
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Different thermal and solubility properties with an identical crystal structure – case of lenalidomide hydrate†
Qi
Zhang
 a,
Yitong
Zhu
a,
Yisheng
Xu
a,
Huaiyu
Yang
*b,
Wei
Li
b and
Xiangyang
Zhang
*a
a,
Yitong
Zhu
a,
Yisheng
Xu
a,
Huaiyu
Yang
*b,
Wei
Li
b and
Xiangyang
Zhang
*a
aState Key Laboratory of Chemical Engineering, East China University of Science and Technology, Shanghai, 200237, China. E-mail: zxydcom@ecust.edu.cn
bDepartment of Chemical Engineering, Loughborough University, Loughborough, LE11 3TU, UK. E-mail: h.yang3@lboro.ac.uk
First published on 26th March 2025
Abstract
Identification of the polymorphism of a medicine is critical for its pharmaceutical properties. Different thermal properties and solubilities indicate different polymorphs. However, in the case of lenalidomide (LDM) hydrate, crystals with different thermal and solubility properties were determined to have an identical crystal structure. LDM, a drug for multiple myeloma and myelodysplastic syndrome, was found to form two dihydrate crystals. The stick-shaped crystals, E1, exhibit a two-step dehydration process, while the rhombic prism-shaped crystals, E2, show a one-step rapid dehydration process. E1 and E2 also showed different dissolution rates. However, powder XRD patterns of E1 and E2 were identical, and their crystal structures were confirmed as identical by single-crystal XRD. Further, investigation into the arrangement of water molecules showed that E1 and E2 contained features of both channel and isolated site hydrates. Water channels parallel to the c-axis were discovered in the hydrated structure, and two dehydration mechanisms were proposed based on the grinding experiments and lattice orientation analysis. E1 and E2 were mainly dehydrated along and perpendicular to the water channels, respectively. In addition, dehydration kinetics and the solid-phase transformation of E1 and E2 were determined.
1. Introduction
The dissolution of active pharmaceutical ingredients (APIs) is a crucial factor in their delivery to the active site through absorption via the gut and circulation in the plasma.1 Hydrates are essential solid forms for improving the dissolution properties of APIs with poor water solubility.2,3 Hydrate formation refers to the interaction of water molecules within the crystal lattice of a drug molecule,4 which can be classified as an isolated site, channel, or ion-associated hydrates depending on the arrangement of water molecules.5,6 It is estimated that almost one-third of APIs in solid-state pharmaceuticals can form hydrates.7,8 These hydrates are influenced by factors such as drug molecule characteristics, solvent type, crystallization method, and solution environment,9 affecting their stability, thermal properties, dissolution properties, quantity of hydrated water molecules, crystal habit, and polymorphism.10–12Thermal dehydration of hydrate crystals is considered a solid-phase reaction,13,14 and studying it provides insight into hydrates. Hydrates can be characterized by powder X-ray diffraction (PXRD) and single-crystal X-ray diffraction (SCXRD) to determine the structural information and by thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) to assess the thermal properties such as melting, dehydration, and transformation.15 For different types of hydrates, the dehydration mechanism has mainly been determined through a combination of structural analysis and computational simulations.16,17 Kang et al. investigated the effect of particle size on hydrates, where narrow and staggered water dehydration channels in the crystal lattice were related to the dehydration process. The dehydration mechanism was confirmed through crystal orientation analysis and slip plane calculations.18 The dehydration kinetic model and dehydration activation energy are also critical in exploring the dehydration mechanism,19,20 and the Flynn–Wall–Ozawa (FWO) method is a model-free method to provide detailed information on different stages of the dehydration process.21,22 Additionally, density functional theory (DFT) and molecular dynamics (MD) simulations have been employed as emerging methods to provide insight into the dehydration properties of hydrates.23–25
Lenalidomide (LDM, CAS No. 191732-72-6), a medicine to treat multiple myeloma and myelodysplastic syndrome,26,27 has poor water solubility with commercial application in the hemihydrate of form B.28 LDM is a typical polymorphic drug system, including nearly 20 forms of LDM and LDM solvates, such as form I,28,29 forms II and III,30 forms A–H,31 and forms 1–7,32 with form 1 as a thermodynamically stable polymorph and form 4 as a metastable polymorph. In addition, two new dihydrates of LDM have been reported.32,33 Some investigations have reported the physicochemical properties and nucleation behaviors of LDM polymorphs.34–36 However, the dehydration mechanism and dissolution properties of LDM hydrates have not been studied.
In this work, LDM dihydrate crystals E1 and E2 have two different shapes. TGA and DSC curves show that the dehydration processes and thermal properties of crystals E1 and E2 are different, and they have different solubilities and dissolution rates. However, the powder XRD of crystals E1 and E2 are almost the same, with almost identical crystal structures determined by SCXRD. The mechanisms of the different dehydration processes were investigated, and the hydrated structures and dehydration models of E1 and E2 are discussed.
