
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of 11C-epoxides, aziridines, and cyclopropanes from structurally modified 11C-sulfur ylides
Joshua
Priest
 a,
Casey J.
McCarthy
ab,
Xia
Shao
a and
Peter J. H.
Scott
*abc
a,
Casey J.
McCarthy
ab,
Xia
Shao
a and
Peter J. H.
Scott
*abc
aDepartment of Radiology, University of Michigan, Ann Arbor, Michigan 48109, USA. E-mail: pjhscott@umich.edu
bDepartment of Medicinal Chemistry, College of Pharmacy, University of Michigan, Ann Arbor, Michigan 48109, USA
cDepartment of Pharmacology, University of Michigan, Ann Arbor, Michigan 48109, USA
First published on 14th November 2025
Abstract
A method for the radiosynthesis of 11C-epoxides, aziridines, and cyclopropanes is described. Through generating 11C-methyl sulfonium salts and corresponding sulfur ylides, the formation of 11C-epoxides is possible in moderate-to-high radiochemical conversions. This method is translated to other 3-membered cyclic species and drug-like scaffolds. Automated radiosynthesis with this method suggests viability for clinical PET radiochemistry.
Positron emission tomography (PET) is a non-invasive imaging technique that enables the in vivo imaging of biological processes to probe disease states. The production of PET radiotracers involves the incorporation of unnatural β+-emitting isotopes into a molecule of interest. Frequently, small molecule PET tracers are labelled with 11C or 18F given these isotopes’ simple production on commercial cyclotrons and the prevalence of carbon and fluorine in drug-like molecules.
11C-Methylating reagents like [11C]CH3I and [11C]CH3OTf have found widespread use in clinical PET tracer production as means to radiolabel nucleophilic functionalities through displacement (O, N, S) and cross-coupling reactions.1 Despite the commonality and operational simplicity of 11C-methylations, [11C]CH3X synthons otherwise generate limited chemical diversity. To maximize the utility and chemical diversity available from [11C]CH3X reagents, we sought novel methods for their incorporation into chemical scaffolds.
We aimed to utilize [11C]CH3I or [11C]CH3OTf with sulfide nucleophiles to form radiolabelled sulfonium salts and generate their sulfur ylides for novel radiolabelling, similar to previously explored 11C-phosphonium salts.2 Sulfur ylides are versatile reagents in cycloadditions, cross couplings, rearrangements, annulations, and carbene precursors.3–6
Given the prevalence of epoxides in drug molecules (Scheme 1A), we were initially interested in whether transformation of a 11C-sulfonium salt to the corresponding ylide, in the presence of a suitable carbonyl compound, would enable a Johnson–Corey–Chaykovsky-type reaction to form 11C-epoxides (Scheme 1B) as ubiquitous intermediates for synthesis and bioactive motifs. Further, we aimed to extend the use of these non-stabilised ylides to imines and activated alkenes to label medicinally valuable aziridines and cyclopropanes.7,8 We believed that a method for the generation of the 11C-isotopologues of these functionalities would serve as a valuable tool for radiochemistry, particularly in cases where 11C-methylation of a target molecule may not be feasible.
 | ||
| Scheme 1 (A) Bioactive compounds featuring epoxide and aziridine motifs. (B) Formation of 11C-epoxides via sulfonium salts. | ||
The trimethyl sulfonium (Me3S+I−) and sulfoxonium iodide (Me3SO+I−) reagents typically employed in analogous cycloadditions provide no selectivity in the group transferred. Correspondingly, the generation of the equivalent 11C-reagent would result in undesirable isotopic dilution and low molar activities of the desired 11C-products. This, combined with the short half-life of 11C (t1/2 = 20.4 min), motivated an alternative approach for translation to radiochemistry. To ensure the exclusive transfer of the 11C–CH3 group, we attempted to synthesise the biaryl sulfonium salt 2a (Scheme 2). We began by exposing sulfide 1a to [11C]CH3OTf at ambient temperature, however this produced multiple products and was not viable in an attempted 11C-epoxide formation. We reasoned that the high reactivity of pure [11C]CH3OTf dissuades direct product formation by promoting favourable side reactions including solvolysis from residual H2O and potential reactivity with MeCN leading to further reactive intermediate formation.9
 | ||
| Scheme 2 [11C]MeOTf and [11C]MeI in formation of sulfonium salt 2a. | ||
We believed that the introduction of AgOTf could aid in product formation through the slow generation of [11C]CH3OTf from less reactive [11C]CH3I. Screening conditions with [11C]CH3I in the presence of AgOTf and 1a successfully produced the desired sulfonium salt at 60 °C over 5 minutes in PhCl. Application of the formed sulfonium salt 2a to the synthesis of 4a produced the desired epoxide in good radiochemical conversion (RCC) when using KOtBu at 120 °C over 5 minutes.
