
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Classic coordination compounds as the inspiration for MOFs: selected catalytic applications
Marco L.
Martínez†
ab,
Antonio
Hernández-Monsalvo†
a,
Pablo
Marín-Rosas†
c,
Ariel
Guzmán-Vargas
b,
Jose Antonio
de los Reyes
d,
Diego
Solis-Ibarra
 a,
Enrique
Lima
*a,
Ricardo A.
Peralta
*c and
Ilich A.
Ibarra
*a
a,
Enrique
Lima
*a,
Ricardo A.
Peralta
*c and
Ilich A.
Ibarra
*a
aLaboratorio de Fisicoquímica y Reactividad de Superficies (LaFReS), Instituto de Investigaciones en Materiales, Universidad Nacional Autónoma de México, Circuito Exterior s/n, CU, Del Coyoacán, 04510, México D.F., Mexico. E-mail: argel@unam.mx; lima@iim.unam.mx
bLaboratorio de Investigación en Materiales Porosos, Catálisis Ambiental y Química Fina, Instituto Politécnico Nacional, ESIQIE-SEPI-DIQI, UPALM Edif. 7 P.B. Zacatenco, GAM, 07738 CDMX, Mexico
cDepartamento de Química, División de Ciencias Básicas e Ingeniería, Universidad Autónoma Metropolitana-Iztapalapa, 09340, Ciudad de México, Mexico. E-mail: rperalta@izt.uam.mx
dDepartamento de Ingeniería de Procesos e Hidráulica, División de Ciencias Básicas e Ingeniería, Universidad Autónoma Metropolitana-Iztapalapa, 09340, Ciudad de México, Mexico
First published on 20th October 2025
Abstract
Coordination chemistry has garnered significant attention across various domains due to its versatility in both academic research and industrial applications, with catalysis being a particularly prominent area of distinction. The exceptional performance of coordination chemistry-based compounds and materials, such as metal–organic frameworks (MOFs), in catalytic processes is ascribed to their formidable physical and structural attributes. Primarily, possessing accessible vacant active metal sites is crucial for promoting the formation of intermediate or transition states involving the species of interest. This feature facilitates effective interactions between reactant molecules and the active sites of these compounds and supramolecular assembled materials. Professor Tilley's contributions have substantially advanced the field of coordination chemistry for catalysis, as he has consistently aimed to enhance the reactivity and selectivity of these compounds in reactions such as hydrosilylation and water oxidation/evolution. This review aims to summarize the catalytic properties of coordination compounds, in which his contributions have pioneered the incorporation of transition metals into catalysts, and compare them with MOFs applied with the same aim, as heterogeneous catalysts, for these reactions.
 From left to right: Ricardo Peralta, Pablo Marín-Rosas, Antonio Hernández-Monsalvo, Enrique Lima, Ilich Ibarra | Ricardo Peralta is a Full Professor at UAM who is interested in novel catalytic application of semi-open metal sites within MOFs. Pablo Marín-Rosas is a PhD Student who is exploring MOF materials for catalytic applications. Antonio Hernández-Monsalvo is working towards a Master's degree by investigating chemically stable MOFs for the sensing of toxic chemicals. Enrique Lima serves as the Vice-Principal of the Institute for Materials Research at UNAM, and his research focuses on bio-applications of MOF materials. Ilich Ibarra is a Full Professor at UNAM working on the use of MOFs for the capture and detection of toxic gases such as SO2 and H2S. |
1. Background
Modern studies on discrete complexes (ca. 1878), also known as coordination compounds, were initiated by Alfred Werner and Sophus Mads Jørgensen, who reconciled their different theoretical approaches to explain the properties of coordination compounds. Initially, Jørgensen tried to elucidate the chemical formula of coordination compounds by proposing a carbon-free analogue of carbon chemistry, which was found to be an incorrect theory. However, some ideas from Jørgensen were pivotal to the progress of coordination chemistry. On the other hand, Werner suggested that metal centres show constant coordination numbers. Additionally, another fundamental contribution from Werner was the rigidity of coordination bonds, where these bonds were entirely fixed in space and therefore, these were treated by the application of structural principles.1 At the beginning of the development of modern coordination compound theory, different characterization techniques were required to elucidate, principally, structural information. For instance, conductance studies and optical techniques were used to validate the ideas of Werner on bonding in coordination compounds.2 Although these techniques were successful in providing valuable insights about the structure of complexes, they “missed” a direct visualization of the atomic distribution in such complexes. Thus, the next big step was performed by Pfeiffer, who was the first person to apply some coordination theory ideas to crystal structures in 1915 for NaCl crystals,2 followed by Wyckoff and Posnjak, who published the first experimental crystallographic study of a coordination compound in 1921.3 Hence, X-ray diffraction (XRD) techniques became one of the most important analytical tools in coordination compounds and, later, in extended coordination-based materials, since the early 1920s. For example, both [Mn2(CO)10] and [Co2(CO)8] complexes, based on transition metals as central atoms and carbonyl groups as ligands, were fully elucidated by X-ray diffraction techniques in 1962 and 1964, respectively.4,5Thus, XRD techniques continued to be used to reveal different and more complicated coordination compounds. Pioneering examples were introduced in 1987 by Professor Tilley and Professor Straus, where Ru–silylene complexes were developed and structurally elucidating, by X-ray single crystal diffraction techniques, relevant transition metals–silicon bonds.6 Currently coordination compounds are extensively used and studied for several applications, for instance, in organic light-emitting diodes (OLEDs),7 biomedical applications (e.g., drug like molecules and medicines),8 analytical chemistry,9 and catalysis, where many commercially available catalytic processes involve coordination compounds, as catalytic reagents.10–12 Specifically, coordination compounds are extensively used in homogeneous catalysis, since these complexes provide metal centres, allowing substrate molecules to be coordinated, as part of a particular catalytic cycle, vide infra. Although these homogeneous processes are, generally speaking, economical and practical, on a large scale, challenges exist due to the costs of separating such catalyst from the products. On the other hand, although common heterogenous catalysts, such as supported nanoparticles, zeolites, and oxides (only to name a few), are relatively easy to separate from products, and present remarkable performances in reactions such as the oxidation of chlorinated volatile organic compounds13 and in oxidation of methane to methanol,14 reactions, which are important in industry and in environmental remediation, these catalysts generally lack on specific control over reaction conditions and selectivity, compared to homogenous catalysts. With these perspectives in mind, it is expected to formulate the following question: is it possible to achieve the best properties of both words (homogenous and heterogenous) in only one catalyst?
To answer this question, first, it is necessary to look at the integration between the natural development of coordination chemistry, especially at the principles of coordination compounds, and supramolecular chemistry. Supramolecular chemistry aims to design and implement functional chemical systems based on molecular components held together by non-covalent intermolecular forces, meaning that this field deals with any interaction like hydrogen bonding, dipolar interactions, halogen bonding, aromatic stacking, and metal coordination interactions. Thus, the transition from discrete complexes to coordination polymers, defined as materials constructed by metal or metal clusters coordinated to molecular-based bridging-ligands, creating an infinite array, can be seen as the logical progression as it is increased the connectivity of the metal centres present in coordination compounds (Fig. 1).15 Metal–organic frameworks (MOFs), a special kind of coordination polymers which exhibit intrinsic porosity, feature high specific surface areas, permanent porosity, high degree of tunability, and, for selected examples, remarkable chemical stability.16 These materials provide the possibility of using them in diverse applications such as energy storage, drug delivery, separation and capture of toxic gases, and, particularly, homogeneous and heterogeneous catalysis. Thus, since MOFs are solid materials, and they can be seen as extended coordination compound networks, it is predictable that these materials could show the advantages of homogeneous and heterogeneous catalysts (see Section 2.2).
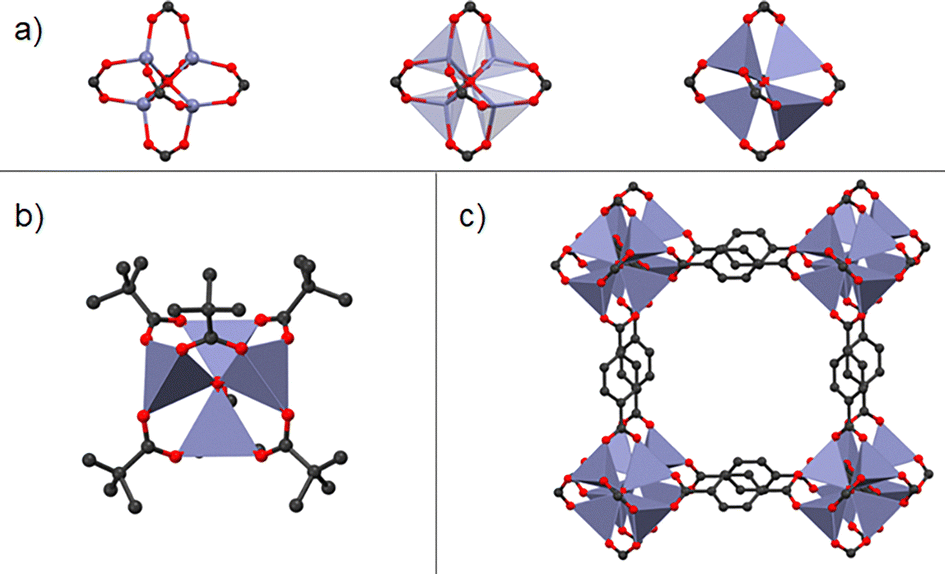 | ||
| Fig. 1 (a) Zn4(O)(CO2)6 cluster based of, (b) Zn4O(O2CC(CH3)3)6, discrete coordination compound, and (c) MOF-5, where terephthalic acid units act as bridging ligand. MOF-5 can be seen as the joint of Zn4(O)(CO2)6 clusters via terephthalic acid, whereas in Zn4O(O2CC(CH3)3)6, Zn4(O)(CO2)6 is just coordinated to pivalate units (no bridging ligand). | ||
Chemical reactivity remains a preeminent area in both academic and applied research. Developing the ability to understand and accurately predict the reactive behavior of various species upon interaction is a skill that constrains a formidable and extensive trajectory of study and practice. This review is presented as an accolade to the significant contributions of Professor T. Don Tilley and his research group. While his expertise spans a wide range of interdisciplinary areas, including polymer, materials, and organometallic chemistry, this work will focus primarily on reactivity studies in novel organometallic systems. Specifically, the synthesis of transition metal coordination complexes that exhibit new chemical properties, with homogeneous catalysis being a centrepiece. It also seeks to compare these findings with metal–organic frameworks (MOFs), porous materials that use the same type of metal–organic coordination interactions but are employed in heterogeneous catalysis. Herein, we aim to relate Professor Tilley's outstanding work to a topic within our research group's domain of expertise by comparing two distinct derivatives of coordination chemistry: metal complexes and porous materials.
