
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLight-driven rheological modulation of solventless polymer networks enabled by precisely architected flexible star polysiloxanes
Satoshi
Honda
 *ab,
Yi
Zhang
a and
Masaru
Tanaka
a
*ab,
Yi
Zhang
a and
Masaru
Tanaka
a
aInstitute for Materials Chemistry and Engineering, Kyushu University, 744 Motooka, Nishi-ku, Fukuoka 819-0385, Japan. E-mail: shonda@ms.ifoc.kyushu-u.ac.jp
bGraduate School of Arts and Sciences, The University of Tokyo, 3-8-1 Komaba, Meguro-ku, Tokyo 153-8902, Japan
First published on 27th October 2025
Abstract
We report the synthesis and photoresponsivity of precisely designed liquid star-shaped poly(dimethyl siloxane) (PDMS) with anthracene end groups. Owing to the efficient end-linking of the flexible PDMS arms upon the first UV irradiation (λ = 365 nm, UV365), the storage modulus (G′) increased from 4.7 Pa to 119 kPa (>25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-fold), and the physical state became a rubbery solid. Subsequent irradiation with UV light at a shorter wavelength (λ = 254 nm, UV254) decreased G′ toward the photostationary state, and viscoelasticity modulation was achieved between the two photostationary states under solvent-free conditions.
000-fold), and the physical state became a rubbery solid. Subsequent irradiation with UV light at a shorter wavelength (λ = 254 nm, UV254) decreased G′ toward the photostationary state, and viscoelasticity modulation was achieved between the two photostationary states under solvent-free conditions.
Smart polymers that respond to external stimuli are emerging as versatile materials for a broad spectrum of applications including adhesives, cosmetics, biomedical devices, and soft robotics.1–5 In particular, photoresponsive polymers have garnered considerable attention for their ability to convert precise, remote, and spatiotemporal optical signals into macroscopic changes in material behavior such as mechanical, morphological, or rheological transformations.6–9 In this context, polymer networks incorporating coumarin moieties have been widely explored for their ability to undergo efficient and reversible [2+2] cycloaddition, (generally under exposure to UV at a wavelength of 365 nm (UV365) to induce dimerization and 254 nm (UV254) to trigger photocleavage), thus enabling dynamic tuning of network stiffness, self-healing properties, and stimuli-responsive adhesion.10–16 Similarly, hexaarylbiimidazole (HABI) derivatives provide another compelling route to photoresponsivity, forming persistent triphenylimidazolyl radicals under UV excitation, which have been harnessed to produce noticeable rheological changes.17–21 These reversible molecular systems are commonly characterized by the scission and formation of covalent bonds and are clearly distinguished from other photoresponsive molecules, such as azobenzenes and stilbenes, which have been employed to cause conformational changes in polymer chains on the basis of trans–cis photoisomerization.22–24
Similar to the coumarin-based photodimerization system, anthracene has also attracted significant attention, in which UV light (>300 nm) induces [4+4] cycloaddition to form small molecular and polymer networks that can be thermally cleaved or exposed to UV light at shorter wavelengths (<300 nm). For small molecules, liquid branched compounds with anthracene moieties and excellent photoresponsivity have recently been reported.25,26 However, many materials used in industry are polymers, and they are inevitably used owing to social demands, cost, and quality standards. As such, achieving such a responsivity with common polymers is meaningful. In this context, anthracene-based photodimerization systems have also been harnessed in various polymer networks,27–32 and have been further extended to photodynamic star polymer networks. Indeed, following the pioneering works of Mengel et al. in 200133 and Zheng et al. in 2002,34 various anthracene-based photodynamic systems for enabling photocontrolled mechanical modulation of hydrogels35 and ion-gels36 have been reported, to mention a few. On the surface, coumarin- and anthracene-based photodimerization systems are quite similar, but the anthracene moieties are obviously bulkier and should form more rigid dimers. Hence, when star polymer networks are formed by the dimerization of anthracene, the resulting network should tend to be more rigid and less flexible, leading to dynamic properties different from those of coumarin-based systems. Moreover, in the synthesis of star polymer networks, it has generally been recognized that the uniformity of their star polymer precursors enhances the mechanical properties of the resulting networks.37,38 However, the limited photoresponse of star polymer networks from their precisely designed precursors suggests the existence of other factors that dictate their photoresponse (Fig. 1a). In contrast, examples of various photoresponsive silicones that exhibit prominent rheological responses15,20 suggest the importance of polymer chain flexibility and compatibility or miscibility between the polymer chain and the photoactive group in rheological responses. However, several long-standing problems in the precision synthesis of polysiloxanes through polymerization reactions have hampered further progress in related fields.39,40 Very recently, these problems have begun to be resolved,20,39,41 opening up the possibility of synthesizing polymer networks from precisely designed networks using flexible polysiloxane. As silicones are flexible, durable, industrially important, and used in a wide range of applications that are irreplaceable with carbon-based polymers, achieving significant photoswitchability with silicone is not only fundamentally but also industrially significant.
 | ||
| Fig. 1 Schematic illustration of (a) conventional design of anthracene-appended polymer networks and (b) precisely architected flexible star-polymer elastomers in this work. | ||
Guided by this research progress and the wealth of aforementioned conventional research on anthracene-appended polymers, here we describe the design and synthesis of novel anthracene-appended star-shaped poly(dimethylsiloxane) (PDMS) with a controlled molecular weight (MW) and narrow dispersity (Đ), and its networks. While previous anthracene- and coumarin-based systems typically achieved approximately 10036–100035-fold increases in the storage modulus (G′), remarkably, the developed anthracene-appended star PDMS realizes a very large initial increase (>25000-fold) upon the first dimerization and a smaller but reproducible decrease in G′ upon subsequent dimer dissociation under solvent-free conditions (Fig. 1b). The large initial response enabled by the marriage of precision manipulation of macromolecular architectures and the flexibility of polysiloxanes demonstrates the potential of the present solventless system for applications requiring finite cycles of tunable properties.
According to the recently developed protocol for the organocatalyzed ring opening polymerization of hexamethylcyclotrisiloxane (D3) initiated from trifunctional silanol (I3), three-armed star-shaped PDMS with hydrosilane end groups (1: Mn = 19800, Mw = 22300, Đ = 1.13) was prepared.41 The star-shaped 1 was then subjected to a hydrosilylation reaction between 9-anthrylmethyl methacrylate in the presence of Karstedt's catalyst (Fig. 2a) to yield three-armed star-shaped PDMS with anthracene end groups (2: Mn = 22000, Mw = 27200, Đ = 1.23), which was thoroughly characterized by the combination of 1H NMR (Fig. S1) and size exclusion chromatography (Fig. S2). Similar to our previous report on coumarin-appended PDMSs,15 the color was slightly yellowish, reflecting the color of 9-anthrylmethyl methacrylate, and the physical appearance of 2 was a transparent oily liquid, which is advantageous for intended dimerization-induced network formation (Fig. 2b).
 | ||
| Fig. 2 (a) Synthesis and (b) photo-induced dimerization and dimer dissociation of anthracene-appended star-shaped PDMS. | ||
The first surprising finding was the sensitivity of 2 to light. As repeatedly reported in previous studies, direct evidence of reversible photodimerization of anthracene in insoluble network materials can be obtained from ultraviolet-visible (UV-VIS) absorption spectra. When UV light at a wavelength of 365 nm (UV365) was irradiated to 2 for 1 min, sandwiched between two quartz glass plates with a spacer to achieve a thickness of 0.1 mm, the absorption at 300–400 nm derived from anthracene drastically decreased and almost disappeared within three minutes (Fig. 3a). Unlike in the solution state, network polymeric materials formed via the photodimerization of anthracene typically require a long time to proceed. For example, in the case of star-PEG networks, a noticeable amount of anthracene-derived absorption remained even after several hours of UV365 irradiation.34,36 Encouraged by the present UV365 response, the same sample was subsequently irradiated with UV light at a wavelength of 254 nm (UV254). As a result, the absorption at 300–400 nm almost reached a photostationary state within one minute, and there was a negligible difference after 3 min (Fig. 3b). The precisely controlled star architecture ensures a uniform distribution of anthracene end groups, which contribute to enhance mechanical properties. Moreover, the flexible nature of PDMS likely provide high segmental mobility, enabling rapid migration and kinetically advantageous [4+4] cycloaddition of anthracene end groups even under solvent-free condition.
 | ||
| Fig. 3 UV-VIS spectra of 2 sandwiched between two quartz glass slides upon sequential irradiation with (a) UV365 and (b) UV254. | ||
Next, the reversibility of our material was evaluated by monitoring the UV-VIS absorption spectra as a function of time under alternating UV365 and UV254 irradiation (Fig. 4). After a significant decrease in absorption during the first UV365 irradiation, the absorbance of 2 at a wavelength of 366 nm varied regularly between 0 and 0.2 upon subsequent UV254 and UV365 irradiation, which is also consistent with previous reports.34 Given the versatility of silicone itself, the ability to achieve reversibility similar to that reported in previous studies but with a solventless system and an extremely quick response would be appealing for a range of applications.
 | ||
| Fig. 4 Absorbance of 2 (λ = 366 nm) monitored by UV-VIS spectroscopy upon UV365 and UV254 irradiation cycles. | ||
A surprising finding introduced at the end of this Communication is the photodynamic rheological properties of 2. Changes in the storage and loss moduli (G′ and G′′) upon cycles of alternating UV365 and UV254 irradiation to 2 under solventless conditions at room temperature (25 °C) were investigated. First, the initial G′ and G′′ (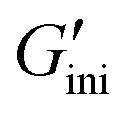 and
and 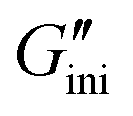 ) values were 4.7 Pa and 8.3 Pa, respectively. In general, the magnitude of the relationship between G′ and G′′ is an indicator used to determine solidity (G′ > G′′) and liquidity (G′ < G′′), and the present result again shows an inherent liquidity of 2. Next, time-dependent plots of G′ and G′′ upon UV365 irradiation revealed a sharp increase in G′ (Fig. 5a, left blue-colored region). The G′ reached 119 kPa, which is more than 25000-fold greater than the
) values were 4.7 Pa and 8.3 Pa, respectively. In general, the magnitude of the relationship between G′ and G′′ is an indicator used to determine solidity (G′ > G′′) and liquidity (G′ < G′′), and the present result again shows an inherent liquidity of 2. Next, time-dependent plots of G′ and G′′ upon UV365 irradiation revealed a sharp increase in G′ (Fig. 5a, left blue-colored region). The G′ reached 119 kPa, which is more than 25000-fold greater than the 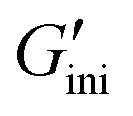 , and the change in G′′ was obviously less prominent. During UV365 irradiation, G′ exceeded G′′, and the resulting network 2D (Fig. 2b bottom) became a rubbery solid. When anthracene groups are introduced into side chains, the presence of only a small number of light-responsive groups, less than 1% of the total, causes dimerization-induced crystallization even with very high-MW polysiloxanes.42 With the developed material, the liquid or rubbery solid state was maintained and no crystalline components were formed throughout the entire process. This noncrystallizable nature is likely due to the presence of anthracene moieties as highly mobile end groups, discouraging crystallization. Upon subsequent UV254 irradiation, the G′ apparently decreased (Fig. 5a, left orange-colored region) but did not revert back to its initial value. Moreover, a gradual decrease in G′ after irradiation with UV light during the cycles of UV365 and UV254 irradiation was observed (Fig. 5a). In addition to the involvement of photostationary states (Fig. 3a and b), the reversibility may be constrained by irreversible side reactions such as photooxidation and photobleaching of anthracene, which are frequently reported in the photochemistry of anthracene-based compounds. Given that the photoirradiation experiments were conducted under ambient atmospheric conditions, the presence of oxygen and humidity may also influence the photoreversibility. To investigate this, a predetermined amount of 2 was stored for 24 h under three different conditions: a nitrogen atmosphere (>99%, 37 °C), an oxygen-rich atmosphere (95%, 37 °C), and a humidified oxygen atmosphere (95% O2, 90% relative humidity, 37 °C). However, the differences in storage conditions resulted in negligible variation in the photoresponsivity monitored with a UV-VIS spectrometer, suggesting that the observed behavior is governed mainly by the presence of photostationary states rather than by the aforementioned side reactions. On the other hand, when the UV365-irradiated network 2D was heated (Fig. 5b, left blue-colored region), G′ decreased to 10 kPa, which is comparable to that of G′′ (3 kPa) (Fig. 5b, right red-colored region). In addition, G′ and G′′ remained unchanged upon subsequent cooling to room temperature (Fig. 5b, central white region), but following UV365 irradiation, the G′ significantly increased to approximately 110 kPa (Fig. 5b, right blue-colored region), demonstrating the manipulability of viscoelasticity by the combination of light and heat stimuli.
, and the change in G′′ was obviously less prominent. During UV365 irradiation, G′ exceeded G′′, and the resulting network 2D (Fig. 2b bottom) became a rubbery solid. When anthracene groups are introduced into side chains, the presence of only a small number of light-responsive groups, less than 1% of the total, causes dimerization-induced crystallization even with very high-MW polysiloxanes.42 With the developed material, the liquid or rubbery solid state was maintained and no crystalline components were formed throughout the entire process. This noncrystallizable nature is likely due to the presence of anthracene moieties as highly mobile end groups, discouraging crystallization. Upon subsequent UV254 irradiation, the G′ apparently decreased (Fig. 5a, left orange-colored region) but did not revert back to its initial value. Moreover, a gradual decrease in G′ after irradiation with UV light during the cycles of UV365 and UV254 irradiation was observed (Fig. 5a). In addition to the involvement of photostationary states (Fig. 3a and b), the reversibility may be constrained by irreversible side reactions such as photooxidation and photobleaching of anthracene, which are frequently reported in the photochemistry of anthracene-based compounds. Given that the photoirradiation experiments were conducted under ambient atmospheric conditions, the presence of oxygen and humidity may also influence the photoreversibility. To investigate this, a predetermined amount of 2 was stored for 24 h under three different conditions: a nitrogen atmosphere (>99%, 37 °C), an oxygen-rich atmosphere (95%, 37 °C), and a humidified oxygen atmosphere (95% O2, 90% relative humidity, 37 °C). However, the differences in storage conditions resulted in negligible variation in the photoresponsivity monitored with a UV-VIS spectrometer, suggesting that the observed behavior is governed mainly by the presence of photostationary states rather than by the aforementioned side reactions. On the other hand, when the UV365-irradiated network 2D was heated (Fig. 5b, left blue-colored region), G′ decreased to 10 kPa, which is comparable to that of G′′ (3 kPa) (Fig. 5b, right red-colored region). In addition, G′ and G′′ remained unchanged upon subsequent cooling to room temperature (Fig. 5b, central white region), but following UV365 irradiation, the G′ significantly increased to approximately 110 kPa (Fig. 5b, right blue-colored region), demonstrating the manipulability of viscoelasticity by the combination of light and heat stimuli.
 | ||
| Fig. 5 Time-course rheological analysis of 2 upon (a) sequential UV365 and UV254 irradiation cycles and (b) sequential UV365 irradiation, heating to 180 °C, cooling, and UV365 re-irradiation. | ||
In conclusion, we have demonstrated the synthesis of a new class of photoresponsive silicone elastomers derived from precisely designed star-shaped PDMS terminated with anthracene moieties. While photodynamic star polymer networks based on the reversible photodimerization of anthracene terminals are now commonplace materials, the combination of PDMS and anthracene enables extremely amplified responses in terms of rheological properties, with a more than 25000-fold increase in G′ upon the first UV365 irradiation. Assisted by the highly flexible nature of PDMS chains, the precisely controlled star architecture allows the formation of a more uniform network structure, likely leading to the present photoresponsivity even under solvent-free conditions. The findings underscore both the opportunities and the limitations of anthracene photodimerization in solvent-free soft materials, and point toward strategies for designing responsive networks, demonstrating how careful selection of photoresponsive molecules and polymers with controlled MW and Đ can be harnessed to amplify their responses to external stimuli. Further systematic studies related to these findings, including MW dependence, synthetic and processing scalability, and durability are currently in progress for extending the generality and practical relevance of the platform in future work.
This work was financially supported by JSPS KAKENHI (Grant Number 23K17337), JST PREST (Grant Number JPMJPR24M9), JST ALCA-Next (Grant Number JPMJAN24C5), and the Fuji Seal Foundation. We thank Mr Kazuki Fuke (The University of Tokyo) for his initial trial and error in synthesizing anthracene derivatives.
Conflicts of interest
There are no conflicts to declare.Data availability
The data underlying this study are available in the published article and its supplementary information (SI). Supplementary information: experimental details, 1H NMR spectrum, Size Exclusion Chromatography traces. See DOI: https://doi.org/10.1039/d5cc04840k.Notes and references
- M. A. C. Stuart, et al. , Nat. Mater., 2010, 9, 101–113 CrossRef PubMed.
- D. Roy, et al. , Prog. Polym. Sci., 2010, 35, 278–301 CrossRef.
- A. Zhang, et al. , Prog. Polym. Sci., 2019, 99, 101164 CrossRef.
- A. J. Rosario and B. Ma, ACS Appl. Polym. Mater., 2024, 6, 14204–14228 CrossRef.
- C. Tsitsilianis, Soft Matter, 2010, 6, 2372–2388 RSC.
- J.-M. Schumers, et al. , Macromol. Rapid Commun., 2010, 31, 1588–1607 CrossRef PubMed.
- J. Lee, et al. , J. Am. Chem. Soc., 2019, 141, 15348–15355 CrossRef PubMed.
- T.-H. Lee, et al. , RSC Adv., 2021, 11, 37392–37402 RSC.
- T.-H. Lee, et al. , ACS Appl. Mater. Interfaces, 2021, 13, 43364–43373 CrossRef PubMed.
- J. P. Chesterman, et al. , Eur. Polym. J., 2018, 105, 186–193 CrossRef.
- K. Auepattana-Aumrung, et al. , Chem. Sci., 2025, 16, 5976–5985 RSC.
- S. R. Trenor, et al. , Chem. Rev., 2004, 104, 3059–3078 CrossRef.
- Y. Chen and K.-H. Chen, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 613–624 CrossRef.
- S. Honda and T. Toyota, Polymer, 2018, 148, 211–216 CrossRef CAS.
- M. Oka, et al. , Macromol. Rapid Commun., 2022, 43, 2200407 CrossRef CAS.
- S. Honda, et al. , J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 9–15 CrossRef.
- D. Ahn, et al. , Chem. Mater., 2017, 29, 7023–7031 CrossRef.
- S. Honda and T. Toyota, Nat. Commun., 2017, 8, 502 CrossRef.
- S.-L. Xiang, et al. , ACS Appl. Mater. Interfaces, 2019, 11, 23623–23631 CrossRef.
- M. Oka, et al. , Adv. Sci., 2021, 8, 2101143 CrossRef PubMed.
- S. Honda, et al. , Angew. Chem., Int. Ed., 2019, 58, 144–148 CrossRef.
- Y. Li, et al. , Nat. Chem., 2024, 16, 446–455 CrossRef.
- H. Akiyama and M. Yoshida, Adv. Mater., 2012, 24, 2353–2356 CrossRef.
- H. Zhou, et al. , Nat. Chem., 2017, 9, 145–151 CrossRef PubMed.
- H. Akiyama, et al. , Macromol. Chem. Phys., 2025, 226, 2400217 CrossRef.
- H. Akiyama, et al. , J. Adhesion, 2018, 94, 799–813 CrossRef.
- C. P. Kabb, et al. , Chem. Sci., 2019, 10, 7702–7708 RSC.
- T. Hughes, et al. , ACS Appl. Mater. Interfaces, 2019, 11, 19429–19443 CrossRef.
- H. Zhang, et al. , Chem. Sci., 2019, 10, 8367–8373 RSC.
- Y. Li, et al. , Sci. Rep., 2020, 10, 20214 CrossRef PubMed.
- P. Froimowicz, et al. , Macromol. Rapid Commun., 2011, 32, 468–473 CrossRef.
- L. A. Connal, et al. , Adv. Funct. Mater., 2008, 18, 3315–3322 CrossRef.
- C. Mengel, et al. , Macromol. Chem. Phys., 2001, 202, 1138–1149 CrossRef.
- Y. Zheng, et al. , Macromolecules, 2002, 35, 5228–5234 CrossRef.
- V. X. Truong, et al. , ACS Macro Lett., 2017, 6, 657–662 CrossRef PubMed.
- A. Saruwatari, et al. , Chem. Commun., 2018, 54, 13371–13374 RSC.
- S. Nakagawa and N. Yoshie, Polym. Chem., 2022, 13, 2074–2107 RSC.
- G. Hild, Prog. Polym. Sci., 1998, 23, 1019–1149 CrossRef.
- L. Shi, et al. , Science, 2023, 381, 1011–1014 CrossRef PubMed.
- J. Goff, et al. , Molecules, 2021, 26, 2755 CrossRef PubMed.
- H. Okamoto, et al. , Commun. Chem., 2024, 7, 61 CrossRef PubMed.
- T. Wright, et al. , Chem. Sci., 2020, 11, 3081–3088 RSC.
| This journal is © The Royal Society of Chemistry 2025 |