2. Materials and methods
2.1 Materials
Methanol (99.8 wt%) and phosphoric acid (99.7 wt%) were purchased from Shanghai Titan Technology Co., Ltd. LDM (99.5 wt%) was provided by Zhejiang Hisun Pharmaceutical Co., Ltd. Ultrapure water was prepared in the laboratory using Milli-Q IQ 7000.2.2 Preparation of LDM E1 and E2
Excess LDM raw material was added to a 10 v% methanol–water mixed solvent and stirred at 25 °C and 300 rpm for 24 h to obtain bulk crystals of E1. A saturated solution of LDM in water at 60 °C was prepared, slowly cooled to room temperature (about 25 °C), and left to obtain single crystals of E1.Bulk crystals of E2 were obtained by adding LDM raw material into a 1 v% phosphoric acid–water mixed solvent to get a saturated solution at 40 °C, then rapidly cooled to 0 °C at 300 rpm for 2 h. The homogeneous saturated solution at 40 °C was slowly cooled to 0 °C and aged to obtain single crystals of E2.
2.3 Characterization methods
The morphologies of the LDM hydrates were analyzed by scanning electron microscopy (SEM, FEI Nova NanoSEM 450). All samples were sputter-coated with a 2 nm Pt/Pd and imaged at an accelerating voltage of 3 kV with an ETD detector at a working distance of 5 mm. Microscope control and data acquisition were conducted using software from FEI.The thermal properties of the LDM hydrates were measured by differential scanning calorimetry (DSC, TA Q2000) and thermogravimetric analysis (TGA, TA Q500). For DSC, 3–5 mg samples were placed in a crucible with a small hole and then heated from 30 °C to 300 °C at 10 K min−1 with a nitrogen flow rate of 50 mL min−1. For TGA, 5–10 mg samples were placed in a crucible with a nitrogen flow rate of 50 mL min−1, and the operating temperature range was set between 30 °C to 350 °C at 10 K min−1 for hydrate characterization or 2, 5, 10, and 15 K min−1 for dehydration kinetics. Different batches of hydrate samples were measured for thermal properties to ensure objectivity.
The spectroscopic properties of the LDM hydrates were characterized by powder X-ray diffraction (PXRD) with a Bruker D8 Advance. Samples were placed in a diffractometer with Cu Kα radiation which collected the patterns in the range of 5–50° at a scanning rate of 10° min−1.
The structural details of LDM forms E1 and E2 were determined by single-crystal X-ray diffraction (SCXRD, Bruker D8 Quest). Single crystals were placed in a diffractometer with Cu Kα radiation at room temperature. Diffraction data were integrated and reduced by APEX3 software. The crystal structures were solved by the direct methods on Olex2 software37 and subsequently refined by F2 full-matrix least-squares with the SHELXL-2018/3 program. The crystallographic data in CIF format of forms E1 and E2 have been deposited in the Cambridge Crystallographic Data Center (CCDC) as No. 2289868 and No. 2383285, respectively.
2.4 Dissolution measurement
An Agilent Technologies 708-DS dissolution tester was used to perform powder dissolution tests for LDM crystals B, E1, and E2. LDM crystals were sieved through 100 mesh sieves to minimize crystal size effects on dissolution outcomes. The amount of each LDM hydrate was higher than the solubility to ensure non-sink conditions resulting in the presence of a solid phase. The samples were suspended in 100 mL of deionized water and stirred at 100 rpm at 37 ± 0.5 °C.0.5 mL samples were withdrawn at predetermined intervals and immediately filtered using a 0.22 μm syringe filter for subsequent high-performance liquid chromatography (HPLC) determination. The concentration of dissolved API was determined by HPLC (Shimadzu LC-20AT, Japan) with a UV detector at 220 nm. A Diamonsil C18 column was used at 40 °C. The mobile phase consisted of a mixture of acetonitrile and 0.05% phosphate buffer (15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :85, v/v) with a flow rate of 1.0 mL min−1. An injection volume of 10 μL was used for analysis. The recovered samples were also characterized using PXRD and DSC. All the experiments were conducted in triplicate to eliminate serendipity.
:85, v/v) with a flow rate of 1.0 mL min−1. An injection volume of 10 μL was used for analysis. The recovered samples were also characterized using PXRD and DSC. All the experiments were conducted in triplicate to eliminate serendipity.
3. Results and analyses
3.1 Different crystal shapes, thermal and solubility properties
Two different shapes of LDM bulk crystals were obtained following the method of section 2.2. Crystals E1 have a stick shape, and crystals E2 have a rhombic prism shape, as shown in Fig. 1a. The particle size distributions were measured to be 80–140 μm for crystals E1 and 40–60 μm for crystals E2, showing that crystals E1 have a larger average crystal size than crystals E2. | ||
| Fig. 1 (a) SEM images, (b) powder dissolution profiles at 37 °C, and (c) TGA and DSC curves of LDM bulk crystals E1 and E2. | ||
The crystals E1 have higher solubility at 60 min than the crystals E2, as shown in Fig. 1b. Solutions with crystals E1 reach saturation around 60 min, reaching concentrations of 0.550 mg mL−1. Solutions containing crystals E2 reach a maximum concentration of 0.600 mg mL−1 around 60 min. PXRD and DSC at 60 min confirm that there is no polymorphic transformation for crystals E1 and E2 (detailed in Fig. S1†). The concentration of the solution with crystals E2 gradually decreases to 0.553 mg mL−1 at 120 min, approaching a similar solubility as the crystals E1. Both crystals E1 and E2 have faster dissolution rates and larger solubilities than the crystals of commercial hemihydrate form B during a 0–120 min period.
The TGA and DSC curves of the crystals E1 and E2 exhibit different thermal behaviors, as shown in Fig. 1c. In the TGA curves, the crystals E1 have a slow two-step dehydration process, showing weight losses of 9.2% and 2.7% corresponding to the removal of 1.5 H2O molecules (fast dehydration rate) and 0.5 H2O molecules (slow dehydration rate), respectively. The crystals E2 have a one-step process with a faster overall dehydration rate with a weight loss of 11.9%, but slower than the 1.5 H2O molecule dehydration rate and faster than the 0.5 H2O molecule dehydration rate in crystals E1. The TGA analysis confirms that the crystals E1 and E2 are both LDM dihydrates. The DSC curves present that crystals E1 and E2 have identical melting peaks around 269 °C, but different endothermic peaks before the melting points. The crystals E1 have two heat-absorption peaks at 97 °C and 167 °C before the melting point. The crystals E2 have a single peak at 113 °C before the melting point, corresponding to the one-step dehydration process. The endothermic peaks before the melting points of crystals E1 and E2 were related to the dehydration and polymorphic transformation processes, which are further analysed in the next section. The thermal properties of crystals E1 and E2 were also compared with those of commercial crystals B, as shown in Fig. S2.†
3.2 Identical powder XRD and single-crystal XRD
PXRD shows that the crystals E1 and E2 share almost the same patterns, as shown in Fig. 2a. The positions of the characteristic peaks of crystals E1 and E2 are identical, such as at 12.1°, 13.8°, 20.0°, 24.7°, and 27.5°, with the strongest peak at about 24.7°, suggesting that crystals E1 and E2 are the same polymorphs before dehydration. In addition, there is no evidence of baseline deflection or significant broadening of diffraction peaks, indicating that the crystallinities between E1 and E2 are almost the same.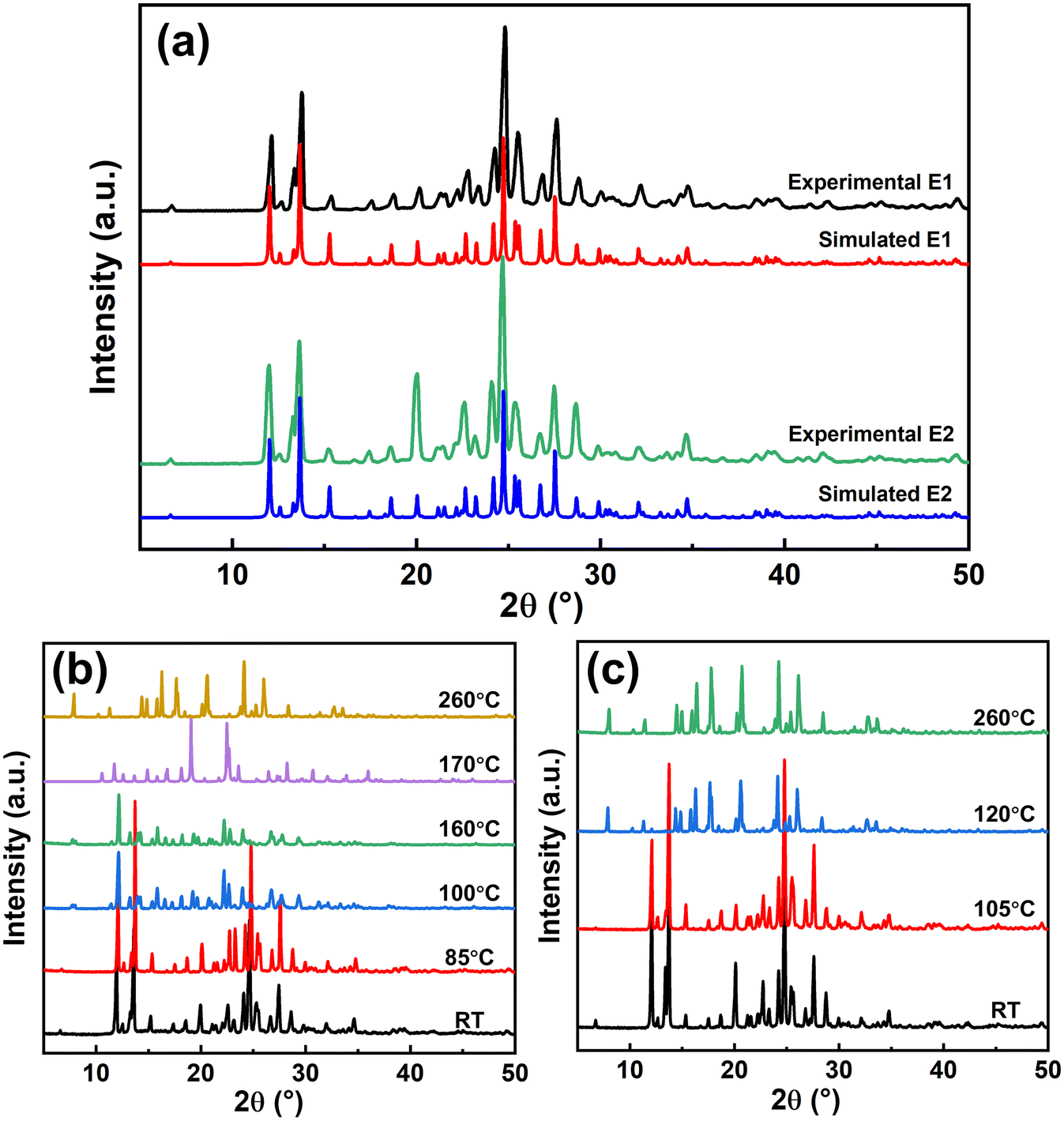 | ||
| Fig. 2 PXRD patterns of (a) LDM E1 and E2 determined from experimental bulk crystals and simulated single crystals and crystals E1 (b) and crystals E2 (c) with solid-phase transformation from room temperature (RT) to melting point. | ||
However, the changes of the PXRD patterns during dehydration are different. At room temperature, the PXRD patterns of crystals E1 and E2 are the same. With increase in temperature, the PXRD pattern of crystals E1 changes between 85 °C and 100 °C, corresponding to the first dehydration step in Fig. 1c, transforming to form B (Fig. 2b). The PXRD pattern further changes between 160 °C and 170 °C to metastable form 4 and stable form 1. This means that the peaks at 97 °C and 167 °C in the DSC curve of crystals E1 represent two different dehydration and polymorphic transformation processes. Comparatively, crystals E2 are dehydrated and transform to stable form 1 between 105 °C and 120 °C, as shown in Fig. 2c. It may be also possible that crystals E2 have an intermediate hydrated structure, but it was not observed due to the rapid transformation. The PXRD pattern of form 1 has no further change before melting, and the peak at 113 °C in the DSC curve of crystals E2 represents the thermal absorption caused by dehydration and polymorphic transformation. Forms 1, 4, and B have already been reported,28,32 and the polymorphic identification of crystals during dehydration is detailed in Fig. S3 and S4.†