2. Catalytic features of coordination compounds and MOFs
In this section, the basic features and current research fields of coordination compounds as catalysts are shown to correlate such features to MOFs and then, fully understand the catalytic mechanisms within MOFs at the molecular level.2.1. Coordination compounds
One fundamental feature by coordination compounds, which allows them to act as efficient catalysts, is the access to vacant coordination sites, granting substrates to coordinate to these unsaturated metal centres. Free coordination sites are fundamental in coordination compounds and, for example, metal centres which typically show an octahedral geometry (i.e., coordinated to six ligands), a missing ligand creates a fivefold coordinated complex and provides a free coordination site. In practice, these coordinative unsaturated metal centres often occur only as intermediary or transition states because of the presence of labile solvent molecules that occupy the free coordination sites.17 Coordination compounds can be classified as kinetically labile and kinetically inert, where the former compounds experience fast ligand exchange, typically, within seconds to a few minutes, contrary to inert complexes which undergo slow ligand exchange (i.e., hours or even days), in where the lability of coordination compounds can be understood in terms of crystal field theory.18 In this context, the lability of complexes has been demonstrated to be responsible for the reactivity and selectivity of coordination compounds.19 Therefore, the metal–ligand bond lability is an essential requirement in order to employ metal atoms as catalytic centers.17,20 Although intrinsic complexes lability generally determines if a complex could be used efficiently as catalyst, excitation via light irradiation can provide fast ligand exchange for inert coordination compounds (e.g., those with Co3+ cations).1,15 On the other hand, non-labile ligands avoid the metal precipitation and assure a correct electronic balance (i.e., electronic density on the metal, trans effect and centre steric effect) permitting all the individual reactions, along the whole catalytic cycle, to go along at necessary rate and selectivity.21 Fundamentally, the catalytic activity is essentially related to the structure of the catalyst, where the electron donating and withdrawing capability of the substituents, defines the electronic effects, while bond length and angle, size of the substituents and atomic radio of transition metals are related to steric effects.22 A remarkable example was accomplished by Werley et al., where they correlated the substituent's effect in (tetraarylcyclopentadienone)iron carbonyl compounds as catalysts in the transfer hydrogenation and dehydrogenation reactions, where the typical proposed catalytic cycle is depicted in Fig. 2a. In such catalytic mechanism, transfer hydrogenation corresponds to moving clockwise, whereas transfer dehydrogenation corresponds to moving counterclockwise. Activation of tricarbonyl compound A with trimethylamine N-oxide in solution forms unsaturated species B, which is catalytically active. Iron hydride C, which is also catalytically active, is generated when B reacts with an alcohol.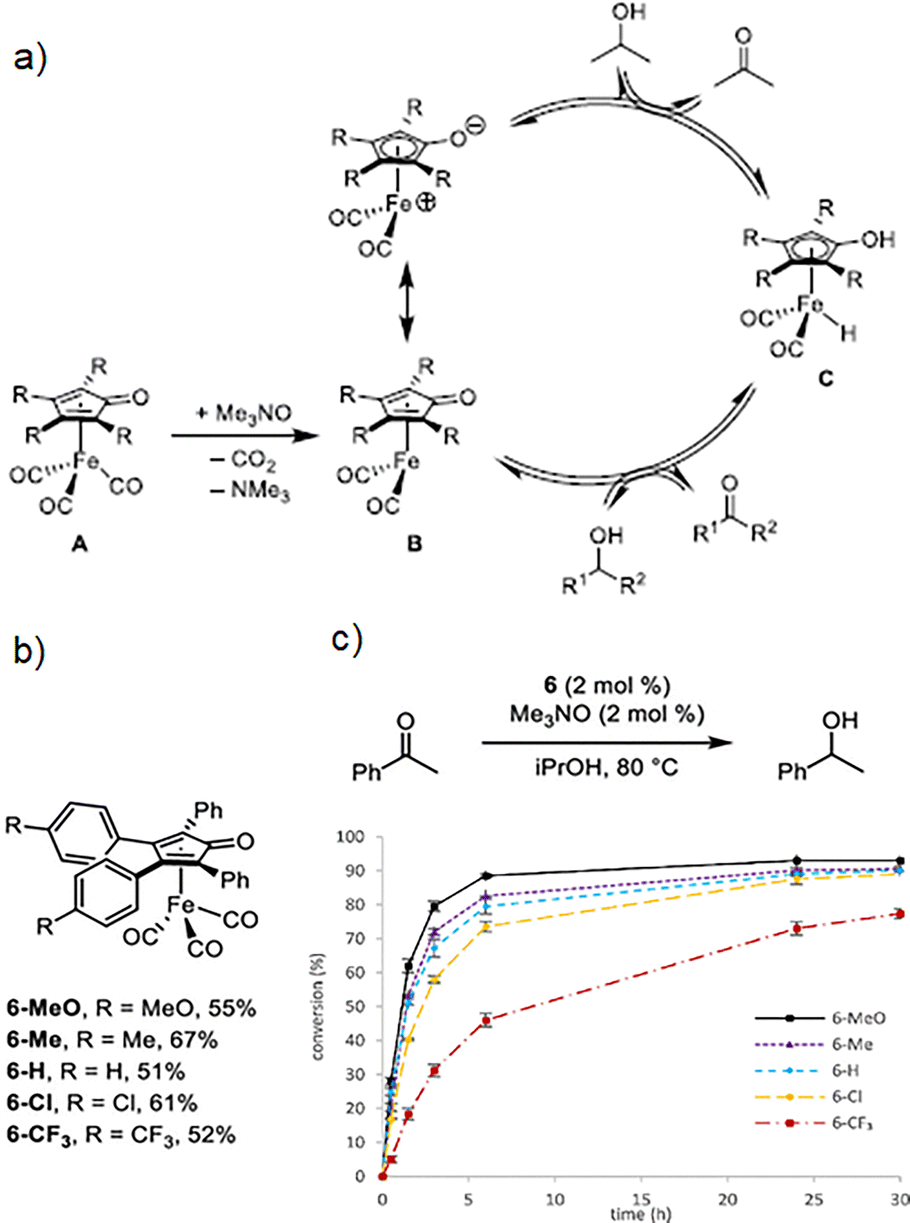 | ||
| Fig. 2 (a) Typical catalytic cycle in transfer hydrogenation and dehydrogenation reactions using compounds based on (cyclopentadienone)iron carbonyl, which are activated with trimethylamine N-oxide, (b) (tetraarylcyclopentadienone)iron carbonyl complexes used as catalysts in transfer hydrogenation and dehydrogenation reactions, and (c) conversion as time function (% vs. h) for the acetophenone reduction using the 5 different catalysts. Reproduced from ref. 23 with permission from American Chemical Society, copyright 2023. | ||
Specifically, they tuned the electron density of the catalyst by selecting electron-rich and electron-withdrawing groups in para positions of the two appended aromatic rings with minimal changes to its total size (Fig. 2b). By this strategy, they demonstrated that the (cyclopentadienone)iron tricarbonyl compounds, with electron-rich substituents, showed better catalytic activity (Fig. 2c), which was confirmed by a linear free-energy relationship.23 Additional to such effects, another important parameter is the so-called ligand bite angle. For example, for the hydroformylation reaction using a bisphosphane ligand, the bite angle can significantly influence the products distribution.18
Recently, it was demonstrated that chemical moderation, far away from the first coordination sphere, imposes a deep influence on catalysts, which is a well-known phenomenon in the action mode of enzymes. Remarkably, enzymes show extremely precise control on catalytic activity and selectivity, mainly by the precise control on the hydrogen bonding interactions, which meticulously adjust the spatial conformation of the substrate to precisely pre-organize it, in a given selective reaction. Thus, the same principles presented by enzyme can be applied to catalysts based on coordination compounds.24 Although the second coordination sphere-based catalysis is often principally associated with hydrogen bonding effects, other supramolecular interactions e.g., ion-pairing, cation-crown ether interactions, p-interactions, and halogen bonding, also show an essential influence on coordination compounds-based catalysis, providing higher activities and selectivity.25 The concepts described above are the basis for describing the properties of coordination compounds as homogenous catalysts. However, some of these concepts have also demonstrated their applicability to some heterogenous systems, creating an approach, often known as surface coordination chemistry, to understand heterogenous catalysts at molecular and atomic levels. Thus, surface coordination chemistry deals with the interdisciplinary integration of heterogenous catalysis with coordination chemistry. Pioneering examples are presented in Ziegler–Natta catalysis for alkene polymerization using a TiCl3 catalyst, in where Ti ions, at the surfaces of TiCl3 crystals, have missing chloride ions, providing coordinatively unsaturated metal centers.26–28 Modern studies concerning surface coordination chemistry have been mostly applied to atomically dispersed metal catalysts on different supports, ultrathin nanostructures, and atomically precise metal nanoclusters.29–31 For example, remarkable examples on the use of surface coordination chemistry are found in single atom catalysis (SAC). Single atom catalysts arise as a natural progress of the use of metal supported catalysts, i.e., metal components finely dispersed on a high-surface-area support. The use of bulk metal particles, metals nanoclusters, and even metal sub-nanoclusters, deals with the problem of irregular morphology, broad particles size, and, consequently, each metal particle may possess multiple active sites with different performances. Therefore, one of the pivotal features which settle the specificity and reactivity of the supported metal catalysts is the size of metal particles. Thus, the size reduction of metal particles benefits the performance of catalysts in: (i) A rise in the presence of unsaturated (low-coordination) metal centres, (ii) quantum size effects, leading to a discrete energy level distributions and a distinctive HOMO–LUMO gap, and (iii) metal–support interactions: chemical-bonding between metal and support, and charge transfer between metals and supports.32 For example, Liu and coworkers successfully prepared atomically dispersed Fe–N–C catalyst for the selective oxidation of the C–H bond, where they established four Fe–Nx (x = 4–6) coordination environments. Particularly, when x = 4, the Fe atoms have a square planar coordination geometry, and the further N coordinated when x = 5 and 6 are coordinated in apical positions. Thus, they found that the FeIIIN5 structure is one order of magnitude more active than the high-spin and low-spin FeIIIN6 and, three times more active than FeIIN4. This can be attributed to the low coordination of Fe, which provides a free coordination site (Fig. 3).33
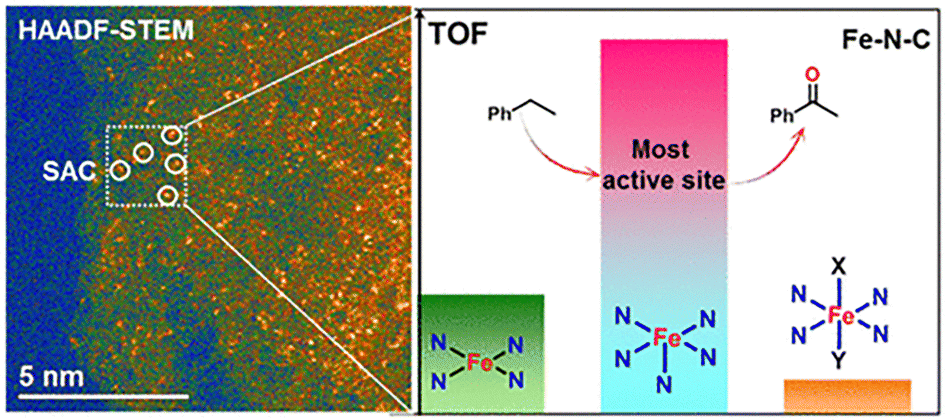 | ||
| Fig. 3 (left) Dispersed SAC via HAADF-STEM (high-angle annular dark-field scanning transmission electron microscopy) technique, and (right) the most active site (FeIIIN5) compared to other catalytic sites. Reproduced from ref. 33 with permission from American Chemical Society, copyright 2017. | ||
2.2. Metal–organic frameworks (MOFs)
