
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLigand exchange of tris(2,4-di-tert-butylphenyl) phosphite: a practical and efficient route to organo gold(I) complexes
David
Vesseur
*a,
Shuo
Li
a,
Sonia
Mallet-Ladeira
b and
Didier
Bourissou
 *a
*a
aUniversité de Toulouse, CNRS, Laboratoire Hétérochimie Fondamentale et Appliquée (LHFA, UMR 5069), 118 Route de Narbonne, 31062 Cedex 09 Toulouse, France. E-mail: d.vesseur@uva.nl; didier.bourissou@utoulouse.fr
bUniversité de Toulouse, CNRS, Institut de Chimie de Toulouse (UAR 2599), 118 Route de Narbonne, 31062 Cedex 09 Toulouse, France
First published on 24th October 2025
Abstract
Tris(2,4-di-tert-butylphenyl)phosphite (2,6-tBu2-PhO)3P is used as a stable, easily displaceable ancillary ligand in the preparation of organo Au(I) complexes. The phosphite gold(I) precursors are bench-stable and readily accessible. They undergo high-yielding ligand exchange with a variety of ligands such as phosphines, N-heterocyclic carbenes and bidentate ligands. This ligand exchange strategy overcomes the limitations associated with traditional gold halide-based synthetic routes and is compatible with different organogold fragments. The protocol is scalable, operationally simple and broadly applicable.
Until the 21st century, gold was widely regarded as catalytically inert and was largely treated as a chemical curiosity. This perception changed in 1998 when Teles et al. demonstrated that Au(I) phosphine complexes could efficiently catalyze the addition of alcohols to alkynes under mild conditions, achieving turnover numbers up to 105.1 Over the following two and a half decades, gold complexes have emerged as exceptionally powerful catalysts, capable of promoting transformations that are often otherwise inaccessible.2 Early developments in the field primarily employed gold complexes bearing simple ligands. However, as the transformations became more sophisticated, so did the ancillary ligands. For many advanced transformations and fundamental studies, the use of more elaborated ligands has become essential. While these ligands enable novel, valuable, and often unprecedented reactivity, the synthesis of the corresponding organogold complexes can present significant challenges.
We recently reported that bidentate ligands can be used to stabilize Au(I) difluorocarbene complexes, which undergo [2+2] and [2+4] cycloadditions with alkenes and 1,3-dienes.3 These Au(I) difluorocarbene complexes were generated via fluoride abstraction from the corresponding Au(I) trifluoromethyl precursors. However, the preparation of these bidentate Au(I) trifluoromethyl complexes in high purity was challenging when using existing protocols for the preparation of Au(I) trifluoromethyl complexes.4,5 Additionally, we were unable to find an effective method for their purification.
By far, the most widely used method for the preparation of organo Au(I) complexes involves a “ligand exchange first” (Fig. 1A). Weakly bound sulphide ligands (e.g., Me2S, THT) are first displaced from gold(I) halide precursors by more strongly coordinating ligands. This exchange is highly favorable due to the low bond dissociation energy (BDE) of the Au–S bond.6 The resulting Au(I) halide complex is then subjected to functionalization conditions to install the desired organic group. However, this sequence can pose challenges when the target ligand or complex is unstable under the conditions required for functionalization. An alternative approach is to functionalize the gold center first and perform ligand exchange subsequently (Fig. 1B).7 This method, though conceptually attractive, is limited by the instability of organogold sulphide intermediates, which are often prone to decomposition and cannot be readily isolated or stored—thereby restricting the scope of accessible organogold species. To overcome these limitations, it is desirable to employ a ligand that forms stable Au(I) complexes while still featuring a sufficiently low BDE to allow facile substitution. Although a few isolated examples of such ligand exchange at Au(I) have been reported (Scheme 1),8 a systematic investigation aimed at developing a general synthetic protocol remains lacking.
 | ||
| Fig. 1 (A) Classical approach for the synthesis of Au(I) complexes. (B) Functionalization of (R2S)Au(I) complexes before ligand exchange. (C) Functionalization of [(RO)3P]AuCl followed by ligand exchange. | ||
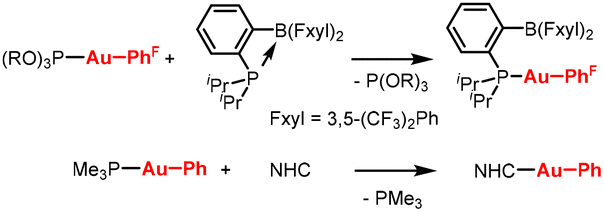 | ||
| Scheme 1 Examples of ligand exchange with phosphite and phosphine ligands. R = 2,4-tBu2-Ph, PhF = 4-(CF3)Ph. | ||
From the few reported examples of ligand exchange, we envisioned that Au(I) complexes featuring the (2,4-tBu2-PhO)3P ancillary ligand could serve as excellent and general organogold sources. While Au(I) phosphite complexes tend to be moderately stable, this is largely offset in the case of (2,6-tBu2-PhO)3P, where the steric bulk contributes to improved stability. Owing to its poor donor properties, this phosphite forms relatively weak Au–P bonds,6 and it is thus expected to be easily displaced by other ligands. Furthermore, this ligand is inexpensive, commercially available, air-stable, and it forms bench-stable Au(I) complexes. Notably, it is also highly soluble in aliphatic solvents, in contrast to most organogold complexes, which greatly facilitates purification after ligand exchange.
Herein, we report a general strategy for the synthesis of organo gold(I) complexes using (2,4-tBu2-PhO)3P as an ancillary ligand that allows for facile ligand exchange (Fig. 1C). [(2,4-tBu2-PhO)3P]AuR complexes are easily prepared and can be stored for prolonged periods. Due to the relatively low BDE of the phosphite and Au(I) centre, the ligand is rapidly and quantitatively displaced by phosphine or carbene ligands, enabling efficient access to a wide variety of Au(I) complexes bearing diverse ligands and functional groups. The displaced phosphite can be readily removed, further simplifying product isolation and purification.
Given our interest in (L)Au(I)CF3 complexes,3 we started our investigation with the phosphite complex (2,4-tBu2-PhO)3PAuCF31, which is readily accessible by reacting (2,6-tBu2-PhO)3PAuCl with AgF and TMSCF3.4b,9 Upon mixing complex 1 with PPh3 in dichloromethane (DCM), ligand exchange was monitored by the disappearance of complex 1 and the appearance of free (2,4-tBu2-PhO)3P in the 31P NMR spectrum. After 30 minutes, complete conversion to the desired product 2a was confirmed by both 31P and 19F NMR spectroscopy (Scheme 2). Complex 2a was isolated in 92% yield by simple washing of the crude mixture with pentane. The displaced phosphite ligand could also be recovered if desired.9 Using the more electron-rich PCy3 ligand, similar results were obtained (2b, 89%). Biaryl ligands, which are often used in Au(I) catalysis and mechanistic studies, also worked very well, delivering complexes 2c–2g in 78–92% yields. Expanding beyond phosphorus-based ligands, a NHC ligand was tested. SIPr proved compatible, yielding complex 2h in 91% isolated yield. Having demonstrated the generality of this approach with monodentate ligands, we then turned our attention to more elaborated ligands where traditional synthetic methods can pose problems. We first investigated phosphine-amine (P^N) ligands, which have become a staple of Au(I)/Au(III) redox chemistry due to their ability to stabilise both oxidation states and facilitate redox cycling.10 Both tert-butyl- and adamantyl-substituted P^N ligands reacted smoothly with complex 1, affording 2i and 2j in 86% and 98% yield, respectively. A phosphine-sulphide ligand11 also performed well, providing 2k in 85% yield. We then explored a phosphine–borane ligand containing a Lewis acidic boron center, known for its rich and unusual coordination properties.12 Previous studies have shown that functionalization of Au(I) complexes bearing such ambiphilic ligands is challenging due to the sensitivity of the borane moiety.7b Using the ligand exchange strategy, the desired complex 2l was readily obtained in 89% yield. Finally, we examined bidentate diphosphine ligands. Mononuclear diphosphine–Au(I) complexes are rarely observed due to the strong preference of gold(I) for linear geometries, often resulting instead in digold species featuring aurophilic Au⋯Au interactions,13 Nevertheless, such monometallic diphosphine complexes have attracted interest for their ability to enhance backdonation from gold, particularly with o-carboranyl-based ligands.3,14,15 Using 1,2-bis(diaminophosphino)-1,2-dicarba-closododecaborane, we successfully obtained complex 2m in 82% yield and high purity. Single-crystal X-ray diffraction (scXRD) analysis showed coordination of a single phosphine donor to the Au(I) center, while NMR spectroscopy indicated rapid exchange in solution. Excitingly, when 1,2-bis(diphenylphosphino)benzene was used, the monometallic complex 2n was obtained with an excellent yield (95%). In the solid state, 2n adopts a T-shape (not a Y-shape) with dissymmetric coordination of the two phosphorus atoms. Possibly, the bulkiness of the (2,6-tBu2-PhO)3P prevents the ligation of a second gold centre. This suggests that the (2,6-tBu2-PhO)3P ligand may enable access to other mononuclear Au(I) complexes with bidentate ligands, which would otherwise be inaccessible with alternative gold sources.
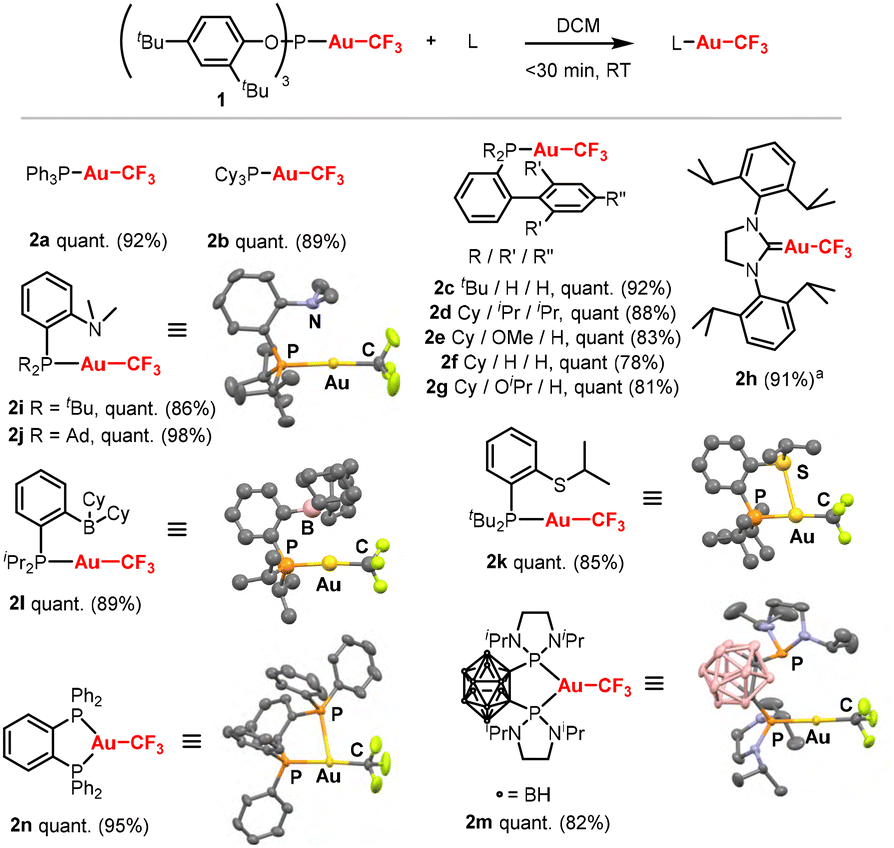 | ||
Scheme 2 Scope of the ligand exchange with (2,4-tBu2-PhO)3PAuCF3. Reaction conditions: (2,4-tBu2-PhO)3PAuCF3 (0.05 mmol, 1.0 eq.), L (0.05 mmol, 1.0 eq.), DCM (0.05 M), rt, <30 minutes. Quantitative conversion determined by 31P NMR spectroscopy, isolated yields in brackets. Molecular structure of 2i, 2k, 2l, 2m and 2n with the hydrogen atoms omitted for clarity. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) Reaction performed in toluene (0.05 M). Reaction performed in toluene (0.05 M). | ||
To demonstrate that the reaction is not limited to trifluoromethyl Au(I) complexes, other organic groups were investigated (Scheme 3). To show the generality of the reaction, five different ligands were tested: the simple phosphines PPh3 and PCy3, JohnPhos, SIPr as a C-based ligand and the o-carboranyl diphosphine as a bidentate ligand. First, the ligand exchange was extended to Au(I) equipped with a pentafluoroethyl group which are underdeveloped compared to their trifluoromethyl analogues. In all cases, the reaction proceeded smoothly, affording complexes 3a–3e in high isolated yields (72–95%). To exemplify further the utility of our ligand exchange methodology, the difluoromethyl substituent was also investigated. A previous study found that while Au(I) difluoromethyl complexes can be synthesized via alternative methods, isolation was often hampered by decomposition during purification.16 The (2,4-tBu2-PhO)3PAuCHF2 precursor itself could be prepared and easily extracted from the reaction mixture with pentane due to its high solubility in aliphatic solvents. Ligand exchange proceeded quantitatively, furnishing the targeted (L)Au(I)CHF2 complexes 4a–4d, which were easily purified by simple pentane washes.9 Then, organic groups without fluorides were tested to demonstrate that the ligand exchange approach goes beyond electron-deficient substituents. The reaction worked well for both methyl-substituted complexes 5a–5e (67–98% yields) and phenyl-substituted complexes 6a–6e (38–95% yields) (Scheme 3).17
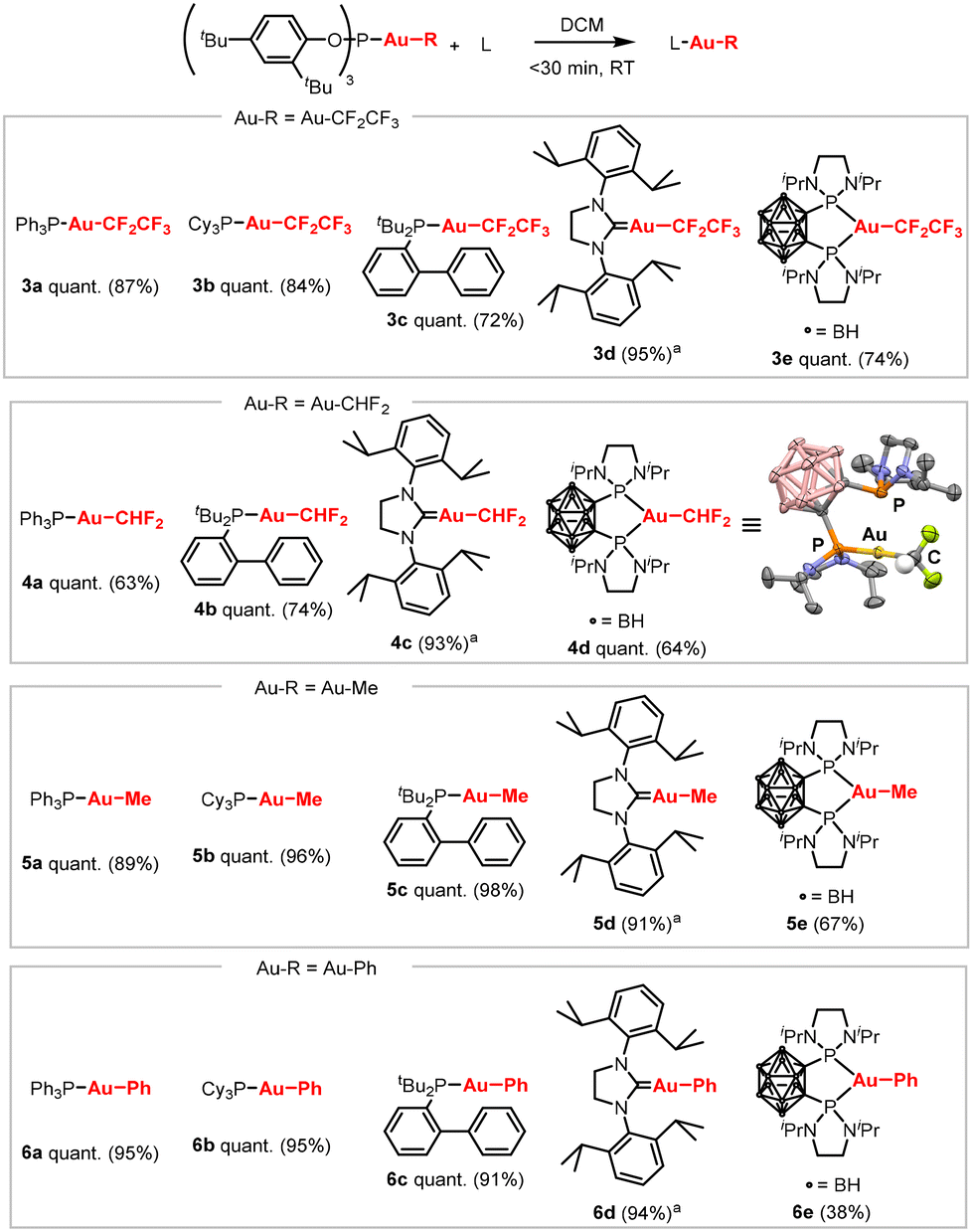 | ||
Scheme 3 Scope of the ligand exchange of (2,4-tBu2-PhO)3PAuR with selected ligands triphenyl phosphine (PPh3), tricyclohexyl phosphine (PCy3), (2-biphenyl)di-tert-butylphosphine (JohnPhos), 1,3-bis(2,6- diisopropylphenyl)-4,5-dihydroimidazol-2-ylidene (SIPr), and o-carborane diphosphine (P^P). Reaction conditions: (2,4-tBu2-PhO)3PAuR (0.05 mmol, 1.0 eq.), L (0.05 mmol, 1.0 eq.), DCM (0.05 M), rt, <30 minutes. Quantitative conversion determined by 31P NMR spectroscopy, isolated yields in brackets. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Reaction performed in toluene (0.05 M). Reaction performed in toluene (0.05 M). | ||
All reactions were carried out in dichloromethane, a solvent in which organo Au(I) complexes are typically stable and highly soluble. It might however be desirable to use alternative solvents. Therefore, the ligand exchange methodology was tested in a range of different solvents, with different polarities and potential coordinating abilities (Scheme 4). In all cases, quantitative conversion was achieved within 30 minutes, as determined by 31P NMR spectroscopy. Lastly, the reaction was successfully scaled up to 1 mmol (0.7 g) scale, yielding the desired complex in 96% yield. These results underscore the robustness and synthetic practicality of the ligand exchange methodology.
 | ||
| Scheme 4 Reaction conditions: (2,4-tBu2-PhO)3PAuCF3 (0.05 mmol, 1.0 eq.), 1,2-bis(diaminophosphino)-1,2-dicarba-closododecaborane (0.05 mmol, 1.0 eq.), solvent (0.05 M), rt, <30 minutes. Ligand exchange of (2,4-tBu2-PhO)3PAuCF3 with the diphosphine carborane ligand in different solvents. Quantitative conversion determined by 31P NMR spectroscopy. Reaction conditions for 1.0 mmol scale: (2,4-tBu2-PhO)3PAuCF3 (1.0 mmol, 1.0 eq.), P^P ligand (1.0 mmol, 1.0 eq.), DCM (1.0 M), rt, <30 minutes. | ||
In conclusion, Au(I) complexes bearing the labile phosphite ligand (2,4-tBu2-PhO)3P have proven to be excellent precursors for the synthesis of organo Au(I) complexes via a straightforward and easy-to-operate ligand exchange protocol. This “ligand exchange last” strategy offers several advantages over the more commonly employed “ligand exchange first” approach, benefiting from the air-stability, low cost and ease of removal of the phosphite (2,4-tBu2-PhO)3P. A broad scope of ligands and organic groups was explored, which afforded the desired Au(I) complexes in high yield and purity. Overall, this ligand exchange approach provides a practical and efficient route to organo gold(I) complexes which would otherwise be difficult to prepare using known synthetic methods.
Financial support from the Centre National de la Recherche Scientifique, the Université de Toulouse, the Agence Nationale de la Recherche (ANR-19-CE07-0037), and the Chinese Scholarship Council (PhD fellowship to S. L.) is gratefully acknowledged. The NMR service of ICT, UAR 2599 (Pierre Lavedan), is acknowledged for assistance with the variabletemperature NMR experiments.
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: details of the experimental methods, NMR spectra and X-ray analyses. See DOI: https://doi.org/10.1039/d5cc05744b.CCDC 2482843 (for 2i), 2482844 (for 2k), 2482845 (for 2l), 2482839 (for 2m), 2492890 (for 2n) and 2482840 (for 4d) contain the crystallographic data.18a–f
Notes and references
- J. H. Teles, S. Brode and M. Chabanas, Angew. Chem., Int. Ed., 1998, 37, 1415–1418 CrossRef CAS PubMed.
- For some reviews, see: (a) A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896–7936 CrossRef PubMed; (b) A. S. K. Hashmi and M. Rudolph, Chem. Soc. Rev., 2008, 37, 1766–1775 RSC; (c) M. Rudolph and A. S. K. Hashmi, Chem. Rev., 2012, 41, 2448–2462 CAS; (d) R. Ciriminna, E. Falletta, C. Della Pina, J. H. Teles and M. Pagliaro, Angew. Chem., Int. Ed., 2016, 55, 14210–14217 Search PubMed; (e) S. A. Shahzad, M. A. Sajid, Z. A. Khan and D. Canseco-Gonzalez, Synth. Commun., 2017, 47, 735–755 Search PubMed; (f) S. Witzel, A. S. K. Hashmi and J. Xie, Chem. Rev., 2021, 121, 8868–8925 CrossRef CAS PubMed; (g) W. Wang, C.-L. Ji, K. Liu, C.-G. Zhao, W. Li and J. Xie, Chem. Soc. Rev., 2021, 50, 1874–1912 RSC.
- D. Vesseur, S. Li, N. Albouy, P. Lavedan, S. Mallet-Ladeira, K. Miqueu and D. Bourissou, Angew. Chem., Int. Ed., 2025, 64, e202515429 Search PubMed.
- (a) M. Blaya, D. Bautista, J. Gil-Rubio and J. Vicente, Organometallics, 2014, 33, 6358–6368 CrossRef CAS; (b) A. G. Tskhovrebov, J. B. Lingnau and A. Fürstner, Angew. Chem., Int. Ed., 2019, 58, 8834–8838 CrossRef CAS PubMed; (c) M. Navarro, L. Delaurenti, A. Pita-Milleiro and J. Campos, JACS Au, 2025, 5, 4604–4610 CrossRef CAS PubMed; (d) Z. R. Wong, B. K. Mai, W. R. Berg, A. Kvitsinski, T. L. F. Correia, Y. Zhang, P. Liu and F. D. Toste, Angew. Chem., Int. Ed., 2025, 64, e202511941 CrossRef CAS PubMed.
- For a review dealing with gold trifluoromethyl complexes, see: J. Gil-Rubio and J. Vicente, Dalton Trans., 2015, 44, 19432–19442 RSC.
- G. C. Fortman and S. P. Nolan, Organometallics, 2010, 29, 4579–4583 CrossRef CAS.
- For selected examples, see: (a) R. Usón, A. Laguna, M. Laguna, B. R. Manzano, P. G. Jones and G. M. Sheldrick, J. Chem. Soc., Dalton Trans., 1985, 11, 2417–2420 RSC; (b) P. Espinet, S. Martín-Barrios, F. Villafañe, P. G. Jones and A. K. Fischer, Organometallics, 2000, 19, 290–295 CrossRef CAS; (c) J. Lloret Fillol, A. Kruckenberg, P. Scherl, H. Wadepohl and L. H. Gade, Chem. – Eur. J., 2011, 17, 14047–14062 CrossRef PubMed; (d) S. Martínez-Salvador, L. R. Falvello, A. Martín and B. Menjón, Chem. – Eur. J., 2013, 19, 14540–14552 CrossRef; (e) L. Gu, Y. Zheng, E. Haldón, R. Goddard, E. Bill, W. Thiel and M. Alcarazo, Angew. Chem., Int. Ed., 2017, 56, 8790–8794 CrossRef CAS PubMed; (f) E. Martín-Encinas, V. Conejo-Rodríguez, J. A. Miguel, J. M. Martínez-Ilarduya, G. Rubiales, B. R. Knudsen, F. Palacios and C. Alonso, Dalton Trans., 2020, 49, 7852–7861 RSC.
- For examples, see: (a) E. Tomas-Mendivil, M. M. Hansmann, C. M. Weinstein, R. Jazzar, M. Melaimi and G. Bertrand, J. Am. Chem. Soc., 2017, 139, 7753–7756 CrossRef CAS; (b) C. A. Theulier, Y. García-Rodeja, S. Mallet-Ladeira, K. Miqueu, G. Bouhadir and D. Bourissou, Organometallics, 2021, 40, 2409–2414 CrossRef CAS; (c) Y. K. Loh, M. Melaimi, D. Munz and G. Bertrand, J. Am. Chem. Soc., 2023, 145, 2064–2069 CrossRef CAS PubMed.
- See SI for details.
- (a) A. Zeineddine, L. Estévez, S. Mallet-Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, Nat. Commun., 2017, 8, 565 CrossRef PubMed; (b) B. Huang, M. Hu and F. D. Toste, Trends Chem., 2020, 2, 707–720 CrossRef CAS PubMed; (c) P. Font, H. Valdés and X. Ribas, Angew. Chem., Int. Ed., 2024, 63, e202405824 CrossRef CAS.
- Y. Luo, K. Ji, Y. Li and L. Zhang, J. Am. Chem. Soc., 2012, 134, 17412–17415 CrossRef CAS.
- (a) S. Bontemps, G. Bouhadir, K. Miqueu and D. Bourissou, J. Am. Chem. Soc., 2006, 128, 12056–12057 CrossRef CAS; (b) G. Bouhadir and D. Bourissou, Chem. Soc. Rev., 2016, 45, 1065–1079 RSC.
- (a) M. C. Gimeno and M. A. Laguna, Chem. Rev., 1997, 97, 511–522 CrossRef CAS; (b) M. A. Carvajal, J. J. Novoa and S. Alvarez, J. Am. Chem. Soc., 2004, 126, 1465–1477 CrossRef CAS PubMed; (c) H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2012, 41, 370–412 RSC.
- (a) M. Joost, L. Estévez, S. Mallet-Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2014, 53, 14512–14516 CrossRef CAS PubMed; (b) A. Zeineddine, F. Rekhroukh, E. D. Sosa Carrizo, S. Mallet-Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2018, 57, 1306–1310 CrossRef CAS PubMed; (c) M. Rigoulet, D. Vesseur, K. Miqueu and D. Bourissou, Angew. Chem., Int. Ed., 2022, 61, e202204781 CrossRef CAS PubMed; (d) D. Vesseur, K. Miqueu and D. Bourissou, Chem. Commun., 2023, 59, 5387–5390 RSC.
- O. Crespo, M. C. Gimeno, A. Laguna and P. G. Jones, J. Chem. Soc., Dalton Trans., 1992, 1601–1605 RSC.
- P. García-Domínguez, Organometallics, 2021, 40, 2923–2928 CrossRef.
- Complexes 5a–5c, 5e, and 6e were found to be highly soluble in pentane. Therefore, the purification method was modified from a pentane wash to extraction with MeCN.
- (a) CCDC 2482839: Experimental Crystal Structure Determination, 2025 DOI:10.5517/ccdc.csd.cc2pbll7; (b) CCDC 2482840: Experimental Crystal Structure Determination, 2025 DOI:10.5517/ccdc.csd.cc2pblm8; (c) CCDC 2482843: Experimental Crystal Structure Determination, 2025 DOI:10.5517/ccdc.csd.cc2pblqc; (d) CCDC 2482844: Experimental Crystal Structure Determination, 2025 DOI:10.5517/ccdc.csd.cc2pblrd; (e) CCDC 2482845: Experimental Crystal Structure Determination, 2025 DOI:10.5517/ccdc.csd.cc2pblsf; (f) CCDC 2492890: Experimental Crystal Structure Determination, 2025 DOI:10.5517/ccdc.csd.cc2pbn7y.
| This journal is © The Royal Society of Chemistry 2025 |