The identical PXRD patterns of crystals E1 and E2 indicate that they are the same polymorph. However, they have different thermal stabilities and dehydration processes as measured by DSC and TGA, and the solid-phase transformation proved different polymorphic transformations during dehydration. SCXRD was used to determine the crystal structures of crystals E1 and E2 for evidence of polymorphisms. Table 1 shows the crystal structure information of LDM crystals E1 and E2, both exhibiting a monoclinic structure with space group P21/c. Each crystal unit cell has four LDM molecules and eight water molecules for both crystals E1 and E2, with nearly identical lattice parameters (all parameters with differences <0.25%). In addition, the bulk and single crystals were identified by PXRD as the same polymorph, as shown in Fig. S5.†
| Parameters | E1 | E2 |
|---|---|---|
| Formula | C13H13N3O3·2H2O | |
| Weight | 295.29 | |
| Crystal system | Monoclinic | |
| Space group | P21/c | |
| a (Å) | 13.4400(3) | 13.4569(3) |
| b (Å) | 8.8402(2) | 8.8327(2) |
| c (Å) | 11.7381(3) | 11.7392(3) |
| α (°) | 90 | 90 |
| β (°) | 99.1610(10) | 99.2310(10) |
| γ (°) | 90 | 90 |
| Z, Z′ | 4, 1 | 4, 1 |
| Volume (Å3) | 1376.84(6) | 1377.33(6) |
| CCDC No. | 2289868 | 2383285 |
The molecular conformations of one LDM molecule and two associated H2O molecules from the crystal unit cells of crystals E1 and E2 were further analysed by Mercury Soft. Fig. 3a shows that the root mean square deviation (RMSD) of molecular overlap between crystals E1 and E2 is 0.0079, which also proves the identical molecular conformation for crystals E1 and E2. Also, both crystals E1 and E2 share an identical packing motif, illustrated in Fig. 3b. The absence of hydrogen bonding interactions between the LDM molecules is observed. However, the O–H⋯O hydrogen bond existing between the two water molecules acts as a bridge between the LDM molecules through the O–H⋯O and N–H⋯O hydrogen bonds to keep structural stability. The crystal structure analysis confirmed the same polymorph for LDM crystals E1 and E2. In addition, the water molecule interacts with adjacent water and LDM molecules by hydrogen bonds, and both H2O molecules in crystals E1 and E2 share two structural features of isolated site and channel hydrates, which is an uncommon hydrated structure. As the crystals E1 and E2 have the same crystal structure, it is expected that the simulated PXRD patterns of crystals E1 and E2 from the single-crystal structures are also identical, as shown in Fig. 2a.
 | ||
| Fig. 3 (a) Molecular overlap and (b) packing motif of LDM and water molecules in the crystal unit cell of crystals E1 and E2. | ||
3.3 Unification of dehydration process after grinding
Crystals E1 and E2 were ground in a ball mill at a rate of 5/S for 20 min until devoid of distinct crystal shapes, as shown in the SEM images in Fig. 4a. Before grinding, crystals E1 and E2 have the same diffraction peaks but different peak intensities, as shown in Fig. 4b and c. After grinding, crystals E1 and E2 have the same diffraction peaks and almost the same peak intensities. | ||
| Fig. 4 (a) SEM images after grinding of crystals E1 and E2; (b) comparison of PXRD patterns of crystals E1 and E2 before and after grinding; (c) differences in diffraction peak intensities of bulk E2 compared to E1, simulated E2 after refining in the (1 0 0) orientation, and ground E2 compared to E1; (d) TGA and DSC curves of ground E1 and E2. | ||
The ground crystals E1 and E2 have similar TGA and DSC curves, shown in Fig. 4d. Ground crystals E1 and E2 have identical dehydration peaks at 122 °C and melting peaks at 269 °C. The dehydration processes tend to be the same, with the loss of two water molecules in one-step amounting to about 11.9%. The ground crystals E1 and E2 exhibit faster dehydration rates, completing their dehydration process at 120 °C, which is faster than the one-step dehydration process of the unground crystals E2.
3.4 Dehydration mechanism of crystals E1 and E2
Why do crystals E1 and E2 have the same PXRD and crystal structure but different thermal properties and the same dehydration process after grinding? The possible reason is that the water molecules in crystals E1 and E2 are dehydrated through different pathways due to the different lattice orientations. After grinding, the dehydration process and thermal properties of crystals E1 and E2 without morphological features become identical due to the absence of a preferred lattice orientation.Fig. 5a shows the supercell structure of crystals E1 or E2 with multiple water channels along the c-axis. These water channels are perpendicular to the ab-plane and parallel to the c-axis, and their radial direction consists of four water molecules. In the ab-plane, both water channels and LDM clusters are arranged in a straight line, forming the blue hydrophilic layers and red hydrophobic layers, respectively, which are sequentially arranged in parallel to form a complete crystal structure. Based on this hydrated structure, there are two potential dehydration pathways for hydrated water molecules to leave the crystal structure.
 | ||
| Fig. 5 (a) Supercell structures along the c-axis of crystals E1 and E2. Schematic diagrams of dehydration channel changes during the dehydration process (b) on E1 crystal face (0 0 1) and (c) on E2 crystal face (1 0 0). | ||
The first pathway related to the dehydration of crystals E1 is represented by crystal face (0 0 1), shown in Fig. 5b. It is noted that crystal planes (3 0 0), (2 2 0), and (4 1 0) are similar to (0 0 1), as they are all parallel to the c-axis. At the beginning of dehydration, it is expected that water molecules leave the crystal structure along the water channels, which corresponds to the first dehydration below 100 °C in the DSC curve in Fig. 1c. Although the detailed dehydration process was not determined, it is possible that water molecules dehydrated along the water channels undergo a slower process of diffusion and leaving, while the LDM molecules that lose the water molecules experience collapse and rearrangement due to the presence of hydrogen bonding between the water and LDM molecules, hindering the release of water molecules inside the crystal structure. This explains why the crystals E1 transform to hemihydrates after the first dehydration in Fig. 2b.
The other dehydration pathway is the radial direction of water channels perpendicular to the c-axis related to crystals E2, for example, crystal face (1 0 0), shown in Fig. 5c. It is noted that the planes (3 1 −2) and (2 2 −2) are similar to (1 0 0), as they are all vertical to the c-axis. It is expected that the water molecules in the channels can leave the crystal structure at once, leading to a faster dehydration process. All water molecules are expected to leave the crystal structure in one step due to the absence of the limitation of the diffusion effect of water channels, consistent with the polymorphic transformation characterization of crystals E2 in Fig. 2c, where the LDM molecules are directly restructured into anhydrous without observing an intermediate hydrated structure. Despite no direct observation of these dehydration pathways and transformation processes, these hypotheses agree with the DSC, TGA and phase transformation results.
It has been demonstrated that the two dehydration pathways are consistent with the dehydrations of the two LDM dihydrates. Fig. 4b presents an overlay of the PXRD patterns of crystals E1 and E2, revealing five characteristic peaks with significant intensity differences (detailed in Table S2†) with their corresponding Miller indices labeled. Several researches have reported that the relative intensity of PXRD diffraction peaks reflects the preferred orientation effect of pharmaceutical solid forms.18,38,39 For crystals E2, the diffraction peaks labeled (3 0 0), (2 2 0), and (4 1 0), which are all parallel to the c-axis, show a significant increase in intensity, while those labeled (3 1 −2) and (2 2 −2), which are all vertical to the c-axis, have lower intensity. Therefore, the lattice orientation becomes stronger on the planes of (3 0 0), (2 2 0) and (4 1 0) parallel to the c-axis but weaker on the other planes perpendicular to the c-axis, indicating that crystals E2 tend to dehydrate in the radial direction of water channels, with a predominance of the second dehydration pathway. Crystals E1 have an opposite lattice orientation change to crystals E2 and therefore tend to dehydrate along the water channels with the first dehydration pathway. Although the crystal faces were not indexed and the dominant crystal surfaces were not identified, comparing the PXRD patterns of E1, E2, ground E1 and ground E2, these (3 0 0), (3 1 −2) and other planes indicate the difference of lattice orientation for E1 and E2, which could lead to the different dehydration processes.
The simulated PXRD pattern of crystals E2 was further calculated using Rietveld refinement with a March–Dollase parameter of 0.7 in Mercury Soft (see Fig. S6†), followed by analyzing the peak intensity changes of E2 in the (1 0 0) preferred orientation, as shown in Fig. 4c. The peak intensities of (1 0 0), (2 0 0) and (3 0 0) increase significantly, indicating that crystal E2 has a clear preferred orientation in (1 0 0). The (4 1 0) peak intensity also increases, corresponding to the higher peak intensity in (4 1 0) for the E2 bulk sample, which confirms that the preferred orientation is the reason why crystals E2 have a different dehydration path from crystals E1.
3.5 Dehydration kinetics of crystals E1 and E2
The dehydration kinetics of LDM crystals E1 and E2 were expressed by the apparent activation energy (Ea) at different dehydration fractions (α). The Flynn–Wall–Ozawa (FWO) method, a non-isothermal isoconversional method to study the kinetic parameters of solid-state interactions,15,21 was used to calculate the Ea, which can be expressed as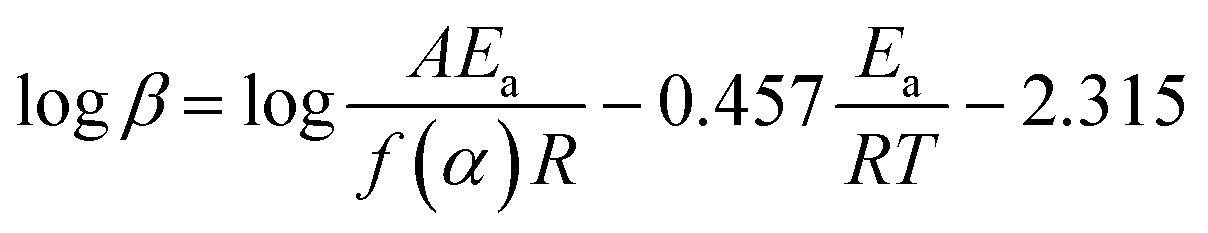 | (1) |
The dehydration kinetics based on the FWO method were performed at the heating rates of 2, 5, 10, and 15 K min−1 using TGA determination, as shown in Fig. S7 and S8.† According to the TGA data, plotting logβ versus −457/RT at different dehydration fractions (α = 10–90%) can yield the Ea from the slope, as shown in Fig. 6. All the plots have a strong positive linear relationship (R2 > 0.99); the Ea of forms E1 and E2 at different α are reported in Table 2. The reaction sequence of the dehydration process can be determined qualitatively by the variation of the Ea with α. With increasing α, the values of Ea for crystals E1 tend to increase and then decrease, indicating that the reaction sequence is a multi-step competitive process complicated by diffusion, which is consistent with the pathway of diffusion and dehydration through water channels. For crystals E2, the values of Ea remain essentially constant within 10% ≤ α ≤ 40% and 50% ≤ α ≤ 90%, indicating that the reaction sequence is a simple first-order reaction, which corresponds to the dehydration pathway of removing all the isolated site type water in one step. The slight increase of Ea within 40% ≤ α ≤ 50% for crystals E2 may be due to the collapse of partial crystal structure during the dehydration process. Obviously, crystals E1 and E2 have different reaction sequences, confirming the different dehydration pathways.
 | ||
| Fig. 6 FWO plots of logβ versus −457/RT at α = 10–90% for the thermal dehydration: (a) crystals E1 and (b) crystals E2. | ||
| α (%) | E a (kJ mol−1) | |
|---|---|---|
| E1 | E2 | |
| 10 | 53.64 | 62.07 |
| 20 | 75.43 | 61.72 |
| 30 | 95.02 | 61.90 |
| 40 | 101.83 | 62.75 |
| 50 | 98.39 | 73.39 |
| 60 | 91.55 | 74.78 |
| 70 | 81.71 | 74.55 |
| 80 | 67.02 | 73.71 |
| 90 | 47.07 | 72.63 |
4. Discussion
A possible explanation of the two pathways for hydrated water molecules to leave the crystal structure is the different lattice orientations. It is reasonable to explain the grinding influence. After grinding, crystals E1 and E2 have almost identical morphologies that are considered the same lattice orientation and therefore have the same dehydration process, as shown in Fig. 4. As the DSC curves show that the dehydration process starts below 100 °C with a one-step process for ground E1 and E2, more water channels are expected to appear on the crystal surface and the dehydration will not be hindered by the LDM molecules' collapse. The overall dehydration rates of the ground crystals are faster than those of the unground crystals E1 and E2 due to more dehydration channels being opened. In addition, the specific lattice orientations are maintained by the grains of bulk crystals E1 and E2 despite their different sizes; thus, they exhibit different dehydration behaviors.There are reports on different dehydration rates due to lattice orientations,18,40,41 and it is reasonable to accept that exposing more water channels in the crystal would accelerate the dehydration process. However, to the best of the authors' knowledge, this is the first report to observe that lattice orientation would induce not only different dehydration rates but also totally different changes in the solid-state transformation which further induce different thermal properties during dehydration. The emergence of this uncommon phenomenon is attributed to the special hydrated structure of LDM dihydrate. The water molecule interacts with both adjacent water and LDM molecules through hydrogen bonding interactions in the crystal unit cell, matching the dual features of channel and isolated site hydrates, which results in the appearance of two different dehydration pathways influenced by the lattice orientation. Nevertheless, the question of why the departure of water molecules from crystals E1 and E2 causes the rearrangement of the crystal structure and the different restructuring processes occurring in crystals E1 and E2 still requires further investigation. Besides, both crystals E1 and E2 remain stable at room temperature, although they have different dehydration temperatures and thermal stabilities. E1 is considered more stable than E2 due to the higher solubility of E2 in the initial dissolution, as evidenced by the greater dissolution rate of E2 compared to that of E1. Therefore, crystals E2 are a promising alternative for solid form LDM.
5. Conclusions
The LDM dihydrates, crystals E1 and E2, have different shapes. Crystals E2 have more stability than crystals E1 by DSC and a higher dissolution rate. TGA shows that crystals E1 have a two-step process of dehydration and restructure to hemihydrate before changing to anhydrous. Crystals E2 have only a one-step process of dehydration and restructure to anhydrous directly. However, the crystals E1 and E2 have the same peak positions in their Powder XRD patterns, and Single-Crystal XRD further proves that crystals E1 and E2 have the same crystal structure.The same crystal structures and polymorphs with different thermal properties and dissolution rates could be due to two distinct dehydration pathways for different lattice orientations in the presence of a specific hydrated structure. It is demonstrated that ground crystals E1 and E2 without the preferred orientations have the same thermal properties and dehydration processes. The two LDM dihydrates, identified as same polymorph, have different thermal and dissolution properties, highlighting the need for a better understanding of thermal properties and structural changes during the dehydration process.
Data availability
Crystallographic data for lenalidomide crystals E1 and E2 have been deposited at the CCDC under No. 2289868 and 2219890. The data supporting this article are included in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by National Key Research and Development Program of the International Scientific and Technological Innovation Cooperation Project among Governments (2021YFE0100400) and Natural Science Foundation of Shanghai Municipality (No. 23ZR1417100). HY is grateful to the Royal Society for its support (IEC\NSFC\201432). The authors thank Pietro Sacchi for helpful discussions.References
- L. H. Emara, R. M. Badr and A. Abd Elbary, Drug Dev. Ind. Pharm., 2022, 28, 795–807 CrossRef PubMed.
- E. Jurczak, A. H. Mazurek, Ł. Szeleszczuk, D. M. Pisklak and M. Zielińska-Pisklak, Pharmaceutics, 2020, 12, 959 CrossRef CAS PubMed.
- J. Zhang, Y. Ying, B. Pielecha-Safira, E. Bilgili, R. Ramachandran, R. Romañach, R. N. Davé and Z. Iqbal, Int. J. Pharm., 2014, 475, 428–437 CrossRef CAS PubMed.
- D. E. Braun, T. Gelbrich, V. Kahlenberg and U. J. Griesser, Mol. Pharmaceutics, 2014, 11, 3145–3163 CrossRef CAS PubMed.
- K. R. Morris and N. Rodríguez-Hornedo, in Encyclopedia of Pharmaceutical Technology, ed. J. Swarbrick and J. Boylan, Marcel Dekker, New York, 1993, vol. 7, pp. 393–440 Search PubMed.
- L. Kumar and A. K. Bansal, Thermochim. Acta, 2011, 525, 206–210 CrossRef CAS.
- V. Koradia, H. L. de Diego, M. R. Elema and J. Rantanen, J. Pharm. Sci., 2010, 99, 3966–3976 CrossRef CAS PubMed.
- X. Ma, F. Müller, S. Huang, M. Lowinger, X. Liu, R. Schooler and R. O. Williams III, Pharmaceutics, 2020, 12, 379 CrossRef CAS PubMed.
- F. Tian, H. Qu, A. Zimmermann, T. Munk, A. C. Jørgensen and J. Rantanen, J. Pharm. Pharmacol., 2010, 62, 1534–1546 CrossRef CAS PubMed.
- A. K. Salameh and L. S. Taylor, J. Pharm. Sci., 2006, 95, 446–461 CrossRef CAS PubMed.
- T. Suzuki and K. Terada, Int. J. Pharm., 2012, 422, 1–8 CrossRef CAS PubMed.
- Z. Pang, Y. Wei, H. Chen, R. Wang, X. Gao, J. Zhang, Y. Gao and S. Qian, Int. J. Pharm., 2021, 597, 120374 Search PubMed.
- A. R. Sheth, D. Zhou, F. X. Muller and D. J. Grant, J. Pharm. Sci., 2004, 93, 3013–3026 CrossRef CAS PubMed.
- J. Sun, C. Xie, X. Zhang, Y. Bao, B. Hou, Z. Wang, J. Gong, H. Hao, Y. Wang, J. Wang and Q. Yin, Org. Process Res. Dev., 2016, 20, 780–785 CrossRef CAS.
- W. H. Hsieh, W. T. Cheng, L. C. Chen and S. Y. Lin, Asian J. Pharm. Sci., 2018, 13, 212–219 Search PubMed.
- Z. Ding, W. Su, X. Huang, B. Tian, X. Cheng, Y. Mao, G. Li, H. Liu and H. Hao, Cryst. Growth Des., 2020, 20, 1150–1161 Search PubMed.
- A. S. Larsen, M. T. Ruggiero, K. E. Johansson, J. A. Zeitler and J. Rantanen, Cryst. Growth Des., 2017, 17, 5017–5022 Search PubMed.
- F. Kang, F. G. Vogt, J. Brum, R. Forcino, R. C. Copley, G. Williams and R. Carlton, Cryst. Growth Des., 2012, 12, 60–74 CrossRef CAS.
- A. Bērziņš and A. Actiņš, CrystEngComm, 2014, 16, 3926–3934 RSC.
- M. Takahashi and H. Uekusa, J. Pharm. Sci., 2022, 111, 618–627 CrossRef CAS PubMed.
- J. H. Flynn and L. A. Wall, J. Polym. Sci. B: Pol. Lett., 1966, 4, 323–328 CrossRef CAS.
- J. Gao, Y. Wang and H. Hao, Front. Chem. Sci. Eng., 2012, 6, 276–281 CrossRef CAS.
- D. E. Braun, T. Gelbrich, V. Kahlenberg and U. J. Griesser, Mol. Pharmaceutics, 2014, 11, 3145–3163 Search PubMed.
- A. S. Larsen, J. Rantanen and K. E. Johansson, J. Pharm. Sci., 2017, 106, 348–355 CrossRef CAS PubMed.
- B. Yang, Y. Li, J. Lei, M. Cai, Z. Hu, Y. Shen and X. Deng, Int. J. Pharm., 2023, 638, 122893 CrossRef CAS PubMed.
- A. A. Guirguis and B. L. Ebert, Curr. Opin. Cell Biol., 2015, 37, 61–67 CrossRef CAS PubMed.
- A. Palumbo, J. Freeman, L. Weiss and P. Fenaux, Expert Opin. Drug Saf., 2012, 11, 107–120 CrossRef CAS PubMed.
- K. Ravikumar and B. Sridhar, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2009, 65, o502–o505 CrossRef CAS PubMed.
- D. P. Konakanchi, B. Gongalla, K. B. Sikha, C. Kandaswamy, K. S. B. R. Adibhatla and V. C. Nannapaneni, WO2011111053A1, 2011.
- R. S. Chen, G. W. Muller, M. S. Jaworsky, M. T. Saindane and L. M. Cameron, WO2005023192A2, 2005.
- R. Yan, H. Yang and Y. Xu, WO2011050590A1, 2011.
- R. Chennuru, P. Muthudoss, R. S. Voguri, S. Ramakrishnan, P. Vishweshwar, R. R. C. Babu and S. Mahapatra, Cryst. Growth Des., 2017, 17, 612–628 CrossRef CAS.
- L. Jia, Z. Li and J. Gong, Pharm. Dev. Technol., 2019, 24, 1175–1180 CrossRef CAS PubMed.
- A. Li, Z. Si, Y. Yan and X. Zhang, J. Mol. Liq., 2021, 330, 115446 CrossRef CAS.
- R. Liang, Y. Zhu, Y. Wu, X. Gu and X. Zhang, CrystEngComm, 2022, 24, 6357–6366 RSC.
- L. Wang, Y. Yan, X. Zhang and X. Zhou, Int. J. Pharm., 2022, 613, 121394 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- S. N. C. Roberts, A. C. Williams, I. M. Grimsey and S. W. Booth, J. Pharm. Biomed. Anal., 2002, 28, 1149–1159 CrossRef PubMed.
- J. L. Crisp, S. E. Dann and C. G. Blatchford, Eur. J. Pharm. Sci., 2011, 42, 568–577 CrossRef CAS PubMed.
- J. Y. Khoo, D. R. Williams and J. Y. Heng, Drying Technol., 2010, 28, 1164–1169 CrossRef CAS.
- A. Joseph, C. E. Bernardes, A. S. Viana, M. F. M. Piedade and M. E. Minas da Piedade, Cryst. Growth Des., 2015, 15, 3511–3524 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ce00798k |
| This journal is © The Royal Society of Chemistry 2025 |