As previously described, metal–organic frameworks (MOFs) are regarded to unite the benefits of both homogenous and heterogenous catalysis, since they show uniform catalytic centres, high activity, and selectivity (i.e., homogenous catalysis), while providing the ease to separate them from the final products, which is the major advantage of heterogenous catalysts.34 Generally, MOFs can promote catalysis via their inorganic nodes, organic linkers, and they can even function as host or support for additional catalytic materials, such as nanoparticles (Fig. 4).35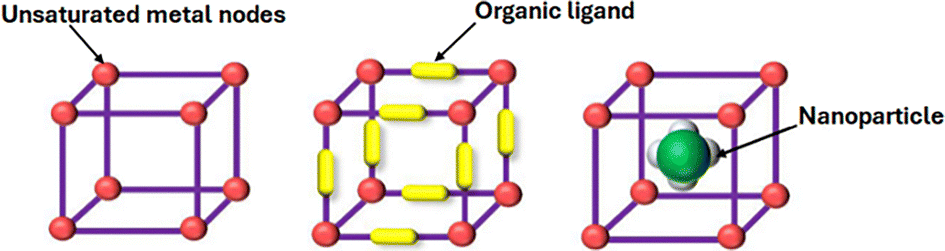 | ||
| Fig. 4 Different possible catalytic sites in metal–organic frameworks. Reproduced from ref. 35 with permission from Elsevier, copyright 2024. | ||
As MOFs are constructed through self-assembly process, the metal atoms coordinate to ligands with a geometry governed by coordination chemistry principles, such as the metal atom size and electronic configuration. Additional to bridging ligands, sometimes metal ions also saturate their coordination sphere by coordinating unidentate labile solvent molecules derived from the synthesis medium. Thus, the as-synthesized MOF will present originally all metal ions in the SBU in their fully coordinatively saturated state, that is, the most stable coordination environment according to their common coordination numbers. These solvent molecules coordinated to the metal ions can be removed during the activation stage (i.e., thermal, chemical or photochemical activation),17 leading to the formation of coordinatively unsaturated sites (CUS) or open metal sites (OMS) within the inorganic nodes, which can perform as Lewis acid in catalytic processes, similarly to coordination compounds-based at homogenous catalysis. Thus, defect engineering and SBU modification have been demonstrated to be powerful strategies to boost the catalytic activity of the inorganic nodes within MOFs. Although the activation step has shown good results to form OMS, this method hardly leads to a full formation of OMS, while it can also modify the structure of the inorganic nodes or even change the oxidation state of the metal centres, negatively impacting their reactivity. Thus, an alternative strategy is to design MOFs that do not contain open metal sites after an activation protocol. To that purpose, it is possible to take advantage of the intrinsic nature of the coordination bond between the inorganic nodes and the organic linkers. Thus, the dynamism of the metal–ligand bonds exhibited in selected MOFs, for example, MFM-300(Sc), which is constructed with [ScO4(OH)2] nodes, coordinated to 3,3′,5,5′-biphenyl-tetracarboxylate (BPTC) ligands. MFM-300(Sc) shows a reversible metal–ligand bond rearrangement induced by the adsorption of guest molecules (e.g., NH3) creating transient open metal sites (Fig. 5a).36 The same hemilabile behaviour (i.e., partial dissociation of bidentate carboxylate donor) also appears by exposing MFM-300(Sc) to H2S, where the Sc3+ centres act as catalytic active sites to produce polysulfides.37 Inspired by these results, Maurin et al. employed MFM-300(Sc) as Lewis acid catalyst for the Strecker reaction between N-benzylideneaniline and trimethylsilyl cyanide (TMSCN) to form 2-phenyl-2-(phenylamino) acetonitrile. Interestingly, the likely mechanism was proposed by density functional theory (DFT) calculations to involve the coordination of CN− to transient open Sc3+ sites (Fig. 5b).38 Furthermore, MFM-300(Sc) was successfully used as catalysis for acetone hydrogenation, where the same hemilabile character of Sc3+ centres, demonstrated to be responsible for the catalytic activity.39
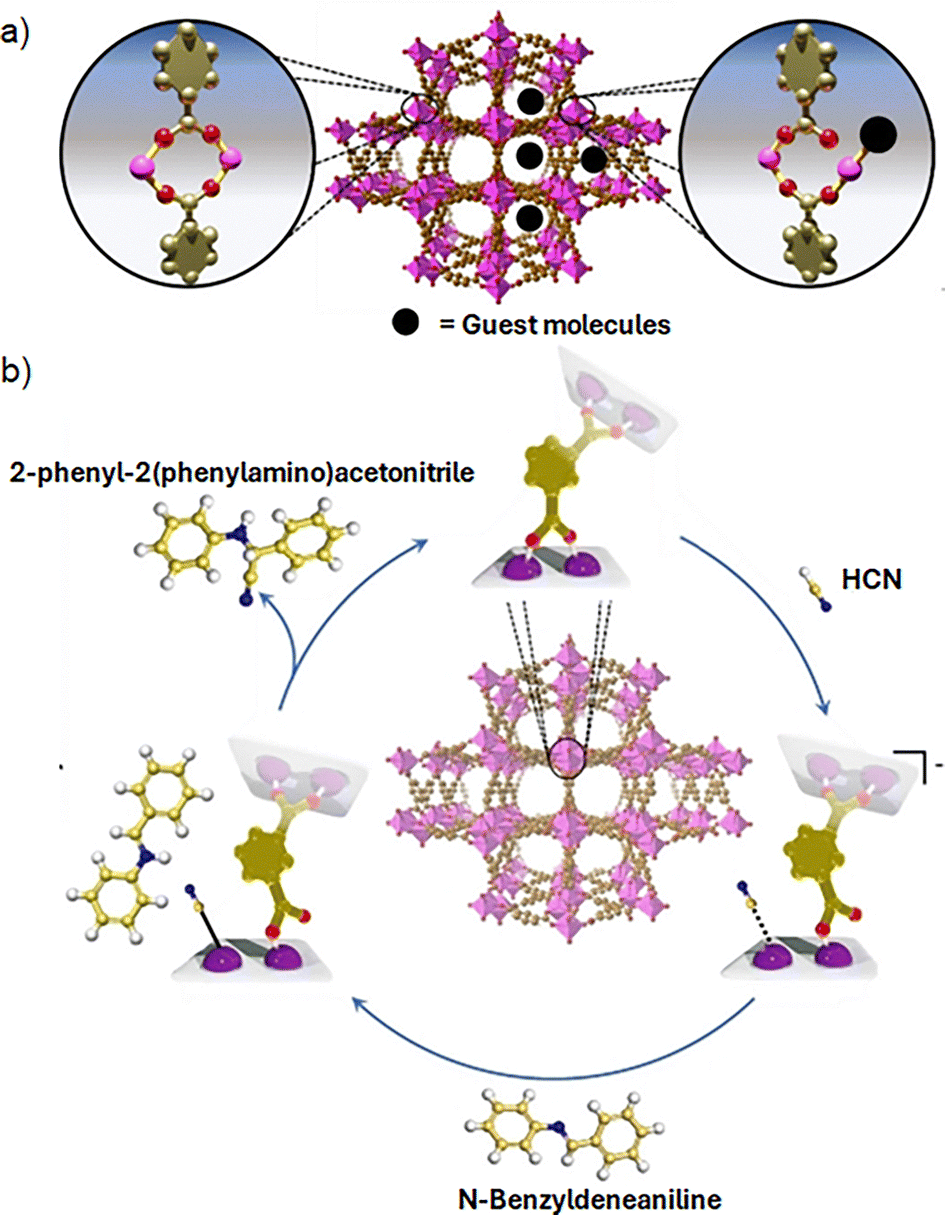 | ||
| Fig. 5 (a) Depiction of the metal–ligand bond hemilability after guest adsorption on Sc(III)-based MFM-300, and (b) the catalytic mechanism to obtain 2-phenyl-2-(phenylamino)acetonitrile as result of the metal-hemilabile linker dynamics. Color code: C = gold, O = red, Sc = pink, N = blue, and H = white. Adapted from ref. 38 with permission from John Wiley and Sons, copyright 2022. | ||
As mentioned in Section 2.1, steric and electronic ligand effects are critical for the activity and stereoselectivity in homogeneous catalysis. Conversely, for conventional heterogenous catalysts, it is more complicated to confirm the structure–activity relationships. However, since MOFs are molecular crystalline materials, i.e., they show defined atomic positions, it is possible to transfer the concept of electronic modulation of the active sites from homogeneous catalysis to heterogenous catalysis. For instance, Vermoortele and coworkers used UiO-66 type MOFs with different benzene-1,4-dicarboxylates (BDC-X; X = H, NH2, CH3, OCH3, F, Cl, Br, NO2) to test the effect of the substituents in the cyclization of citronellal, where they demonstrated that the selectivity for the isopulegol isomers as well as the activity strongly depend on the Lewis acidity of the active site (Fig. 6a).40 Thus, the catalytic conversion rate was significantly increased by electron-withdrawing groups on the linker, where UiO-66-NO2 is the most active material, indicating that the electronic active site modulation, is similar to characteristic complexes in homogenous catalysis. This enhanced activity was attributed to the nitro-substituent linkers, which concede a strong Lewis acid behaviour to Zr(IV). Additionally, citronellal is strongly adsorbed on the nitro-modified cluster, since the NO2 groups strongly interact with some hydrogen atoms from the adsorbate in the pre-reactive complex, the transition state, and adsorbed-product state, reducing the free energy along the whole catalytic cycle (Fig. 6b).
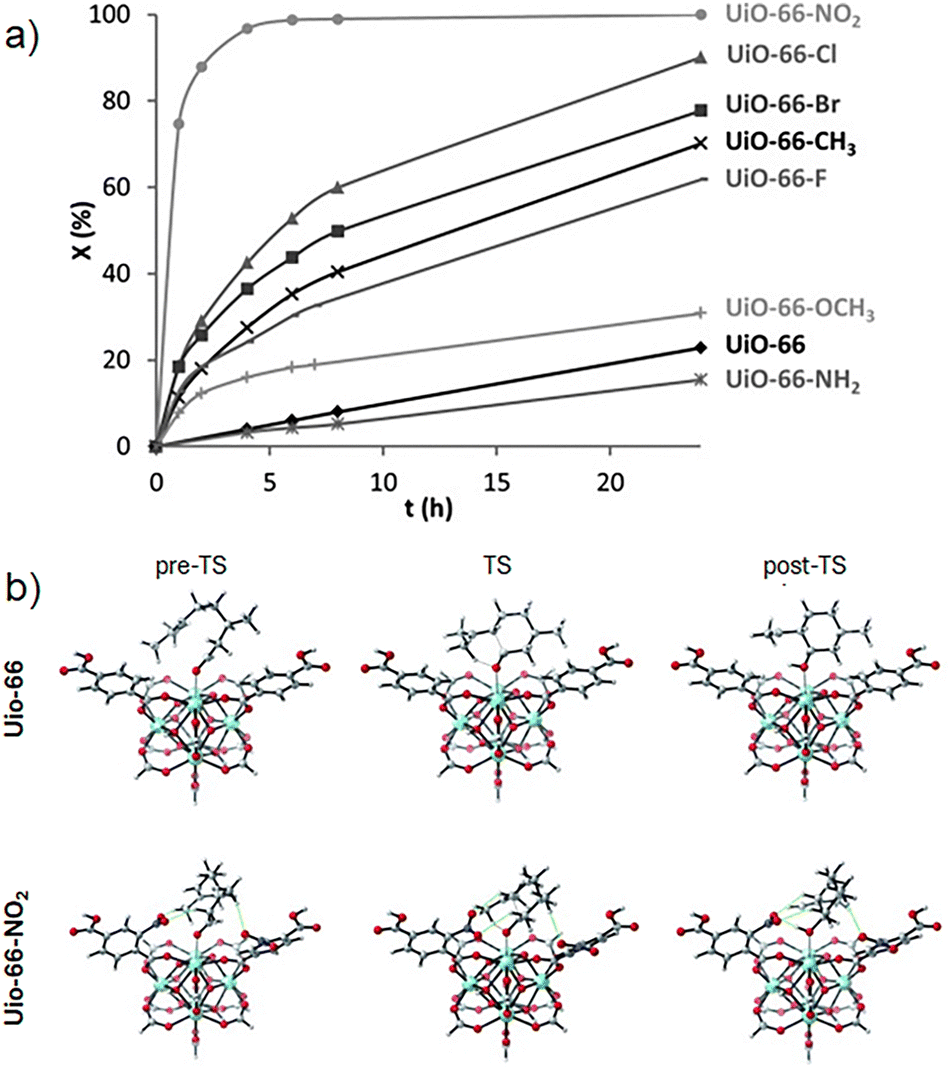 | ||
| Fig. 6 (a) Citronellal conversion (%) over UiO-66-X against time (h), and (b) UiO-66 and UiO-66-NO2 critical states across the reaction profile from citronellal to isopulegol. Pre-TS, TS, and post-TS stands for the adsorption of citronellal, the cyclization transition state, and the adsorbed isopulegol state, respectively. Green lines indicate the interactions between the nitro groups and the hydrogen atoms of citronellal, which are shorter than 3 Å. Adapted from ref. 40 with permission from John Wiley and Sons, copyright 2012. | ||
MOFs can also act as organometallic platforms. Concretely, the MOF structure and tunability provide the possibility to immobilize organometallic catalysts within the framework structure, leading to the creation of a protective effect, which is one of the most significant advantages of using a MOF. Confinement of the active species in a pore can provide protection from other reactive species, a phenomenon which is challenging to achieve in the homogenous phase by ligand engineering alone. Furthermore, immobilization of the catalytic site within the framework avoids catalyst deactivation pathways through aggregation and/or self-association. Additionally, the tunability of the catalyst's microenvironment might extend the lifetime, which translates into higher turnover numbers. Although this approach has demonstrated remarkable catalytic features, it presents challenges in synthetic complexity and characterization difficulty.20,41,42
The synthesis of green MOFs has been an increasing field in recent years; the use of linkers derived from natural sources and green methodologies could represent an advance in MOF synthesis. SU-101 was reported in 2020,43 the synthesis is carried out at room temperature using water as solvent, Bi3+ as metal centre and ellagic acid as linker. This material has been used in energy storage,44 pollutant separation,45 detection46 and catalysis.47 Another green MOF was presented by Wu and collaborators for the selective capture of radioactive iodine.48 The material was prepared using γ-cyclodextrin and NaOH in an environmentally friendly approach, testing four different solvents (DMA, DMF, MeOH, and EtOH). Characterization vía PXRD, FT-IR, TG, BET and SEM demonstrated the correct synthesis of the different CD-MOFs. Materials were tested for the capture of volatile iodine at 75 °C, MOFs synthetized with DMA and DMF (named CD-Na-DMA and CD-Na-DMF) presented the best volatile iodine adsorption of 2.25 and 1.87 g g−1, respectively. Kinetic studies revealed that the adsorption process takes place in a dual model, indicating a process governed by chemisorption and additional physical adsorption interactions. The recyclability of the CD-Na-DMA and CD-Na-DMF materials was tested over five adsorption–desorption cycles, presenting good stability and minimal activity loss. The adsorption capacity was studied in liquid phase using cyclohexane solutions with different iodine concentrations, where CD-Na-DMA and CD-Na-DMF presented maximum adsorption of 709.2 and 701.2 mg g−1, respectively. This work presented an alternative for MOFs in the capture of volatile iodine and the future of green linkers for the synthesis of new MOFs.
The use of other porous materials, such as COFs or POPs, can be an interesting alternative since these materials could be used in catalysis as support of active molecules in reactions of interest.
In 2023, Cui et al. reported the synthesis of phosphine–M complexes (M = Ir and Ru) in covalent organic frameworks (COFs). The 25.0–25.2 wt% ratio at which the coordination complex loadings were coupled to the porous material was sufficient to incorporate the catalytically active centres of the metal complexes, without compromising the high surface area of the material (178–163 m2 g−1). These new composites proved to be effective heterogeneous catalysts for the hydrogenation of quinolines, an important reaction to produce enantiomerically pure heterocyclic amines. These catalysts achieved efficiency yields greater than 90% in 72 h, with identical product stereochemistry compared to the homogeneous systems. They also demonstrated the maintenance of their catalytic activity and selectivity even after 10 catalytic cycles, along with their excellent reactivity and enantioselectivity.49
Another similar example is the union, by metalation, between copper-based coordination complexes ([Cu′Mes]n) and porous organic polymers (POPs), resulting in large surface area active materials (1190–1350 m2 g−1) called Cu-CatPOPs. In 2023, Nguyen et al. reported the synthesis of Cu-CatPOPs and demonstrated their excellent catalytic activity in the selective oxidation of alcohols. Using CatPOPs, faster oxidation rates were obtained compared with the use of non-Cu-containing species, with which no significant reaction occurs, reaching efficiency percentages between 60 and 85% after a reaction time of 20 minutes.50 While these examples may be more sophisticated alternatives, the porous nature of coordination-based materials like MOFs themselves allows for the same qualities without the extra step of bonding the two materials together.
3. Hydrosilylation reaction
Hydrosilylation stands as the most prevalent catalytic reaction for the addition of silicon–hydrogen (Si–H) binding across the existing double or triple bonds found in unsaturated organic compounds. When the silicon group attaches to the terminal carbon, it's categorized as an anti-Markovnikov addition, whereas a Markovnikov addition involves its placement on the more substituted carbon (Fig. 7). This transformation holds significant utility at both laboratory and industrial scales for synthesizing organosilicon compounds, which are highly valued in organic and polymer chemistry.51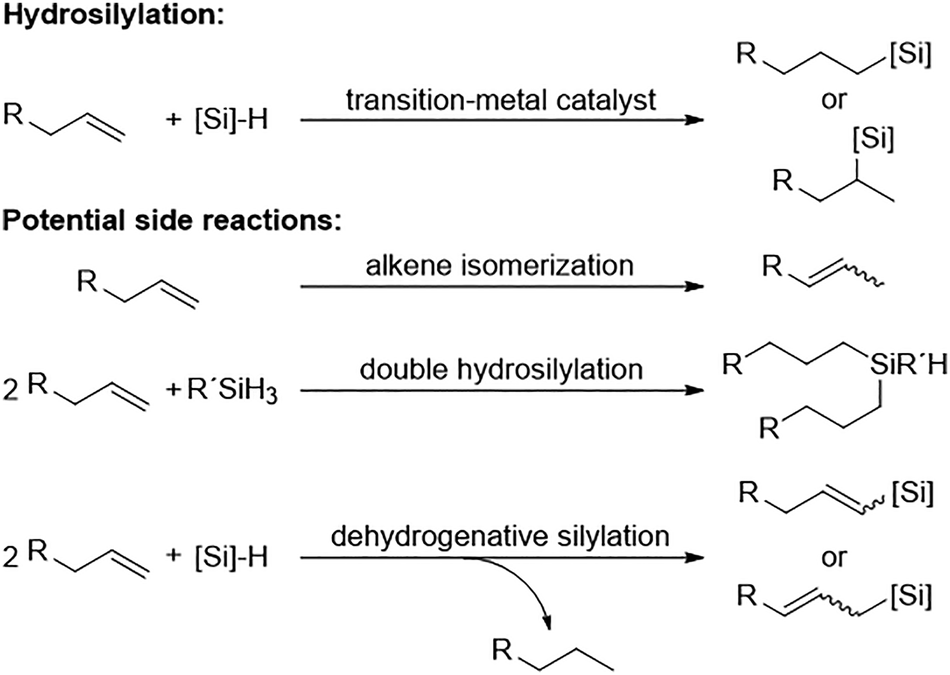 | ||
| Fig. 7 Scheme of alkene hydrosilylation reaction catalysed by transition metal. Adapted from ref. 53 with permission from American Chemical Society, copyright 2017. | ||
Hydrosilylation is typically mediated by homogeneous metal catalysts, operating via a Chalk–Harrod mechanism. This pathway initiates with the oxidative addition of the hydrosilane to the metal catalyst, which is followed by the coordination of the unsaturated organic compound. Subsequently, reductive elimination occurs, yielding the desired product. The application of alkenes and alkynes in this reaction furnishes alkylsilanes and vinylsilanes, respectively; aldehydes and ketones are transformed into silyl ethers; and the incorporation of esters results in the formation of mixed alkylsilyl acetals.52
The predominant catalysts used for this reaction are based on expensive rare metals, including Rh, Pt, and Ru. Specifically, two Pt complexes, Speier's catalyst and Karstedt's catalyst, currently dominate commercial hydrosilylation reactions, particularly for alkenes. These Pt-based catalysts exhibit exceptionally high activity, good anti-Markovnikov selectivity, broad functional group compatibility, and are easy to handle.53 However, their major drawbacks are their non-reusability, the persistence of trace toxic metallic impurities in organic products after use, and the inability to meet the massive demand for these rare metals. Consequently, the development of safer and more abundant metal catalysts has become a central focus of research.54
Inexpensive transition metals like Co, Fe, and Ni have quickly emerged as viable alternatives in the field. Given their significantly greater abundance and comparable effectiveness to their precious counterparts, the aim has been to solidify the integration of these metals into cost-effective and highly active catalysts.53
Bicoordinate bis(amido) metal complexes (M[N(SiMe3)2]2) have been established as effective precursors in the hydrosilylation catalysis of carbonyl compounds. In 2010, Yang and Tilley described the use of a simple Fe2+ silylamide, (Fe[N(SiMe3)2]2), and its outstanding performance for the catalysis of carbonyl groups hydrosilylation in organic compounds. Under mild conditions, aldehydes and ketones underwent catalytic hydrosilylation with the addition of the silane Ph2SiH2 (1.6 equiv.) using catalyst loadings between 0.03 and 2.7 mol% in C6D6. This yielded the corresponding silyl ethers with conversion rates exceeding 90% at varying reaction times (Fig. 8). For the unsaturated substrates investigated in that study (0.49–1.47 mmol), the hydrosilylation turnover numbers were reported between 2200 and 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 min−1. This homogeneous catalytic system demonstrated efficient hydrosilylation of carbonyl functional groups at room temperature using a simple Fe complex, proving to be an accessible catalytic precursor for this reaction. Notably, no special neutral donor ligand or activator was required.54
000 min−1. This homogeneous catalytic system demonstrated efficient hydrosilylation of carbonyl functional groups at room temperature using a simple Fe complex, proving to be an accessible catalytic precursor for this reaction. Notably, no special neutral donor ligand or activator was required.54
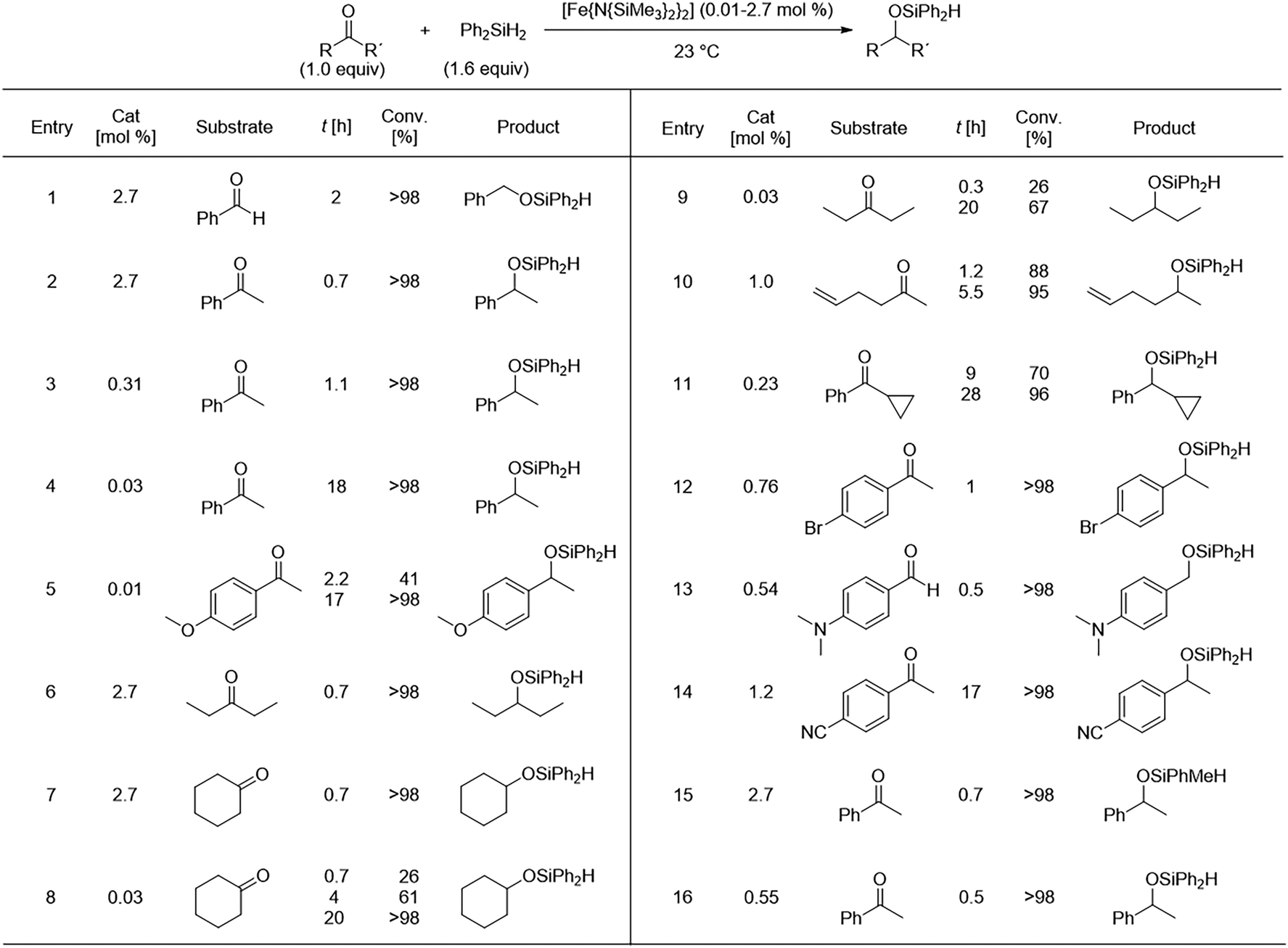 | ||
| Fig. 8 Hydrosilylation of ketones under the catalysis of [Fe[N(SiMe3)2]2. Adapted from ref. 54 with permission from John Wiley and Sons, copyright 2010. | ||
At the time, other metals like Ni were lagging in their use for developing similar two-coordinate bis(amido) metal complexes. However, Lipschutz and Tilley reported in 2012, the successful synthesis and characterization of a stable Ni2+-based complex of this nature, which was quite groundbreaking at the time. This complex, dubbed “1”, was evaluated as a precursor for various reactions. Notably, it was used to form Ni-based compounds with fascinating chemistry and geometries. It also demonstrated its utility in the homogeneous catalysis of olefin hydrosilylation with secondary silanes. Upon reacting complex “1” with 1-octene (55 equiv.) and diphenylsilane (50 equiv.) in d6-benzene, the formation of the product (n-octyl)diphenylsilane was observed at room temperature after 2 hours. This occurred via an anti-Markovnikov addition, achieving a yield greater than 95%. In this manner, the Ni2+ complex “1” was established as an adequate catalyst for several intriguing chemical transformations, particularly excelling in the hydrosilylation of olefins.55
Subsequent research into these types of complexes has been instrumental in introducing heterogeneous catalysts for hydrosilylation that are based on abundant transition metals. Metal–organic frameworks (MOFs) present a novel alternative for the integration of simpler metal moieties as the active sites for the catalytic hydrosilylation of organic substrates. As promising heterogeneous catalysts, MOFs offer the advantage of facile separation from reaction mixtures. Their robust structures further enhance stability and recyclability compared to some homogeneous catalysts. Furthermore, MOFs can be precisely engineered to possess desired porosity and surface area, alongside specific metal centres and organic linkers. This tunability allows for the deliberate tailoring of their catalytic activity and selectivity, thereby facilitating the production of preferred isomers or reaction products.56 Mainly the focus of MOFs based on carboxylates and abundant transition metals have distinguished themselves in recent years for their excellent performance in the hydrosilylation of unsaturated substrates. The metal ions present in the nodes of these MOFs act as accessible reaction sites, something not possible with the simple and direct exposure of a ligand-free salt.57 A good degree of transformation and exhibiting good tolerance to various functional groups are characteristics that have made them applicable for catalysing hydrosilylation across a wide range of alkenes in latest years.
In 2019, the first Ni-carboxylate-based MOF catalyst, designated Ni-L4-1, was reported for the anti-Markovnikov hydrosilylation of alkenes.56 This material, at a loading of 2.5 mol%, displayed to effectively promote the selective terminal hydrosilylation of various alkenes (0.5 mmol) with diphenylsilane (1.05 equiv.) in THF, at temperatures ranging from 40 to 80 °C with varying reaction times. Mostly presenting yields greater than 70% (Fig. 8). Furthermore, this catalyst demonstrated excellent recyclability; simple centrifugation allowed for its separation, and it maintained its efficiency undiminished for 8 cycles (Fig. 9). It also exhibited very good tolerance towards different functional groups. The scalability of the reaction was also demonstrated, with merely 0.01 mol% of the catalyst proving sufficient to catalyse a 150 mmol-scale synthesis, achieving a 95% yield after 40 hours. This corresponds to an impressive turnover number of 9500. The proposed mechanism for this silane addition posits that the metal centre forms a Ni–hydride (Ni–H) intermediate, with the assistance of Na methoxide (NaOMe), which promotes the addition of the silicon group to its desired position (anti-Markovnikov). This clearly showcases the significant synthetic utility of this catalyst for the hydrosilylation of alkenes.
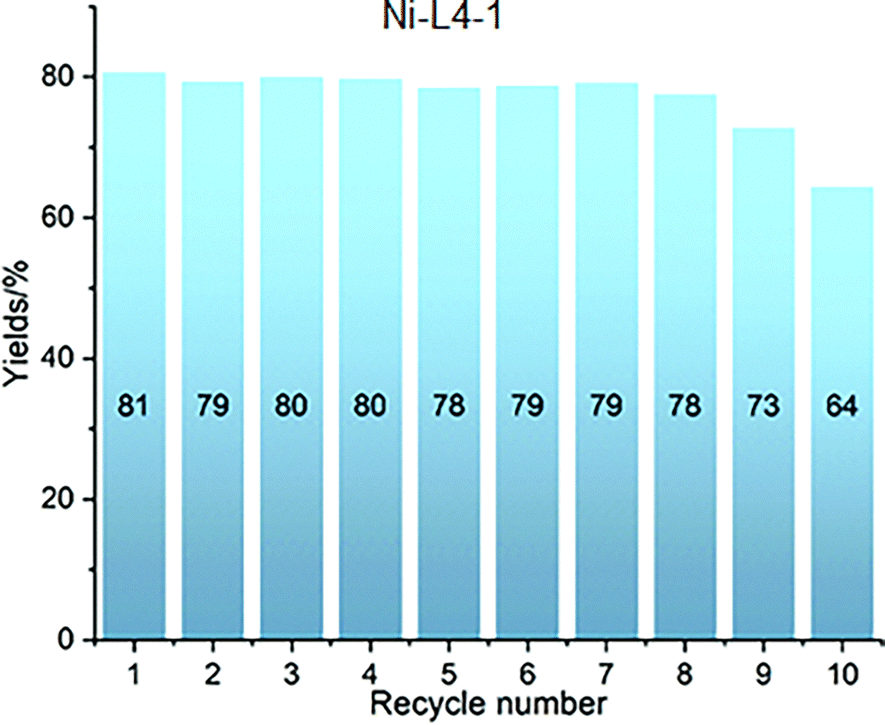 | ||
| Fig. 9 Recycling experiments for nickel carboxylate-based MOF Ni-L4-1. Reproduced from ref. 56 with permission from Royal Society of Chemistry, copyright 2019. | ||
In 2022, the hydrosilylation of olefins catalysed by a Co-carboxylate-based MOF, namely [Co(cptpy)NO3]m, was reported under comparable reaction conditions.57 This material exhibited excellent stability during the hydrosilylation reaction. At 90 °C, a mere 0.1 mol% catalyst loading was sufficient to convert various olefins and styrene (4.0 mmol) into their corresponding linear alkylsilanes with conversions exceeding 90%, utilizing a 1.1:1 H2SiPh2/substrate molar ratio (Fig. 10). The catalyst's exceptional recyclability was also demonstrated; it could be separated by simple gravity after each cycle and was successfully reused 5 times without any additional treatment, retaining both its conversion percentage and structural integrity. Similarly, the proposed mechanism for the Co-based MOF is based on the formation of an intermediate, likely a metal-hydride, obtained from the interaction between the metallic site and the silane (in this case, the reducing agent NaBEt3H assists in the creation of the active hydride species), which promotes the addition. This ultimately leads to the formation of the hydrosilylation adduct, achieving the desired regioselectivity for the silicon group and regenerating the metallic catalyst for the subsequent cycle.
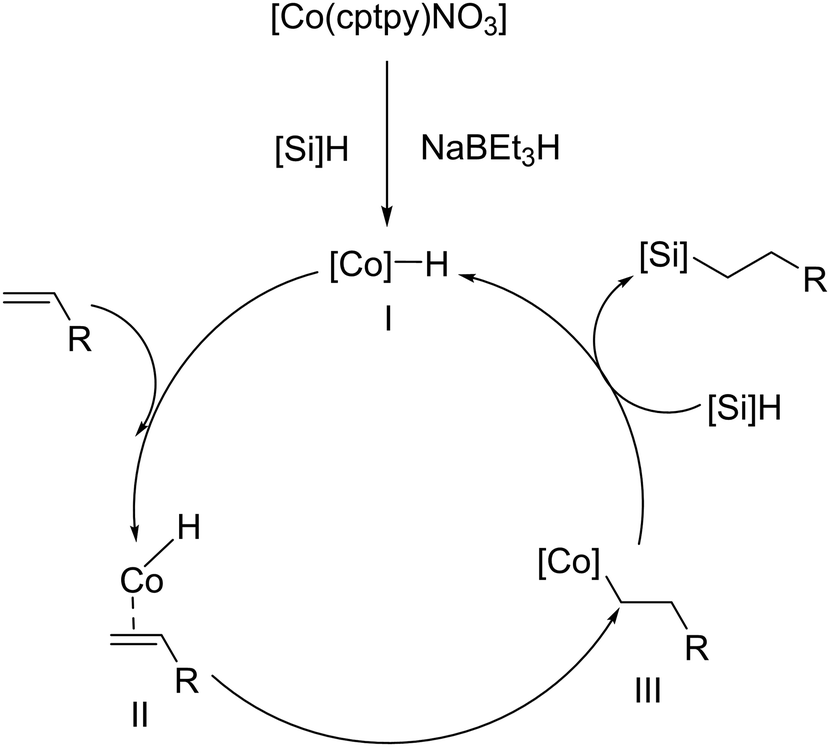 | ||
| Fig. 10 Proposed mechanism for the Co-MOFs catalysed alkene hydrosilylation. Reproduced from ref. 57 with permission from John Wiley and Sons, copyright 2022. | ||
In both the Ni and Co MOFs discussed, the mechanism of action of the active site appears to be quite analogous. This evidence strongly suggests that, despite the current limitations in the range of applicable substrates, the intrinsic chemical and structural properties of MOFs are highly conducive to their use in the hydrosilylation of organic unsaturated substrates. The use of MOFs in catalytic hydrosilylation can be interpreted as a subsequent step to the use of coordination complexes, which is further underscored by the highlighted features of these materials, including their excellent recyclability, synthetic utility, and impressive tolerance to various functional groups. When comparing catalysts (Table 1) based on active metal sites, regardless of whether they are heterogeneous (like MOFs) or homogeneous (like complexes), it is generally acknowledged that the proposed dynamic mechanism for the catalytic process often follows the same fundamental steps. This can even lead to similar conversion percentages for certain applications. However, the nature of the specific catalytic process itself dictates the precise requirements necessary to select the optimal catalyst conditions.
| Catalyst | Catalyst loading (mol%) | Effective yield (%) | TOFa/TONb | Recyclability | Ref. |
|---|---|---|---|---|---|
| a Turnover frequency (TOF). b Turnover number (TON). | |||||
| Fe[N(SiMe3)2]2 | 0.03–2.7 | >90 | 2200–10000 min−1a |
— | 54 |
| Ni2+ bis(amido) complex “1” | 2 | >95 | — | — | 55 |
| Ni-L4-1 | 2.5 | >70 | 9500b | 8 cycles | 56 |
| [Co(cptpy)NO3]m | 0.1 | >90 | 9500b | 5 cycles | 57 |
Research focused on incorporating abundant transition metals as active sites for the catalytic hydrosilylation of unsaturated organic compounds continues to represent a significant area of opportunity for future development. The ongoing shift from precious metals to earth-abundant alternatives like Co, Ni and Fe is driven by considerations of sustainability, cost, and availability. However, all the work carried out to date on catalysts, spanning from early homogeneous complexes to modern MOFs, has undeniably contributed to a clearer perspective on the fundamental nature of this highly important reaction in organic and polymer chemistry. This collective knowledge has also illuminated the pressing needs that must be addressed and their order of priority in the ongoing evolution of efficient and cost-effective catalysts for hydrosilylation, paving the way for more sustainable chemical processes.
4. Water oxidation/oxygen evolution reaction
Environmental problems have been a great concern over the past two decades, and clean energy has become the focus of many investigations. The synthetic route of photosynthesis attracted significant attention since this reaction can produce valuable chemical products. However, the water oxidation step is considered a challenge for this process, since the reaction requires the transfer of protons from the catalytic site and the formation of molecular oxygen (O2). Thus, the catalysis of water oxidation has been the focus of multiple studies, and the use of earth-abundant metals presents an interesting alternative.58,59 In 2009, Tilley et al., presented the use of cubic Co oxide spinel [Co3O4] nanoparticles for alkaline water electrolysis.60 The research proved the relation between the particle size and the catalytic activity of the material. Nanoparticle size was changed using different ratio of an aqueous ammonium hydroxide solution in a water/ethanol solvent system, in the absence of surfactants or stabilization groups that could inhibit the catalytic activity of the material. Additionally, TEM and PXDR studies showed three different particle sizes: small, medium, and large (5.9, 21.1, and 46.9 nm). BET surface areas were lower than estimated, but this was due to possible aggregation problems. [Co3O4] nanoparticles were tested in Ni foam supports as anodes for water oxidation in alkaline solution (1.0 M KOH), and high activity was present for anodes loaded with 1 mg cm−2 of the synthesized material. The current density of 10 mA cm−2 was achieved at overpotentials of 328, 363, and 382 mV when the particle size was increased. The electrochemical overpotential was plotted against the BET surface area, suggesting that the activity of the catalyst can increase as a function of the area in the material.In 2013, Tilley and coworkers presented the synthesis of Co metaphosphate [Co(PO3)2] nanoparticles for water oxidation at neutral pH.61 The material was synthesized using the TMP (thermolytic molecular precursor) method and deposited on the surface of a Ni-foam electrode. Since [Co(PO3)2] presented low stability in strong acid or basic conditions, the tests for water oxidation were performed under phosphate-buffered water (pH = 6.4). The nanoparticles showed electroactivity at an overpotential of 310 mV, which presented an advantage over other Co catalysts ([Co3O4], [Co3(PO3)2] thin films). In addition, post-catalytic characterization of the material indicates a restructuring phenomenon occurring in the catalyst, since CoOx species were present on the surface of the material. The authors noted a synergistic effect between the surface-bound species, which play a significant role in the water oxidation catalysis, as different electrocatalytic results were observed across various Co materials. Later, in 2015, Tilley and co-workers studied a Co cubane complex [Co4O4(OAc)4py4] (py = pyridine, OAc = acetate).62 Electrochemical experiments were performed over a wide pH range, demonstrating that the cubane complex reacts with H+ and OH− to give observable intermediates. Experiments also showed that Co complexes must be oxidized to a state of [Co4+Co33+] to be active. To regenerate the [Co34+] complex, sodium hydroxide must be added, leading to the formation of O2. Additionally, characterization and isotopic labeling experiments suggested that the formation of the O–O bond occurs via terminal oxo ligands (Fig. 11). This implies that the cubane core remains unchanged during the oxygen evolution reaction (OER). The findings presented in this work provided a mechanistic understanding that can lead to the design and development of catalysts for water oxidation reactions.
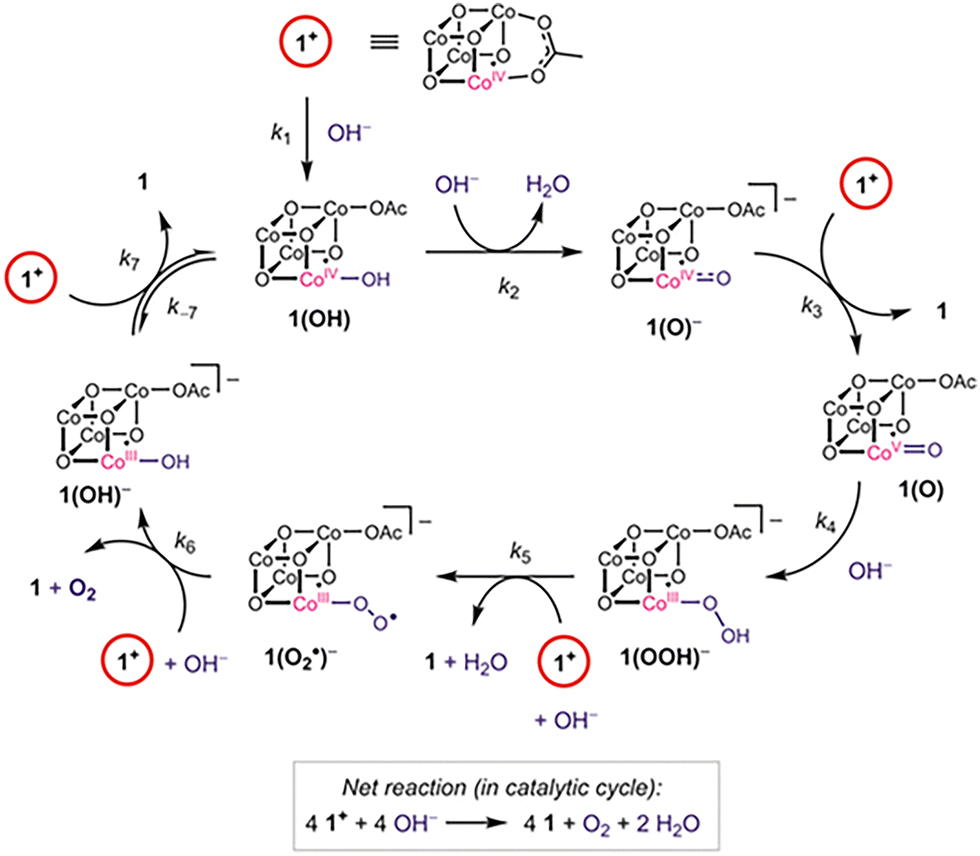 | ||
| Fig. 11 Proposed mechanism for the cubane complex in the oxygen evolution reaction. Reproduced from ref. 62 with permission from American Chemical Society, copyright 2015. | ||
In a subsequent study, the performance of an Fe-doped Ni (oxy)hydroxide [FexNi1−xOOH] catalyst was studied.63 The addition of Fe3+ into the NiOOH catalyst showed a decrease in faradaic resistance, and the charge relaxation became more favourable. The material demonstrated excellent electrocatalytic activity for the oxygen evolution reaction (OER) when Fe doping was approximately 35% [Fe0.35Ni0.65OOH]. It is suggested that the formation of charged intermediates and structural considerations play a significant role in the current observed for the reaction. Monometallic Fe and Ni materials presented low catalytic activity due to poorly stabilized intermediates. The activation energy and faradaic resistances obtained for both materials are higher than on the [Fe0.35Ni0.65OOH] surface; this could be related to strong binding between the OER intermediates and the pure metallic sites. As presented by Tilley, the valence state in the metal plays an important role in the activity of the material. Zhang and collaborators explored the effect of valence states in Mn-nanocatalysts, using the CO hydrogenation to aromatics as a model reaction.64 Different Mn oxides were prepared containing different oxidation states, where the +2 state presented the best performance, showing high selectivity (82.9%) to aromatic hydrocarbons with enhanced stability compared to other catalysts (MoZr, CeZr and Zn-oxides). Additional computational studies were crucial for proposing reaction pathways that are important to understanding the reaction and avoiding undesired products. These results present an important opportunity for the design and optimization of catalysts that can transform small molecules into valuable products.
Metal–organic complexes that are supported by organic ligands can also serve as catalysts in EOR reactions. For instance, Wang et al., reported the induction of coordinatively unsaturated metal sites (CUMSs) in a metal–organic complex consisting of phytic acid–Co2+ (referred to as Phy–Co2+) using plasma technology at room temperature.65 This process is clean, requires no additional reagents, and takes only a few minutes. The high energy involved can etch the surface of the material, generating defects like oxygen vacancies. However, plasma treatment partially destroyed the coordination geometry of the Co–O bond, thereby facilitating the accessibility of more CUMSs. Several characterization techniques, including electron spin resonance and XAS (X-ray absorption spectra), provided substantial evidence that the coordination geometry of the treated material, now called P–Phy–Co2+, was modified when CUMSs were formed. The properties presented were expected to produce high OER activity since the unsaturated sites can act as active centres for the reaction. The required overpotential to reach 10 mA cm−2 for P–Phy–Co2+ (306 mV) presented an improvement over the Phy–Co2+ (383 mV) sample, indicating that the CUMSs induced by the plasma treatment enhanced the activity of the material. This strategy was further applied to a bimetallic system, where the Phy–Co2+/Fe3+ only needed an overpotential of 265 mV to reach the same 10 mA cm−2; this result presented is superior to other commercial materials. This work presents a novel approach in the creation CUMSs in metal–organic complexes, improving their application and performance in OER reactions.
In 2023, Budagumpi and co-workers presented the use of N-heterocyclic carbene (NHC) ligands to obtain Ni and Co complexes for OER applications.66 The synthesized materials presented excellent overpotentials of 373–424 mV to reach 50 mA cm−2 of current density. However, the electrochemical activity was tested with the addition of conductive carbon materials; highly conductive graphitic carbon was added to the synthesized materials. The overpotentials of the composite-complexes improved to 323–373 mV to achieve the same 50 mA cm−2 of current density. The high activity presented could be due to several factors, including the selected metal or the coordination of the complex, as well as the π-conjugated systems in the ligands. However, the complex and the graphitic carbon material presented stable π–π stacking interactions that could also play an important role in the OER activity of the catalysts (Fig. 12).
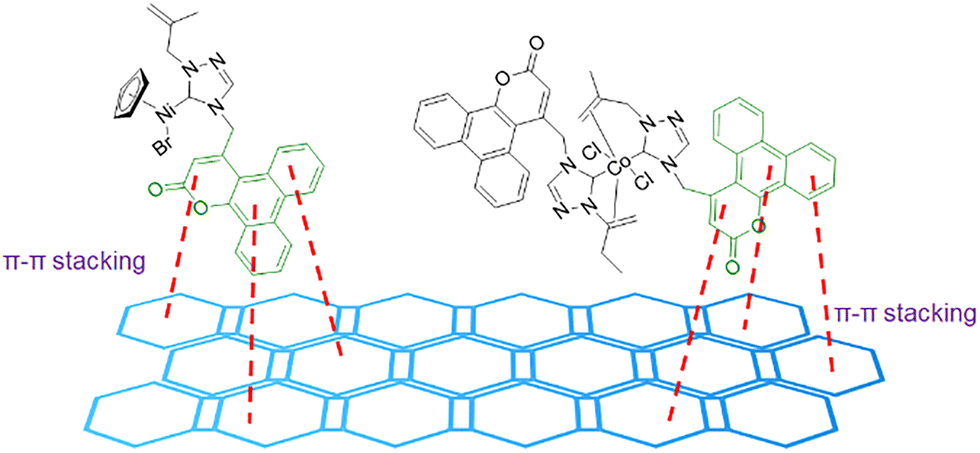 | ||
| Fig. 12 π–π stacking interactions between the coumarin and the graphitic carbon material. Adapted from ref. 66 with permission from Royal Society of Chemistry, copyright 2024. | ||
Over the years, Ru complexes have been widely studied due to their high catalytic activity in water oxidation. However, these homogeneous catalysts face several challenges, including poor reusability, difficulties in isolation, and intermolecular degradation. MOFs offer a promising alternative since their porous properties can facilitate the diffusion of reactants and gases through the crystalline structure. Additionally, MOFs can distribute metal complexes uniformly within their frameworks, with these sites serving as active catalytic sites. For instance, Morris et al. presented the synthesis of MOF UiO-67 thin films doped with a Ru complex [Ru(tpy)(dcbpy)OH2]2+ (tpy = 2,2′:6′,2′′-terpyridine, dcbpy = 5,5′-dicarboxy-2,2′-bipyridine) (Fig. 13a).67 Characterization confirmed the correct synthesis of UiO-67 and the presence of the Ru linker in the structure. This material, referred to as Ru-UiO-67, was tested for water oxidation electrocatalysis. The Ru-UiO-67 thin films presented a steady catalytic current of 71 μA cm−2 and a faradaic efficiency of 55%. Control experiments (undoped UiO-67 and FTO thin films) demonstrated that the incorporation of Ru plays a critical role in the electrochemical water oxidation reaction. The Ru-doped MOF produced 0.7 mmol of O2, with a calculated TOF of 0.2 s−1, and the material exhibited structural and electrochemical stability for 1 h. This study provides a novel approach to immobilizing electroactive Ru catalysts within MOFs for water splitting applications.
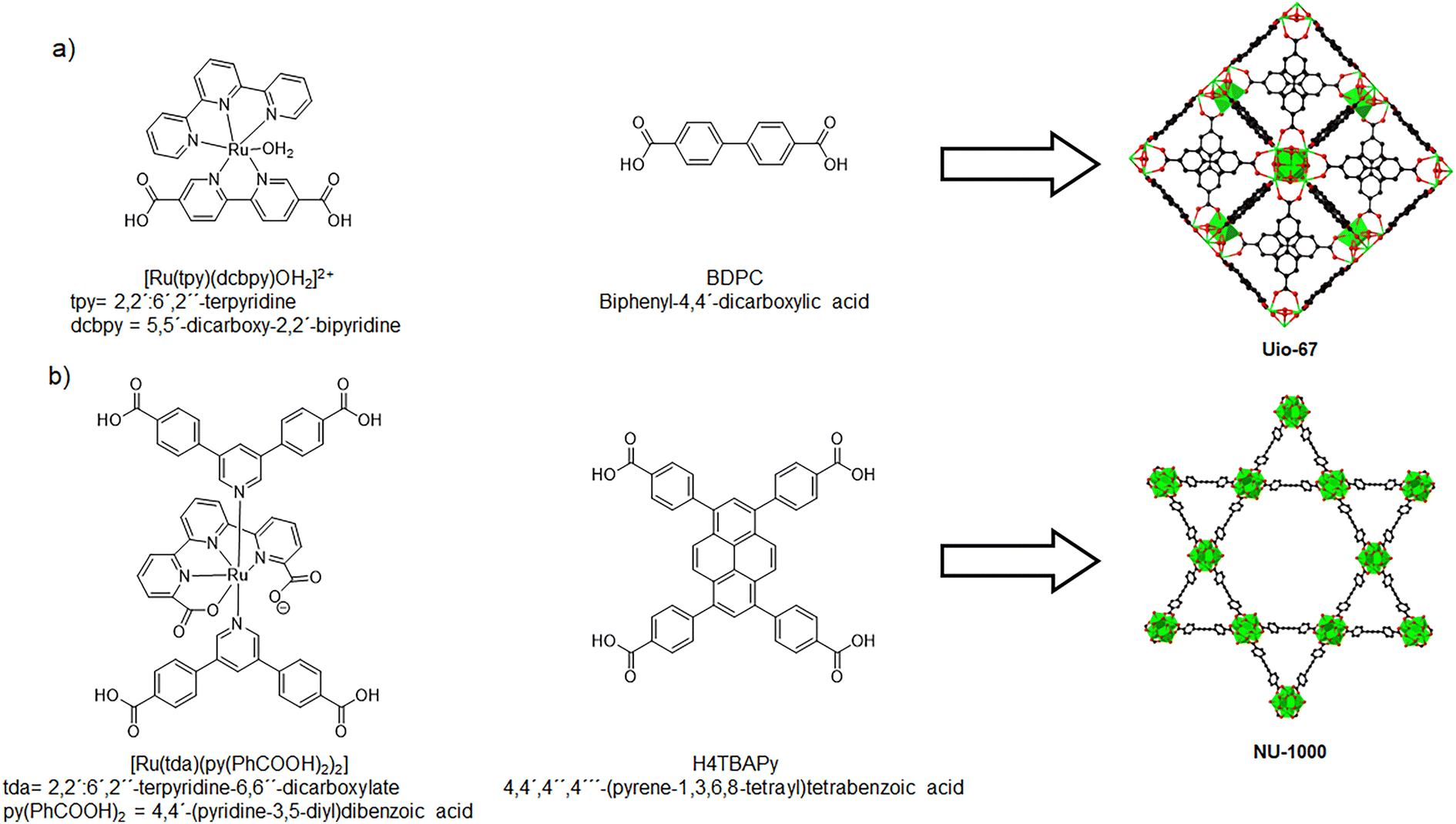 | ||
| Fig. 13 Ru linkers that present geometric similarities with existing MOF linkers. (a) Mixed linkers used for the synthesis of Ru-Uio-67. (b) Mixed linkers used for the synthesis of NU-1000-Ru. Adapted from ref. 67 and 68 with permission from John Wiley and Sons and Royal Society of Chemistry, copyright 2016 and 2022. | ||
In 2022, a variation of NU-1000 was synthesized using a mixture of linkers, (Fig. 13b).68 A new Ru metallo-linker [Ru(tda)(py(PhCOOH)2)2] (tda = 2,2′:6′,2′′-terpyridine-6,6′′-dicarboxylate, py(PhCOOH)2 = 4,4′-(pyridine-3,5-diyl)dibenzoic acid) was prepared, which has similarities with H4TBAPy, the native linker of the MOF. The mixed linker MOF was synthesized using solvothermal methods and characterized by PXRD and SEM. Additional FTIR and EDX experiments confirmed the presence of the Ru linker within the MOF, where the characteristic signals of the [Ru(tda)(py(PhCOOH)2)2] were present. The maximum proportion of the Ru linker used was 30% (the resulting material was named NU-1000-Ruhigh). Nevertheless, it was found that the final Ru content was only 5.7%. This could be attributed to several factors, like different pKa of the coordinating carboxylates or poor solubility of the Ru linker. The cyclic voltammograms for NU-1000-Ruhigh presented new waves, which are from the metallo-linkers that are in close contact with the electrode. Nevertheless, it was discovered that most of these Ru linkers are not directly accessible; the process occurs indirectly through prior oxidation of the TBAPy linkers. The material was tested as a catalyst in the electrochemical oxidation of water. A potential of 1.30 V was applied to the sample, giving a low faradaic efficiency of 37%. This could be due to the oxidative transformation of the TBAPy and Ru linkers, which takes place at the same potential in the aqueous electrolyte. It is important to note that a control experiment was conducted with the NU-1000 material, obtaining negligible amounts of oxygen in the reaction, demonstrating the importance of the Ru linker in the MOF as a water oxidation catalyst.
However, the use of precious metals is not viable for large-scale applications due to their abundance and value. Thus, the development of MOFs using only earth-abundant metals (Co, Ni, Fe) is the focus of several groups. Sun et al. reported the synthesis of an efficient electrocatalyst based on a Ni-MOF nanosheet array on Ni foam (Ni-MOF/NF).69 PDRX and FTIR characterizations were used to confirm the correct synthesis of the Ni-MOF, while SEM confirmed that the Ni-MOF nanosheet completely covered the Ni foam substrate. The material was tested for electrocatalytic OER activity, needing an overpotential of 320 mV to achieve 100 mA cm−2, presenting favorable results compared to other Ni-based catalysts. Ni-MOF/NF showed a high turnover frequency (TOF) of 0.25 mol O2 s−1 and an excellent stability working for 20 h without significant loss in its catalytic activity. This work provided one of the first alternatives for MOFs as electrocatalysts in the oxygen evolution reaction, presenting new opportunities in this field.
A promising approach is the use of bimetallic MOFs; the synergic effect between metals can be beneficial for electrocatalysis applications. In 2021, Zhao and coworkers presented a NiFe-MOF deposited in graphene nanosheets.70 The material (named NiFe-MOF/G) was synthesized with a 4:1 Ni:Fe molar ratio, characterization demonstrated homogeneous dispersion of the MOF grains in the graphene nanosheets. The catalytic activity of the material was tested, OER experiments were performed, obtaining low overpotentials of 258 mV to achieve 10 mA cm−1, a faradaic efficiency of 98.6%, and it was confirmed no side reactions occur through the process. The NiFe-MOF/G nanocomposites presented stability for more than 30 h of continuous activity; the material presented excellent performance compared to the monometallic and bulk NiFe-MOF catalysts. These results suggested the importance of doping the catalyst with Fe cations; the simulations revealed an increase in the accessibility of the active sites in the material and a decrease in the free energy for the rate-determined OER step (Fig. 14). This research highlights the importance of the role of metal centres in MOFs for electrocatalysis and water oxidation applications. The development of catalysts that are highly active towards water oxidation is a pivotal step in the quest for new technologies and clean energy. Over the years, numerous materials have been put to the test, yielding intriguing results (Table 2) and deepening our understanding of the reaction mechanism. While nanoparticles and metal complexes have shown promise, their reusability remains a challenge. In this context, MOFs stand out as a potential solution, given their unique properties. However, more relevant studies are needed to enhance their activity for water oxidation applications. Don Tilley's investigations inspired other groups and laid the bases for the use of these materials in reactions like the oxygen evolution reaction.
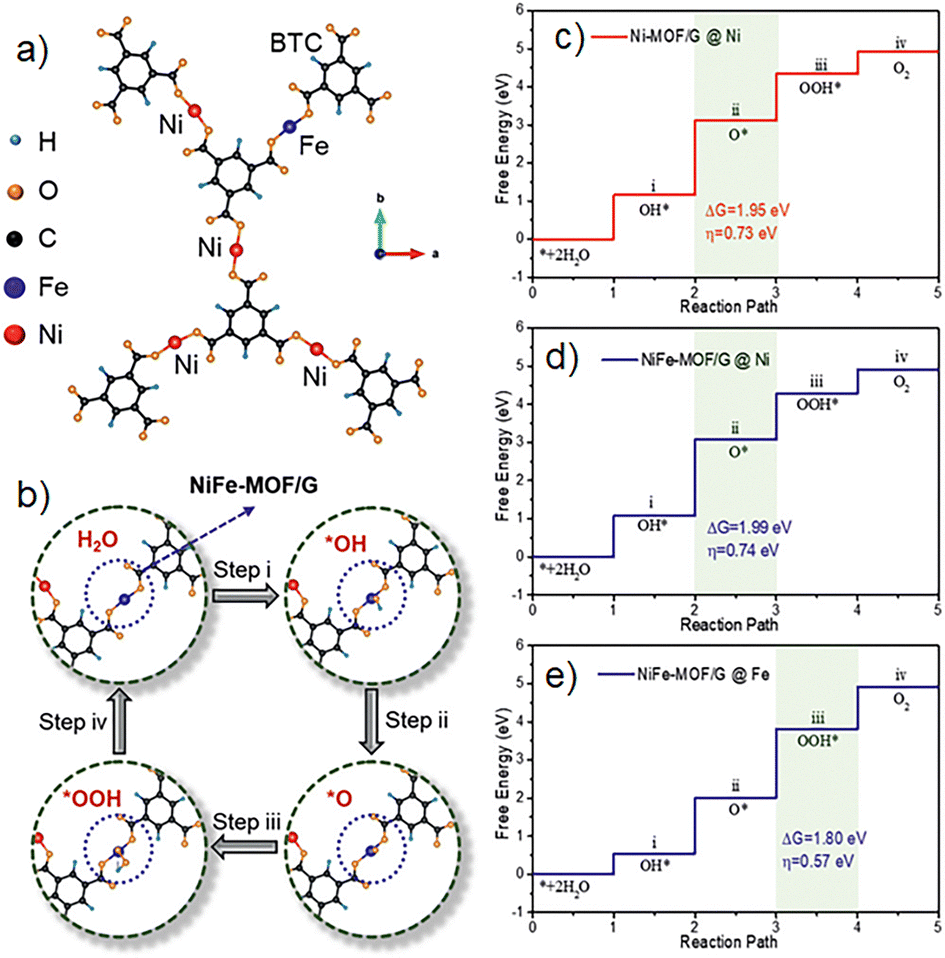 | ||
| Fig. 14 (a) Simulation of the NiFe-MOF/G model, (b) active site and steps for the OER. Free energy diagram for four steps on (c) Ni-MOF/G, (d) NiFe-MOF/G@Ni and (e) NiFe-MOF/G@Fe. Reproduced from ref. 70 with permission from John Wiley and Sons, copyright 2021. | ||
| Catalyst | Overpotential (mV) | Current density | TOF | Recyclability | Ref. |
|---|---|---|---|---|---|
| [Co3O4] | 328, 363, and 382 | 10 mA cm−2 | 1.87 × 10−2 s−1 | — | 60 |
| [Co(PO3)2] | 310 | 0.17 mA cm−2 | 0.002–0.005 s−1 | — | 61 |
| [Co4O4(OAc)4py4] | 728 | 2 mA cm−2 | 0.2 s−1 | — | 62 |
| [Fe0.35Ni0.65OOH] | 325 | 10 mA cm−2 | — | — | 63 |
| Phy–Co2+/Fe3+ | 265 | 10 mA cm−2 | — | 2000 cycles | 65 |
| Ni–NHC, Co–NHC | 323, 373 | 50 mA cm−2 | — | — | 66 |
| Ni-MOF/NF | 320 | 100 mA cm−2 | 0.25 s−1 | 500 cycles and 20 h | 69 |
| Ni–Fe MOF/G | 258 | 10 mA cm−1 | 1.80 s−1 | 32 h | 70 |
5. Conclusions
The development of highly active catalysts has been essential to both industrial processes and environmental remediation. In the pursuit of novel technologies, catalysts built upon the coordination of abundant transition metals have consistently demonstrated their ability to achieve favourable and impactful results. This tribute review to Professor Tilley, focuses on the pivotal role that these supramolecular compounds have developed for reactivity research in which molecules of interest and the active metal sites interact to boost the catalysis for a wide range of reactions.Don Tilley's foundational contributions to coordination complex catalysis have undeniably marked a turning point in the development of the field. The incorporation of abundant transition metals into coordination complexes, rather than costly noble metals, presented an opportunity for new materials such as metal–organic frameworks to improve their performance as catalysts in different reactions. The incorporation of MOFs, which are porous crystalline materials rooted in highly stable coordination chemistry, can be understood as a consequent advance to the use of coordination complexes. The implementation of these materials as heterogeneous catalysts seeks to simplify the recovery of the material and improve their reuse, without compromising efficiency or selectivity. This shift towards more sustainable and viable solutions was a significant step forward in catalysis.
The current generation of catalysts is now expected to serve as a source of inspiration to encourage oncoming generation of researchers. To explore novel research lines and broaden their engagement in the development of innovative alternatives and the use of these materials for the solution of future challenges. Coordination chemistry and chemical reactivity are two fields that still offer a vast number of starting points for new ideas. Especially those delving into merging new technologies, such as machine learning or optimization simulations, for hybrid and increasingly comprehensive works.
Author contributions
All authors contributed in writing the original draft, reviewing and editing the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
All data is available in the main text and we asked for the permission for the figures.Supplementary information (SI) is available. See DOI: https://doi.org/10.1039/d5cc04776e.
Acknowledgements
M. L. M. and P. M. R., thank SECIHTI for the PhD fellowships (1080048 and 1277642). A. H. M. thank SECIHTI for the MSc fellowship (1336791). We would like to express our gratitude to the Metropolitan Autonomous University of Mexico for providing financial support for the maintenance of equipment.References
- James E. Huheey, Ellen A. Keiter and R. L. Keiter, Inorganic Chemistry Principles of Structure and Reactivity, HarperCollins College Publishers, New York, 4th edn, 1993 Search PubMed.
- G. B. Kauffman, Encyclopedia of Inorganic Chemistry, 2005 DOI:10.1002/0470862106.ia048.
- Ralph W. G. Wyckoff and E. Posnjak, J. Am. Chem. Soc., 1921, 43, 2292–2309 CrossRef CAS.
- L. F. Dahl and R. E. Rundle, Acta Crystallogr., 1963, 16, 419–426 CrossRef CAS.
- G. G. Sumner, H. P. Klug and L. E. Alexander, Acta Crystallogr., 1964, 17, 732–742 CrossRef CAS.
- Daniel A. Straus, T. D. Tilley, Arnold L. Rheingold and S. J. Geib, J. Am. Chem. Soc., 1987, 109, 5872–5873 CrossRef CAS.
- C.-M. Che, C.-C. Kwok, S.-W. Lai, A. F. Rausch, W. J. Finkenzeller, N. Zhu and H. Yersin, Chem. – Eur. J., 2010, 16, 233–247 CrossRef CAS PubMed.
- H. Shamran Mohammed and V. D. Tripathi, J. Phys.: Conf. Ser., 2020, 1664, 012070 CrossRef.
- Rehab H. Elattar, Samah F. El-Malla, Amira H. Kamal and F. R. Mansour, Coord. Chem. Rev., 2024, 501, 215568 CrossRef CAS.
- B. Cornils, W. A. Herrmann and R. W. Eckl, J. Mol. Catal. A: Chem., 1997, 116, 27–33 CrossRef CAS.
- A. W. Parkins, in Studies in Inorganic Chemistry, ed. F. R. Hartley, 1991, ch. 5, vol. 11, pp. 106–123 Search PubMed.
- B. Cornils and W. A. Herrmann, J. Catal., 2003, 216, 23–31 CrossRef CAS.
- F. Zhou, Q. Xin, Y. Fu, Z. Hua, Y. Dong, M. Ran, H. Song, S. Liu, R. Qu, Y. Yang, X. Zhang, C. Zheng and X. Gao, Chem. Eng. J., 2023, 464, 142471 CrossRef CAS.
- L. Wang, J. Jin, W. Li, C. Li, L. Zhu, Z. Zhou, L. Zhang, X. Zhang and L. Yuan, Energy Environ. Sci., 2024, 17, 9122–9133 RSC.
- J. W. Steed and J. L. Atwood, Supramolecular Chemistry, 3rd edn, 2022 Search PubMed.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- U. Kokcam-Demir, A. Goldman, L. Esrafili, M. Gharib, A. Morsali, O. Weingart and C. Janiak, Chem. Soc. Rev., 2020, 49, 2751–2798 RSC.
- C. E. Housecroft and A. G. Sharpe, Quimica inorganica Housecroft, Pearson, 5th edn, 2018 Search PubMed.
- G. A. Bhat and D. J. Darensbourg, Coord. Chem. Rev., 2023, 492, 215277 CrossRef CAS.
- R. A. Peralta, M. T. Huxley, J. D. Evans, T. Fallon, H. Cao, M. He, X. S. Zhao, S. Agnoli, C. J. Sumby and C. J. Doonan, J. Am. Chem. Soc., 2020, 142, 13533–13543 CrossRef CAS PubMed.
- D. Astruc, Organometallic Chemistry and Catalysis, Springer, Berlin, Heidelberg, 1st edn, 2007 Search PubMed.
- A. A. Malik, W. Yang, Z. Ma and W.-H. Sun, Catalysts, 2019, 9 Search PubMed.
- B. K. Werley, X. Hou, E. P. Bertonazzi, A. Chianese and T. W. Funk, Organometallics, 2023, 42, 3053–3065 CrossRef CAS PubMed.
- J. N. H. Reek, B. de Bruin, S. Pullen, T. J. Mooibroek, A. M. Kluwer and X. Caumes, Chem. Rev., 2022, 122, 12308–12369 CrossRef CAS PubMed.
- J. Trouve and R. Gramage-Doria, Chem. Soc. Rev., 2021, 50, 3565–3584 RSC.
- P. Cossee, J. Catal., 1964, 3, 80–88 CrossRef CAS.
- E. J. Arlman, J. Catal., 1964, 3, 89–98 CrossRef CAS.
- E. J. Arlman and P. Cossee, J. Catal., 1964, 3, 99–104 CrossRef CAS.
- P. Liu and N. Zheng, Nat. Sci. Rev., 2018, 5, 636–638 CrossRef CAS.
- R. Qin, K. Liu, Q. Wu and N. Zheng, Chem. Rev., 2020, 120, 11810–11899 CrossRef CAS PubMed.
- B. C. Gates, Chem. Sci., 2024, 15, 16821–16843 RSC.
- X.-F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS PubMed.
- W. Liu, L. Zhang, X. Liu, X. Liu, X. Yang, S. Miao, W. Wang, A. Wang and T. Zhang, J. Am. Chem. Soc., 2017, 139, 10790–10798 CrossRef CAS PubMed.
- P. Marín-Rosas, N. S. Portillo-Vélez, J. G. Flores, E. González-Zamora, A. Islas-Jácome, L. D. Herrera-Zúñiga, I. A. Ibarra, M. Viniegra, N. Martín-Guaregua, R. A. Peralta and J. Aguilar-Pliego, J. Mex. Chem. Soc., 2025, 69, 259–267 CrossRef.
- Y. Zhang, X. Yu, Y. Hou, C. Liu, G. Xie and X. Chen, Mol. Catal., 2024, 555, 113851 CAS.
- P. Lyu, A. M. Wright, A. López-Olvera, P. G. M. Mileo, J. A. Zárate, E. Martínez-Ahumada, V. Martis, D. R. Williams, M. Dincă, I. A. Ibarra and G. Maurin, Chem. Mater., 2021, 33, 6186–6192 CrossRef CAS.
- J. G. Flores, J. A. Zarate-Colin, E. Sanchez-Gonzalez, J. R. Valenzuela, A. Gutierrez-Alejandre, J. Ramirez, V. Jancik, J. Aguilar-Pliego, M. C. Zorrilla, H. A. Lara-Garcia, E. Gonzalez-Zamora, G. Guzman-Gonzalez, I. Gonzalez, G. Maurin and I. A. Ibarra, ACS Appl. Mater. Interfaces, 2020, 12, 18885–18892 CrossRef CAS PubMed.
- R. A. Peralta, P. Lyu, A. Lopez-Olvera, J. L. Obeso, C. Leyva, N. C. Jeong, I. A. Ibarra and G. Maurin, Angew. Chem., Int. Ed., 2022, 61, e202210857 CrossRef CAS.
- J. L. Obeso, A. Lopez-Olvera, C. V. Flores, R. A. Peralta, I. A. Ibarra and C. Leyva, Chem. Commun., 2023, 59, 3273–3276 RSC.
- F. Vermoortele, M. Vandichel, B. Van de Voorde, R. Ameloot, M. Waroquier, V. Van Speybroeck and D. E. De Vos, Angew. Chem., Int. Ed., 2012, 51, 4887–4890 CrossRef CAS.
- V. Pascanu, G. Gonzalez Miera, A. K. Inge and B. Martin-Matute, J. Am. Chem. Soc., 2019, 141, 7223–7234 CrossRef CAS PubMed.
- F. Chen, H. F. Drake, L. Feng, J. A. Powell, K.-Y. Wang, T.-H. Yan and H.-C. Zhou, Inorganics, 2021, 9, 27 CrossRef CAS.
- E. S. Grape, J. G. Flores, T. Hidalgo, E. Martinez-Ahumada, A. Gutierrez-Alejandre, A. Hautier, D. R. Williams, M. O'Keeffe, L. Ohrstrom, T. Willhammar, P. Horcajada, I. A. Ibarra and A. K. Inge, J. Am. Chem. Soc., 2020, 142, 16795–16804 CrossRef CAS.
- R. A. Marquez, J. L. Obeso, R. R. Vaidyula, V. B. López-Cervantes, R. A. Peralta, P. Marín Rosas, J. A. de los Reyes, C. B. Mullins and I. A. Ibarra, J. Mater. Chem. A, 2024, 12, 32735–32744 RSC.
- J. Piątek, T. M. Budnyak, S. Monti, G. Barcaro, R. Gueret, E. S. Grape, A. Jaworski, A. K. Inge, B. V. M. Rodrigues and A. Slabon, ACS Sustainable Chem. Eng., 2021, 9, 9770–9778 CrossRef.
- V. B. Lopez-Cervantes, J. L. Obeso, J. G. Flores, A. Gutierrez-Alejandre, R. A. Marquez, J. A. de Los Reyes, C. V. Flores, N. S. Portillo-Velez, P. Marin-Rosas, C. A. Celaya, E. Gonzalez-Zamora, D. Solis-Ibarra, R. A. Peralta and I. A. Ibarra, Chem. Sci., 2025, 16, 5483–5492 RSC.
- J. L. Obeso, J. G. Flores, C. V. Flores, V. B. Lopez-Cervantes, V. Martinez-Jimenez, J. A. de Los Reyes, E. Lima, D. Solis-Ibarra, I. A. Ibarra, C. Leyva and R. A. Peralta, Dalton Trans., 2023, 52, 12490–12495 RSC.
- Y. Cai, J. Li, Q. Zhang, C. Liu, C. Wang, H. Shi, L. Jiang and D. Wu, Sep. Purif. Technol., 2025, 376, 134078 CrossRef CAS.
- Q. Sun, B. Aguila, J. Perman, L. D. Earl, C. W. Abney, Y. Cheng, H. Wei, N. Nguyen, L. Wojtas and S. Ma, J. Am. Chem. Soc., 2017, 139, 2786–2793 CrossRef CAS PubMed.
- Y. Zhu, D. Mukherjee, T. R. Helgert and S. T. Nguyen, CCS Chem., 2023, 5, 445–454 CrossRef CAS.
- S. J. Clarson, Silicon, 2009, 1, 57–58 CrossRef CAS.
- B. Marciniec, Hydrosilylation, Springer Dordrecht, 1st edn, 2010 Search PubMed.
- X. Du and Z. Huang, ACS Catal., 2017, 7, 1227–1243 CrossRef.
- J. Yang and T. D. Tilley, Angew. Chem., Int. Ed., 2010, 49, 10186–10188 CrossRef PubMed.
- M. I. Lipschutz and T. D. Tilley, Chem. Commun., 2012, 48, 7146–7148 RSC.
- Z. Zhang, L. Bai and X. Hu, Chem. Sci., 2019, 10, 3791–3795 RSC.
- Z. Yu, Z. Song, C. Lu, Y. Bai, J. Li, J. Liu, P. Liu and J. Peng, Appl. Organomet. Chem., 2022, 36, e6648 CrossRef.
- M. A. Asraf, H. A. Younus, M. Yusubov and F. Verpoort, Catal. Sci. Technol., 2015, 5, 4901–4925 RSC.
- M. Kondo, H. Tatewaki and S. Masaoka, Chem. Soc. Rev., 2021, 50, 6790–6831 RSC.
- A. J. Esswein, M. J. McMurdo, P. N. Ross, A. T. Bell and T. D. Tilley, J. Phys. Chem. C, 2009, 133, 15068–15072 CrossRef.
- H. S. Ahn and T. D. Tilley, Adv. Funct. Mater., 2012, 23, 227–233 CrossRef.
- A. I. Nguyen, M. S. Ziegler, P. Ona-Burgos, M. Sturzbecher-Hohne, W. Kim, D. E. Bellone and T. D. Tilley, J. Am. Chem. Soc., 2015, 137, 12865–12872 CrossRef PubMed.
- J. R. Swierk, S. Klaus, L. Trotochaud, A. T. Bell and T. D. Tilley, J. Phys. Chem. C, 2015, 119, 19022–19029 CrossRef.
- Z. Li, D. Liao, G. Tian, X. Fan, X. Chai, W. Chang, Y. Gao, B. Yuan, Z. Li, F. Wei and C. Zhang, J. Am. Chem. Soc., 2025, 147, 32548–32559 CrossRef PubMed.
- D. Yan, C.-L. Dong, Y.-C. Huang, Y. Zou, C. Xie, Y. Wang, Y. Zhang, D. Liu, S. Shen and S. Wang, J. Mater. Chem. A, 2018, 6, 805–810 RSC.
- M. Vijayakumar, Z. Yhobu, J. G. Małecki, D. H. Nagaraju, R. S. Keri and S. Budagumpi, Catal. Sci. Technol., 2024, 14, 2489–2502 RSC.
- S. Lin, Y. Pineda-Galvan, W. A. Maza, C. C. Epley, J. Zhu, M. C. Kessinger, Y. Pushkar and A. J. Morris, ChemSusChem, 2017, 10, 514–522 CrossRef CAS PubMed.
- A. Howe, T. Liseev, M. Gil-Sepulcre, C. Gimbert-Surinach, J. Benet-Buchholz, A. Llobet and S. Ott, Mater. Adv., 2022, 3, 4227–4234 RSC.
- Q. Liu, L. Xie, X. Shi, G. Du, A. M. Asiri, Y. Luo and X. Sun, Inorg. Chem. Front., 2018, 5, 1570–1574 RSC.
- Y. Wang, B. Liu, X. Shen, H. Arandiyan, T. Zhao, Y. Li, M. Garbrecht, Z. Su, L. Han, A. Tricoli and C. Zhao, Adv. Energy Mater., 2021, 11, 2003759 CrossRef CAS.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |