsmith, R. Ouyang, C. B. Musgrave, L. M. Ghiringhelli and M. Scheffler, Sci. Adv., 2018, 2, 1–10 Search PubMed.
- D. Weber, Z. Naturforsch., B, 1978, 33, 1443–1445 CrossRef.
- N. Ikeda, K. Teshima and T. Miyasaka, Chem. Commun., 2006, 1733–1735 RSC.
- A. Kojima, ECS Meeting Abstracts, 2008, vol. MA2008-02, p. 27.
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed.
- A. K. Jena, A. Kulkarni and T. Miyasaka, Chem. Rev., 2019, 119, 3036–3103 CrossRef CAS PubMed.
- Z. Xiao, Z. Song and Y. Yan, Adv. Mater., 2019, 31, 1–22 Search PubMed.
- L. Gollino and T. Pauporté, Sol. Energy, 2021, 5, 2000616 CAS.
- W. Ke, C. C. Stoumpos and M. G. Kanatzidis, Adv. Mater., 2019, 31, 1803230 CrossRef CAS PubMed.
- J. M. Kadro and A. Hagfeldt, Joule, 2017, 1, 29–46 CrossRef.
- C. R. Groom and F. H. Allen, Angew. Chem., Int. Ed., 2014, 53, 662–671 CAS.
- F. Thétiot, I. Sasaki, C. Duhayon and J. P. Sutter, J. Chem. Crystallogr., 2009, 39, 225–227 Search PubMed.
- L. A. Paton and W. T. A. Harrison, Angew. Chem., Int. Ed., 2010, 49, 7684–7687 CAS.
- W. Y. Zhang, Y. Y. Tang, P. F. Li, P. P. Shi, W. Q. Liao, D. W. Fu, H. Y. Ye, Y. Zhang and R. G. Xiong, J. Am. Chem. Soc., 2017, 139, 10897–10902 CAS.
- H. Zhang, CSD Private Communication, 2019.
- Q. Cui, N. Bu, X. Liu, H. Li, Z. Xu, X. Song, K. Zhao and S. F. Liu, Nano Lett., 2022, 22, 5973–5981 CAS.
- C. Shi, H. Yu, Q. Wang, L. Ye, Z. Gong, J. Ma, J. Jiang, M. Hua, C. Shuai, Y. Zhang and H. Ye, Am. Ethnol., 2020, 132, 173–177 Search PubMed.
- R. D. Shannon, Acta Crystallogr., Sect. A, 1976, 32, 751–767 Search PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. C:Struct. Chem., 2015, 71, 3–8 Search PubMed.
- U. Muller, in Inorganic Structural Chemistry, John Wiley & Sons Ltd., 2006, pp. 166–180 Search PubMed.
- R. Søndenå, S. Stølen, P. Ravindran, T. Grande and N. L. Allan, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 184105 Search PubMed.
- Y. Tanaka, T. Kojima, Y. Takata, A. Chainani, S. W. Lovesey, K. S. Knight, T. Takeuchi, M. Oura, Y. Senba, H. Ohashi and S. Shin, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 144104 Search PubMed.
- M. Nowak, B. Kauch and P. Szperlich, Rev. Sci. Instrum., 2009, 80, 46107 CAS.
- W. Shockley and H. J. Queisser, J. Appl. Phys., 1961, 32, 510–519 CAS.
- J. Li, J. Duan, X. Yang, Y. Duan, P. Yang and Q. Tang, Nano Energy, 2021, 80, 105526 CAS.
- S. Fujita, Jpn. J. Appl. Phys., 2015, 54, 30101 CrossRef CAS.
- S. L. Shinde, S. Senapati and K. K. Nanda, Adv. Nat. Sci.:Nanosci. Nanotechnol., 2014, 6, 15002 Search PubMed.
- A. H. M. Smets, K. Jäger, O. Isabella, R. A. Swaaij and M. Zeman, Solar Energy: The physics and engineering of photovoltaic conversion, technologies and systems, UIT Cambridge, 1st edn, 2015 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: CCDC 2381380–2381383, 2381385–2381387, 2381389, 2381390. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ce00907j |
| This journal is © The Royal Society of Chemistry 2025 |