
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Silylene–copper(I) catalysis: regioselective protoboration of terminal alkynes
Sandeep H.
Kaulage
 a,
Brij Kumar
Shah
a,
Rishukumar
Panday
a,
Himanshu
Sharma
bc,
Kumar
Vanka
bc and
Shabana
Khan
*a
a,
Brij Kumar
Shah
a,
Rishukumar
Panday
a,
Himanshu
Sharma
bc,
Kumar
Vanka
bc and
Shabana
Khan
*a
aDepartment of Chemistry, Indian Institute of Science Education and Research Pune, Dr Homi Bhabha Road, Pashan, Pune-411008, India. E-mail: shabana@iiserpune.ac.in
bPhysical & Materials Chemistry Division, CSIR-National Chemical Laboratory, Pune, Dr Homi Bhabha Road, Pashan, Pune-411008, India
cAcademy of Scientific and Innovative Research (AcSIR), Sector 19, Kamla Nehru Nagar, Ghaziabad 201002, Uttar Pradesh, India
First published on 12th November 2025
Abstract
Herein, we report an efficient regioselective protoboration of the terminal alkynes catalyzed by newly synthesized silylene–copper(i)–aryl complexes. This method offers a broad substrate scope, good functional-group compatibility, and a gram-scale synthetic ability. The insight into the mechanistic cycle is also provided with the support of experimental and theoretical studies.
N-heterocyclic silylenes (NHSis) have emerged as powerful σ-donor ligands over the past two decades due to their unique electronic properties, which modulate the electronic environment at the metal centre, offering promising avenues in homogeneous catalysis.1 While the coordination chemistry of silylenes with a wide range of transition metals has been increasingly explored, their chemistry with coinage metals (Cu, Ag, and Au) remains comparatively underdeveloped.2 Nevertheless, recent efforts have led to the successful synthesis and characterization of several structurally well-defined silylene–coinage metal complexes, opening avenues for their applications in catalysis.1–4 Notably, Stalke and co-workers demonstrated the first application of NHSi–Cu(I) clusters in CuAAC catalysis.3 Following this pioneering work, our group introduced several NHSi–Cu(I) complexes, demonstrating their enhanced catalytic efficiency in various organic transformations.4 Very recently, we have shown the synthetic versatility and facile bond activation with NHSi–Cu(I)–aryl complexes.4d In contrast to the N-heterocyclic carbene (NHC)-coinage metal complexes,5 NHSi–Cu(I) complexes have been rarely employed in homogeneous catalysis. Among the extensively studied reactions involving NHC–copper complexes, the protoboration of terminal alkynes with diboron reagents and a proton source stands out due to its direct applicability in synthetic chemistry.5,6 However, to achieve the desirable regio- and stereoselective vinyl boronate esters via the protoboration of alkynes, a tailor-made ligand coordinated to the copper(I) centre is essential. There are some examples of NHC–copper-catalysed protoboration reactions,7 but the use of silylene Cu(I) complexes has surprisingly not been investigated to date. Motivated by the abovementioned fact, we targeted the exploration of the NHSi–Cu(I) complexes for the protoboration of terminal alkynes.
Herein, we report the synthesis, characterization, and catalytic application of silylene–Cu(I) complexes in the regioselective protoboration of terminal alkynes. To the best of our knowledge, this represents the first example of silylene-supported Cu(I) catalysis in protoboration chemistry without the utility of base. Our findings highlight the ability of silylenes to serve as spectator ligands and give regioselective products with low catalyst loading.
The silylene-based copper(I) halide complexes (C1–C4)8,9c were synthesized via the complexation of ligands (L1–L4)9 with CuCl as metal precursors (Scheme 1). Further, we focused on the synthesis of NHSi-coordinated Cu(I)–aryl complexes and isolated the complexes C5–C8 (Scheme 1) via a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 reaction of L1, L2, L3, and L4 with (CuMes)n, respectively, in good yields. All the newly synthesized complexes were characterized with single-crystal X-ray diffraction studies (Fig. 1) and routine NMR spectroscopy (see SI, S4). The 29Si{1H} NMR shifts of all the newly synthesized complexes are compared in Table S1 (See SI, S3).
:1 reaction of L1, L2, L3, and L4 with (CuMes)n, respectively, in good yields. All the newly synthesized complexes were characterized with single-crystal X-ray diffraction studies (Fig. 1) and routine NMR spectroscopy (see SI, S4). The 29Si{1H} NMR shifts of all the newly synthesized complexes are compared in Table S1 (See SI, S3).
 | ||
| Scheme 1 Synthesis of C1–C10, Ar = 2,6-diisopropyl phenyl, Mes = 2,4,6-trimethylphenyl. | ||
 | ||
| Fig. 1 The molecular structures of C1, C5, and C6. The anisotropic displacement parameters are depicted at the 50% probability level. Hydrogen atoms and solvents are omitted for clarity. Selected bond distances (Å) and angles (°): For (C1) P1–N1 1.740(3), N1–Si1 1.761(3), Si1–Cu1 2.184(1), Cu1–Cl1 2.32(1); Si1–Cu1–Cl1 132. 4(3), P1–N1–Si1 118.2(2); for (C5) P1–N1 1.747(5), N1–Si1 1.752(6), Si1–Cu1 2.232(2), Cu1–C40 1.946(6); Si1–Cu1–C40 173.5(2); For (C6) P1–N1 1.739(6), N1–Si1 1.748(6), Si1–Cu1 2.219(3), Cu1–C34 1.936(8); Si1–Cu1–C1 175.4(3). | ||
After the characterization of the complexes, we went for optimization of reaction conditions for the protoboration of terminal alkynes.8,9 We have screened various bases and solvents using phenyl acetylene as a model substrate for optimization purposes (Table S2). In this case, B2pin2 serves as a boron source and MeOH as a proton source (Table S2). We began our study by employing NHSi–copper halide-based catalyst, C1 as a catalyst (4 mol%), 1.1 equiv. of B2pin2 and KOtBu in toluene at room temperature (rt) for 12 h, which afforded the corresponding vinyl boronate ester (2a) in 80% yield (entry 1, Table S2). An increase in the yield was observed for 2a within 8 h when the solvent was changed to acetonitrile (entry 2, Table S2), revealing the effect of the solubility of the base. Moreover, the screening of other analogous catalysts (C2–C4) also afforded the desired product in good yields.
To achieve the base-free methodology, we envisioned using NHSiCu(I)–aryl catalysts (C5–C8) for further optimization (Table S2), which were expected to react directly with the boron source due to the highly reactive Cu–aryl bond. It was found that among the various catalysts, C6 worked well and demonstrated the highest yield (99%) and regioselectivity without a base (See Table S2, entry 13). To understand the catalytic efficiency of our newly developed catalyst C6, we compared it with the previously reported NHC–Cu(I) catalysts [IPr–CuCl (C9) and IPr–CuMes (C10)] for the protoboration of phenylacetylene (see Table 1 and Table S2).9d Interestingly, NHSi–Cu catalyst C6 outperforms the NHC–Cu catalysts (C9 and C10) in terms of both isolated yield and reaction time. The rationale behind selecting ligands L1–L4 lies in their steric and electronic variations, aimed at analyzing their effects on the protoboration of terminal alkynes. All the ligands were found to favor the formation of the anti-Markovnikov product (Table S5), suggesting that ligand variation has minimal influence on regioselectivity. However, a significantly enhanced product yield was observed with the silylene ligands, particularly with the C6 system. This is presumably due to improved stabilization of the active catalyst species through the strong σ-donating ability of the NHSi ligand.
| With base | Without base | ||
|---|---|---|---|
| Catalyst | Yield % | Catalyst | Yield % |
| a Reaction conditions: 0.5 mmol of 1a, 0.55 mmol of B2pin2, 2 mol% of Cat, 1.1 equiv. of MeOH, and 2 mL ACN; reaction time 6 h. b Reaction time 12 h. #Yields of products (α + β) and regioselectivity (±2%) were determined by 1H NMR using CH2Br2 as the internal standard. | |||
| C1 | 91 | C5 | 95 |
| C2 | 93 | C6 | 99 |
| C3 | 89 | C7 | 77 |
| C4 | 90 | C8 | 87 |
| C9 | 84 | C10 | 88 |
Moreover, the choice of the catalyst and solvent played a critical role in modulating the reaction outcome. Among the boron sources, bis(pinacolato)diboron (B2pin2) was found to be the most efficient, offering high conversion and excellent selectivity under mild conditions (Table 2). With the optimized conditions in hand, we next explored the substrate scope of the protoborylation reaction and screened both aliphatic and aromatic terminal alkynes.
|
|
||||
|---|---|---|---|---|
| S. no | C6 (mol%) | B-Source | Yield % |
α:β:β(cis) |
| a Reaction conditions: 0.5 mmol of alkynes, 0.55 mmol of B2pin2, 2 mol% of C6, 1.1 equiv. of MeOH, time 6 h, and 2 mL ACN. nd = not determined. | ||||
| 1 | 2 | B2cat2 | 72 | nd:72:28 |
| 2 | 2 | B2pin2 | 99 | 01:99:nd |
| 3 | 2 | B2neop2 | 78 | 44:46:10 |
| 4 | 2 | pinB-Bdan | 69 | 77:23:nd |
A diverse set of terminal alkynes bearing electron-donating, electron-withdrawing, and sterically demanding substituents was examined (Scheme 2). Electron-rich alkynes (2b, 2c, 2g, and 2j) underwent smooth protoborylation to afford the corresponding vinylboronates in excellent yields (up to 82%). Electron-deficient alkynes (2d, 2e, 2f, 2h, and 2k) also reacted efficiently, albeit with excellent yields (70–94%). The product 2l can be isolated as three regio-isomers, but in our case, we observed only the formation of two (branched-branched and linear-linear) (α![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) :β = 6:94). The linear-branched isomer was not observed in the NMR spectrum of 2l.
:β = 6:94). The linear-branched isomer was not observed in the NMR spectrum of 2l.
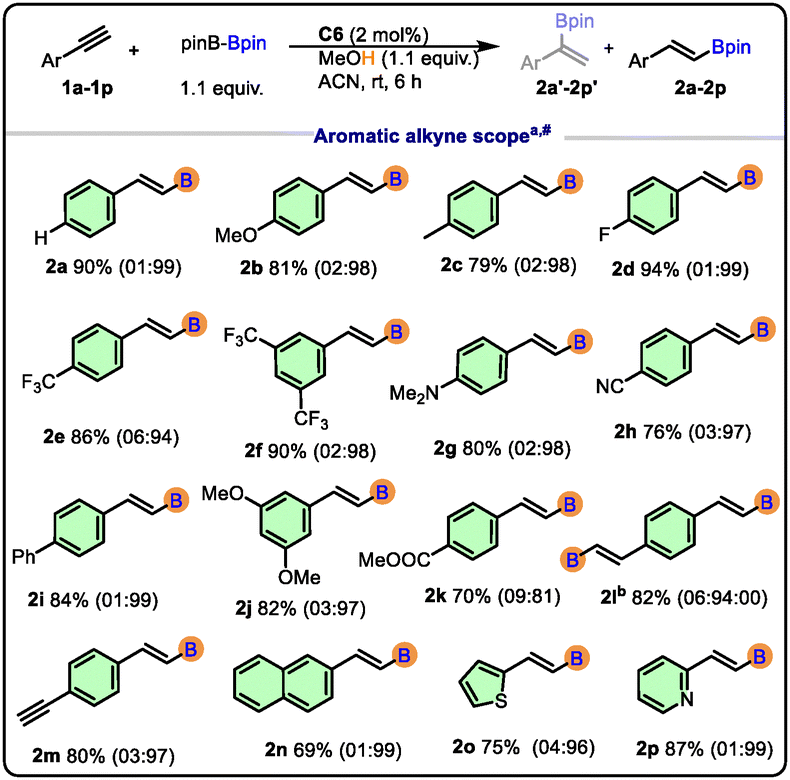 | ||
| Scheme 2 Substrate scope of aromatic terminal alkynes. aReaction conditions: 0.5 mmol of alkynes, 0.55 mmol of B2pin2, 2 mol% of C6, 1.1 equiv. of MeOH, time 6 h, and 2 mL ACN. b1.1 mmol of B2Pin2, 4 mol% of C6, 2.1 equiv. of MeOH, 12 h, B = Bpin. #Yields of products (α + β) and regioselectivity (±2%) were determined by 1H NMR using CH2Br2 as the internal standard. | ||
Further, we explored protoboration of aliphatic terminal alkynes. Notably, Boc-protected amine and ether functionality on alkynes led to the excellent yields (3e, 3g, and 3h). Aliphatic terminal alkynes having functional groups like ester, alcohol, and silanes also underwent protoborylation smoothly, highlighting the broad applicability of the methodology (Scheme 3). In all cases, the reactions proceeded with excellent regioselectivity, furnishing predominantly the anti-Markovnikov product.
 | ||
| Scheme 3 Substrate scope of aliphatic terminal alkynes. aReaction conditions: 0.5 mmol of alkynes, 0.55 mmol of B2pin2, 2 mol% of C6, 1.1 equiv. of MeOH, time 6 h, and 2 mL ACN. #Yields of products (α + β) and regioselectivity (±2%) were determined by 1H NMR using CH2Br2 as the internal standard; B = Bpin. | ||
Further, we explored the scope of various boron sources by screening four different diboron reagents (Table 2). Among them, B2cat2 exhibited high β-selectivity, similar to B2pin2, while B2neop2 led to a mixture of regio-isomers. Notably, the use of the unsymmetrical diboron reagent pinB-Bdan resulted in the formation of branched vinyl boronates as the major product, incorporating the -B(dan) moiety, in accordance with the Lewis acidity trend of the boron sources employed.10
We further demonstrated the synthetic utility of our method by synthesizing the borylated compounds on a gram scale (Scheme 4). The production of 2a with 78% yield on a gram scale shows the feasibility of the reaction to scale up to a bulk amount.
 | ||
| Scheme 4 Gram-scale synthesis of 2a. | ||
To understand the reaction mechanism, we performed the stoichiometric experiments of C1 along with base and B2pin2 (Section S7.2). Based on that, we propose that the intermediate I was formed as a first step, which, upon σ-bond metathesis with a diboron reagent, leads to the formation of the thermally unstable copper boryl species(Int-II).11a The intermediate I was characterized by 1H, 31P{1H} and 29Si{1H} NMR spectra (Section S7.2). Alternatively, the intermediate II can also be prepared by reacting NHSiCu(I)-mesityl species with B2pin2 at −78 °C (11B{1H} NMR = δ 33.79 ppm), which is only stable at low temperature.4d,11 Based on these experiments and previous reports, we can say that the Cu–boryl species is the key intermediate for this reaction, which undergoes syn-addition across the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bond of the alkyne.11b The resultant vinyl–copper intermediate III undergoes protonation, forming the β-vinylboronate (Fig. 2).
C bond of the alkyne.11b The resultant vinyl–copper intermediate III undergoes protonation, forming the β-vinylboronate (Fig. 2).
 | ||
| Fig. 2 Plausible mechanistic Cycle with base and without base pathways. (L = PhC(NtBu)2 SiN(PPh)2(2,6-iPr2-C6H4)). | ||
To further validate this mechanism, deuterium-labelling experiments were performed using MeOD as a proton source. The incorporation of deuterium at the β-position of the vinyl boronate confirmed the involvement of a protonation step (Section S7.7) following the migratory insertion of the alkyne into the Cu–B bond.
To validate the proposed mechanism and β-product selectivity, DFT calculations were performed using the B3LYP-D3 functional (See SI for details).12 We initiated the study from NHSiCu(I)-Bpin, which forms exergonically from either NHSi–CuCl (with base, –9.9 kcal mol−1) or NHSiCu-Mes (without base, –4.2 kcal mol−1). Next, terminal alkyne coordinates in both α- and β-modes with comparable energies; however, the subsequent Bpin transfer step is the key step: the β-pathway requires only 5.9 kcal mol−1 to cross the transition state (TSβ), while the α-pathway demands 10.8 kcal mol−1. This 4.9 kcal mol−1 barrier difference decisively favours β-selectivity, leading to Int-III (Fig. 3), which further reacts with MeOH to give the final β-product (–13.2 kcal mol−1). These results explain the exclusive formation (∼99%) of the β-isomer under the reaction conditions. To confirm that the selectivity was not an artifact of the chosen functional, we recalculated the data at PCM-PBE0-D3/def2-TZVP//PCM-PBE0-D3/def2-SVP (Fig. S19), which supports that the β-selectivity is intrinsic and not functional-dependent.
 | ||
| Fig. 3 The free energy profile for the protoboration of a terminal alkyne is shown here. Level: PCM(acetonitrile)-B3LYP-D3/def2-TZVP//PCM(acetonitrile)-B3LYP-D3/def2-SVP. | ||
In summary, we have shown the utility of NHSiCu(I)-Mes complexes as an efficient catalyst for the base-free protoboration of terminal alkynes with high regioselectivity as compared to traditional NHCs. These reactions are selective in the presence of several functional groups, furnishing vinyl boronate esters in high yields and are scalable up to the gram scale, paving the way for NHSi–copper complexes in several other important transformations.
S. K. thanks the SERB-CRG grant (CRG/2021/000395) for the financial support. S. K. also thanks DST-FIST for the single-crystal X-ray diffractometer. S. H. K. thanks IISER Pune for the fellowship. B. K. S. H. S. and R. P. thank UGC for the fellowship. K. V. is grateful to DST-SERB (CRG/2021/003255) and FBR060307 for funding.
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: experimental details, X-ray data, NMR spectra, and computational details. See DOI: https://doi.org/10.1039/d5cc06125c.CCDC 2464517–2464519 contain the supplementary crystallographic data for this paper.13a–c
Notes and references
- (a) Y. P. Zhou and M. Driess, Angew. Chem., Int. Ed., 2019, 58, 3715–3728 CrossRef CAS PubMed; (b) S. Raoufmoghaddam, Y. P. Zhou, Y. Wang and M. Driess, J. Organomet. Chem., 2017, 829, 2–10 CrossRef CAS; (c) B. Blom, M. Stoelzel and M. Driess, Chem. – Eur. J., 2013, 19, 40–62 CrossRef CAS PubMed; (d) Z. Hendi, M. K. Pandey, S. K. Kushvaha and H. W. Roesky, Chem. Commun., 2024, 60, 9483–9512 RSC.
- M. Ghosh and S. Khan, Dalton Trans., 2021, 50, 10674–10688 RSC.
- A. N. Paesch, A. K. Kreyenschmidt, R. Herbst-Irmer and D. Stalke, Inorg. Chem., 2019, 58, 7000–7009 CrossRef CAS PubMed.
- (a) N. Parvin, J. Hossain, A. George, P. Parameswaran and S. Khan, Chem. Commun., 2019, 56, 273–276 RSC; (b) J. Hossain, J. S. Gopinath, S. Tothadi, P. Parameswaran and S. Khan, Organometallics, 2022, 41, 3706–3717 CrossRef CAS; (c) N. Parvin, B. Mishra, A. George, M. Neralkar, J. Hossain, P. Parameswaran, S. Hotha and S. Khan, Chem. Commun., 2020, 56, 7625–7628 RSC; (d) M. Ghosh, K. Gaurav, P. Panwaria, R. Panday, S. Tothadi and S. Khan, Chem. Sci., 2025, 16, 14518–14533 RSC.
- (a) C. M. Crudden and D. P. Allen, Coord. Chem. Rev., 2004, 248, 2247–2273 CrossRef CAS; (b) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS PubMed.
- (a) T. Tsushima, H. Tanaka, K. Nakanishi, M. Nakamoto and H. Yoshida, ACS Catal., 2021, 11, 14381–14387 CrossRef CAS; (b) A. H. Cherney, N. T. Kadunce and S. E. Reisman, Chem. Rev., 2015, 115, 9587–9652 CrossRef CAS PubMed.
- (a) H. Yoshida, ACS Catal., 2016, 6, 1799–1811 CrossRef CAS; (b) J. Yun, Asian J. Org. Chem., 2013, 2, 1016–1025 CrossRef CAS; (c) E. A. Romero, R. Jazzar and G. Bertrand, Chem. Sci., 2016, 8, 165–168 RSC; (d) A. J. J. Lennox and G. C. Lloyd-Jones, Chem. Soc. Rev., 2014, 43, 412–443 RSC.
- M. Stollenz and F. Meyer, Organometallics, 2012, 31, 7708–7727 CrossRef CAS.
- (a) S. H. Kaulage, N. Parvin, K. V. Khopade and S. Khan, Chem. Commun., 2024, 60, 9958–9961 RSC; (b) J. A. Cabeza, P. García-Álvarez and L. González-Álvarez, Chem. Commun., 2017, 53, 10275–10278 RSC; (c) N. Parvin, R. Dasgupta, S. Pal, S. S. Sen and S. Khan, Dalton Trans., 2017, 46, 6528–6532 RSC; (d) L. Kuehn, A. F. Eichhorn, D. Schmidt, T. B. Marder and U. Radius, J. Organomet. Chem., 2020, 919, 121249 CrossRef CAS.
- (a) T. Tsushima, M. Nakamoto and H. Yoshida, ACS Catal., 2024, 14, 12694–12703 CrossRef CAS; (b) H. Yoshida, Y. Takemoto and K. Takaki, Chem. Commun., 2014, 50, 8299–8302 RSC.
- (a) R. S. C. Charman, J. A. Hobson, R. A. Jackson, M. F. Mahon, S. E. Neale and D. J. Liptrot, Chem. – Eur. J., 2024, 30, e202302704 CrossRef CAS PubMed; (b) P. M. Rutz and C. Kleeberg, Chem. – Asian J., 2024, 19, e202400286 CrossRef CAS PubMed.
- (a) H. Sharma, T. Tewari, S. H. Chikkali and K. Vanka, J. Organomet. Chem., 2023, 986, 122621 CrossRef CAS; (b) H. Liang and J. P. Morken, J. Am. Chem. Soc., 2025, 147(16), 13126–13130 CrossRef CAS PubMed.
- (a) CCDC 2464517: Experimental Crystal Structure Determination, 2025 DOI:10.5517/ccdc.csd.cc2nqjkh; (b) CCDC 2464518: Experimental Crystal Structure Determination, 2025 DOI:10.5517/ccdc.csd.cc2nqjlj; (c) CCDC 2464519: Experimental Crystal Structure Determination, 2025 DOI:10.5517/ccdc.csd.cc2nqjmk.
| This journal is © The Royal Society of Chemistry 2025 |