We next examined the effect the sulfonium salt and its respective ylide species had on epoxidation (Table 1). Exposing sulfides 1a–1e to identical conditions in the formation of 2a, we compared their conversion to 4a. While all the sulfides formed their sulfonium salts equally effectively by rTLC, they differed significantly in conversion to 4a. The biaryl sulfides 1a–1c (Table 1, entries 1–3) showed a preference for electron dense aryl substituents with 2b performing most effectively, while tricyclic sulfides 1d and 1e (Table 1, entries 4 and 5) were substantially less effective in the formation of 4a. In all, this suggests that the rigidity and electron density of the operant sulfur ylide influence the nucleophilicity toward attack on the carbonyl of 3a.
|
|
|||
|---|---|---|---|
| Entry | Sulfide | 4a (rTLC) (%) | 4a (rHPLC) (%) |
| 1 | 1a | 93 | 75 |
| 2 | 1b | 92 | 86 |
| 3 | 1c | 87 | 60 |
| 4 | 1d | 77 | 42 |
| 5 | 1e | 65 | 21 |

|
|||
Due to the electron rich nature of the most successful sulfide 1b, we investigated the synthesis of its respective sulfonium salt 2b under milder conditions (Table 2). While this revealed that the AgOTf loading could be modestly reduced without detriment, attempts to form 2b in the absence of AgOTf, even at 120 °C, were unsuccessful. Furthermore, MeCN was not a compatible solvent for 2b formation.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | T (°C) | AgOTf | Time | 2b (rHPLC) |
| 1 | MeCN | 80 | 1.0 eq. | 15 min | Trace |
| 2 | PhCl | 120 | 0 eq. | 5 min | Trace |
| 3 | PhCl | 60 | 1.0 eq. | 5 min | 91% |
| 4 | PhCl | rt | 1.0 eq. | 5 min | 25% |
| 5 | PhCl | 60 | 0.75 eq. | 5 min | 89% |
| 6 | PhCl | 60 | 0.5 eq. | 5 min | 56% |
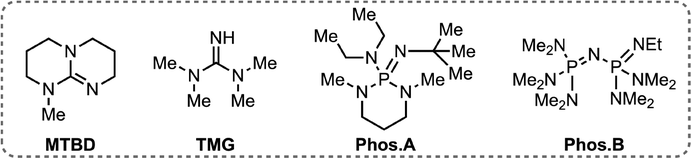
We proceeded to explore the range of bases that could be employed alongside 2b to form the operant ylide species (Table 3). The pKa of 2b is unknown, though similar sulfonium species display pKas 16–18, prompting us to explore strong organic and inorganic bases.10 KOtBu provided improved RCC to 4a over KOH, and comparable results at lower equivalency (Table 3, entries 1–3). Reducing the temperature of the reaction to 80 °C gave a similar result to 120 °C for 4a (Table 3, entry 4), however significantly reduced RCCs were noticed with substrates 4b and 4g at 80 °C (vide infra). Phosphazene bases successfully promoted the reaction with P2-Et similarly effective to KOH though alternative organic bases were ineffective (Table 3, entries 5–8) perhaps on account of their hygroscopicity.
Applying the optimized protocol to a range of substrates featuring aldehydes and ketones, we discovered that those featuring electron withdrawing or neutral substituents performed well (4b, 4c, 4e, 4g, 4h) with aldehyde substrates requiring only 1 equivalent of KOtBu to give high conversions (Fig. 1). 4d unfortunately only gave complex mixtures which can be attributed to the known instability of electron-rich epoxides and their propensity to form polymers.11 Heterocyclic examples 4i and 4j were formed in modest RCC, as were spirocyclic epoxides 4k and 4l. Further, example 4f and its precursor 3f feature a competing acidic benzylic site which was tolerated in low RCCs.
 | ||
Fig. 1 Conditions-[11C]CH3I (in PhCl) added to 1b (20 µmol) and AgOTf (1 eq.) (300 µL PhCl), heated to 60 °C for 5 min to form 2b; 200 µL of 2b solution added to vial containing base, treated with 3a–o (20 µmol, 200 µL MeCN) and stirred at 120 °C for 5 min before filtering and rHPLC analysis. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Reaction performed at 100 °C. Reaction performed at 100 °C. | ||
Substrates 4b and 4g feature ester groups which may be subject to transesterification in the presence of potassium tert-butoxide. This prompted us to change to the non-nucleophilic P2-Et as an alternative, affording both epoxides in good RCCs.
To complement the synthesis of 11C-epoxides, we aimed to expand this protocol to other valuable functionalities accessible from sulfur ylides including aziridines and cyclopropanes. Electron rich aziridine 4m was challenging with only low RCCs achieved regardless of temperature or base employed, however N-Ts aziridine 4n could be formed in fair RCCs albeit with notable variability.
Extension of the protocol to activated alkenes was also feasible, with 4o formed successfully in moderate RCC. Notably, the trifluoromethyl substituted cyclopropane present in 4o can be employed as a replacement for metabolically labile tert-butyl groups in medicinal compounds.8
Further, we wanted to examine this method in the context of biologically active, complex substrates as a feasible means to clinically relevant 11C-products. The successful synthesis of 4o prompted us to attempt the synthesis of the T-type calcium channel blocker [11C]apinocaltamide (ACT-709478) 4p using alkene precursor 3p which was accessed in 5 steps from commercially available starting materials in high yields (Fig. 2A). Subsequently, we managed to successfully form 4p in modest RCC (Fig. 2B), following our standard conditions with 2 equivalents of KOtBu.
 | ||
| Fig. 2 (A) Synthesis of precursor 3p. (B) Radiosynthesis of [11C]Apinocaltamide 4p. | ||
Apart from being privileged motifs in select bioactive molecules, epoxides can be versatile intermediates for further chemical differentiation.12 To illustrate this, we attempted a ring-opening hydrolysis of 4a (Fig. 3). Transformation of epoxide 4a to diol 5 was achieved by introducing H2O and TFA after completion of the epoxide formation. This transformation confirms the presence of epoxide 4a as an intermediate product and provides a novel route to radiolabelled diols.
 | ||
| Fig. 3 Radiolabelling of diol 5 from hydrolysis of epoxide 4a. | ||
Using [11C]CH3OTf in the formation of our sulfonium salts would simplify the reaction protocol and avoid the addition of silver. We therefore reinvestigated the use of [11C]CH3OTf with aliphatic cyclic sulfide 1f in effort to prevent any possible side reactions (Scheme 3). We first subjected 1f to [11C]CH3I using our standard conditions, successfully forming 2f and proceeding to form epoxide 4b in modest RCC. Unfortunately, while the analogous formation of 2f using [11C]CH3OTf appeared successful by rTLC, rHPLC revealed a much broader peak than expected for 2f and, although subsequent formation of epoxide 4b was possible, the RCC was very low. The reasons for differing results when using [11C]CH3OTf and [11C]CH3I are likely a result of intractable byproduct formation from highly reactive [11C]CH3OTf the identity of which remain unclear and need to be further investigated in the future.9
 | ||
| Scheme 3 Reactivity of sulfide 2f formed using [11C]CH3I or [11C]CH3OTf. | ||
Finally, we attempted an automated procedure using higher activities of [11C]CH3I (>500 mCi) adapted from the manual conditions. Automation of the method provided a set of challenges relative to the manual reactions. The formation 2b from [11C]CH3I proceeded successfully, however it was not fully consumed in the second step unless the amount of P2-Et base was increased relative to the manual reactions with 3b. Additionally, the subsequent step at 120 °C produced a new major signal by rHPLC, which had been observed only as a minor byproduct previously, with only trace 4b observed. Lowering the temperature of the reaction allowed us to favour epoxide formation over byproduct, suggesting both significant differences between the manual and automated heating efficiencies and the thermal instability of 4b. Nevertheless, with modified conditions, we successfully isolated 22 mCi of 4b in 99% radiochemical purity from 600 mCi of [11C]CH3I (3.65% non-decay corrected activity yield) in 19 minutes with a molar activity >710 mCi µmol−1 (n = 2). Analysis of the crude reactions showed only a modest decrease in RCC relative to the manual reactions (59%).
In summary, we have produced a range of radiolabelled sulfonium salts, formed their respective sulfur ylides, and explored their reactivity under conditions amenable to 11C-radiolabelling. Synthesis of simple and complex (hetero)aryl epoxides from aldehydes and ketones has been explored and extension of the protocol to the formation of aziridines and trifluoromethyl substituted cyclopropanes was also accomplished. The process can be automated using a radiochemistry synthesis module and is amenable for the 11C-labelling of biologically active, complex substrates that are relevant for clinical PET imaging applications (Scheme 4).
 | ||
| Scheme 4 Automated radiosynthesis of 4b from aldehyde 3b. | ||
Financial support of this work from NIH (R01EB021155 to PJHS) is gratefully acknowledged. The authors also thank Prof. Melanie Sanford and her lab, as well as Eric Webb, Gregory Bowden, Jay Wright and Allen Brooks for helpful discussions.
Conflicts of interest
The authors declare no conflicts of interest.Data availability
The data supporting this article are all available in the supplementary information (SI). Supplementary information: experimental/synthetic protocols, characterization data, radioTLC and HPLC traces. See DOI: https://doi.org/10.1039/d5cc05951h.Notes and references
- (a) M. Müller, V. Shalgunov, L. Hvass, J. T. Jørgensen, V. Kramer, M. Staudt, U. M. Battisti, A. Kjaer and M. M. Herth, Bioorg. Med. Chem. Lett., 2023, 80, 129088 CrossRef PubMed; (b) K. Bamminger, V. Pichler, C. Vraka, T. Limberger, B. Moneva, K. Pallitsch, B. Lieder, A. S. Zacher, S. Ponti, K. Benčurová, J. Yang, S. Högler, P. Kodajova, L. Kenner, M. Hacker and W. Wadsak, J. Med. Chem., 2024, 67, 4036–4062 CrossRef CAS PubMed; (c) M. Honer, A. Polara, H. Kuwabara, H. Jacobsen, A. Pähler, T. Hartung, A. Caruso, D. Esterhazy, M. Stoffel, R. F. Dannals, D. F. Wong, E. Borroni and L. C. Gobbi, J. Labelled Compd. Radiopharm., 2023, 66, 222–236 CrossRef CAS PubMed; (d) Y. Xu, Y. Xu, S. Biby, B. Kaur, Y. Liu, F. A. Bagdasarian, H.-Y. Wey, R. Tanzi, C. Zhang, C. Wang and S. Zhang, J. Med. Chem., 2024, 67, 555–571 CrossRef CAS PubMed; (e) T. Luo, N. Sang, Y. Liu, Y. Zhou, R. Wu, F. A. Bagdasarian, H.-Y. Wey, J. Lang, C. Wang and P. Bai, Bioorg. Chem., 2024, 146, 107279 CrossRef CAS PubMed.
- (a) T. Kihlberg, P. Gullberg and B. Långström, J. Labelled Compd. Radiopharm., 1990, 28, 1115–1120 CrossRef CAS; (b) M. Björkman and B. Långström, J. Chem. Soc., Perkin Trans. 1, 2000, 3031–3034 RSC.
- (a) T.-B. Hua, Y.-H. Ma, X.-Y. He, L. Wang, J.-Y. Yan and Q.-Q. Yang, Org. Chem. Front., 2022, 9, 3558–3564 RSC; (b) Q.-Q. Cheng, L. A. Massey, B. S. Willett, Y. Deng, H. Arman and M. P. Doyle, Angew. Chem., Int. Ed., 2018, 57, 10343–10346 CrossRef CAS PubMed; (c) X. Wang, J. Yu, M. Xu, H. Mao, Y. Shan, X. Lv and L. Zhou, Org. Lett., 2022, 24, 5896–5901 CrossRef CAS PubMed; (d) C. Li, K. Jiang, Q. Ouyang, T.-Y. Liu and Y.-C. Chen, Org. Lett., 2016, 18, 2738–2741 CrossRef CAS PubMed.
- (a) J. Vaitla, K. H. Hopmann and A. Bayer, Org. Lett., 2017, 19, 6688–6691 CrossRef CAS PubMed; (b) J. D. Neuhaus, R. Oost, J. Merad and N. Maulide, Top. Curr. Chem., 2018, 376, 15 CrossRef PubMed.
- (a) C. A. D. Caiuby, M. P. de Jesus and A. C. B. Burtoloso, J. Org. Chem., 2020, 85, 7433–7445 CrossRef CAS PubMed; (b) A. C. B. Burtoloso, R. M. P. Dias and I. A. Leonarczyk, EurJOC, 2013, 2013, 5005–5016 CAS.
- (a) D. Kaiser, I. Klose, R. Oost, J. Neuhaus and N. Maulide, Chem. Rev., 2019, 119, 8701–8780 CrossRef CAS PubMed; (b) D. Antoniak and M. Barbasiewicz, Org. Lett., 2022, 24, 516–519 CrossRef CAS PubMed; (c) C. A. D. Caiuby, L. G. Furniel and A. C. B. Burtoloso, Chem. Sci., 2022, 13, 1192–1209 RSC.
- (a) H. J. Dequina, C. L. Jones and J. M. Schomaker, Chem, 2023, 9, 1658–1701 CrossRef CAS PubMed; (b) F. M. D. Ismail, D. O. Levitsky and V. M. Dembitsky, Eur. J. Med. Chem., 2009, 44, 3373–3387 CrossRef CAS PubMed.
- (a) J. Decaens, S. Couve-Bonnaire, A. B. Charette, T. Poisson and P. Jubault, Chem. – Eur. J., 2021, 27, 2935–2962 CrossRef CAS PubMed; (b) M. Bos, T. Poisson, X. Pannecoucke, A. B. Charette and P. Jubault, Chem. – Eur. J., 2017, 23, 4950–4961 CrossRef CAS PubMed; (c) D. Barnes-Seeman, M. Jain, L. Bell, S. Ferreira, S. Cohen, X.-H. Chen, J. Amin, B. Snodgrass and P. Hatsis, ACS Med. Chem. Lett., 2013, 4, 514–516 CrossRef CAS PubMed.
- (a) R. G. Giles, H. Heaney and M. J. Plater, Tetrahedron, 2015, 71, 7367–7385 CrossRef CAS; (b) D. N. Kevill and S. W. Anderson, J. Org. Chem., 1986, 51, 5029–5032 CrossRef CAS; (c) B. L. Booth, K. O. Jibodu and M. F. J. R. P. Proença, J. Chem. Soc., Perkin Trans. 1, 1983, 1067–1073 RSC.
- J.-P. Cheng, B. Liu and X.-M. Zhang, J. Org. Chem., 1998, 63, 7574–7575 CrossRef CAS.
- (a) A.-L. Brocas, C. Mantzaridis, D. Tunc and S. Carlotti, Prog. Polym. Sci., 2013, 38, 845–873 CrossRef CAS; (b) J. Park, A. Kim and B.-S. Kim, Nat. Commun., 2023, 14, 5855 CrossRef CAS PubMed; (c) V. Franzen and H.-E. Driesen, Chem. Ber., 1963, 96, 1881–1890 CrossRef CAS.
- (a) S. Meninno and A. Lattanzi, ACS Org. Inorg. Au, 2022, 2, 289–305 CrossRef CAS PubMed; (b) A. R. da Silva, D. A. dos Santos, M. W. Paixão and A. G. Corrêa, Molecules, 2019, 24, 630 CrossRef PubMed; (c) B. H. Rotstein, S. Zaretsky, V. Rai and A. K. Yudin, Chem. Rev., 2014, 114, 8323–8359 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |