
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBoron monoxide is a one-dimensional polymer
Joseph F.
Thuma
 a,
Rana
Biswas
bc,
Carl F.
Fleischer
III
d,
Levi
Stanley
a,
Wenyu
Huang
ad and
Frédéric A.
Perras
*ad
a,
Rana
Biswas
bc,
Carl F.
Fleischer
III
d,
Levi
Stanley
a,
Wenyu
Huang
ad and
Frédéric A.
Perras
*ad
aDepartment of Chemistry, Iowa State University, Ames, IA 50011, USA
bDivision of Materials Sciences and Engineering, Ames National Laboratory, Ames, IA 50011, USA
cDepartment of Physics and Astronomy, Electrical & Computer Engineering, Microelectronics Research Center, Iowa State University, Ames, IA 50011, USA
dChemical and Biological Sciences Division, Ames National Laboratory, Ames, IA 50011, USA. E-mail: fperras@ameslab.gov
First published on 25th September 2025
Abstract
It was recently reported that boron monoxide (BO) is formed through the cross-linking of B4O2 structural building units. Multiple theoretical phases agree with this description. Using pycnometry, multidimensional 17O NMR spectroscopy, and plane-wave DFT calculations we determined the likely polymorph to be a one-dimensional polymer initially proposed in 1955.
Among the binary oxides, boron monoxide (BO) is perhaps unique in having evaded attempts to determine its structure for nearly a century. The material was initially reported in 1940,1,2 however, the first synthesis for the preparation of a single-phase BO material was described in 1955 by Wartik and Apple.3 Their synthesis employed the condensation of tetrahydroxydiboron (B2(OH)4, Fig. 1) at high temperature and while it was soon discovered that the boron–boron bonds were preserved in the resulting material,4,5 the long-range structure was never determined. Several potential model structures were reported,6–8 most exhaustively by Claeyssens et al.,9 however, none were supported by experimental evidence.
 | ||
| Fig. 1 The synthesis of BO involves the condensation of B2(OH)4 to form structural building units composed of a six-membered B4O2 rings that then interlink into a polymeric material. The structural building units can organize into either one-dimensional (A), two-dimensional (B) and (C), or three-dimensional (D) and (E) structures. Three-dimensional structures (D) and (E) feature twisting of the B2O4 moieties, which is inconsistent with the observation of symmetry-amplified J splittings. Darker shades indicate moieties in the foreground. | ||
Recently, some of us applied multidimensional 11B NMR spectroscopy to revisit this structural conundrum.10 Key observations included (1) the detection of a single crystallographically unique boron site; (2) observation of symmetry-amplified 11B–11B J couplings;11–13 (3) observation of the collinearity of closest B–B bonds from 11B–11B–11B triple-quantum correlations; and (4) observation of diffraction signals that suggested the formation of a layered structure. The work strongly suggested that the main structural building unit was a B4O2 ring with local D2h symmetry.10 This agrees with the isolation of a B4O2(OH)4 intermediate by Carmalt et al.14 Owing to the observed diffraction patterns we proposed that the material was two-dimensional, which could have important practical implications;15–21 however, a great deal of uncertainty on the structure remained. B4O2 rings may cross-link in a number of patterns, as evident from the structures compiled by Claeyssens and listed in Fig. 1.9 We thus sought to apply 17O NMR spectroscopy to further narrow the possible structures for BO. While most materials are indistinguishable from 11B NMR (with the exception of structure D and E, which do not have local D2h symmetry), structure A features a single unique 17O site while the others feature intraring and interring oxygens that may be distinguishable. All five models further differ in their B–B–O–B dihedral angles that could be probed using 17O{11B} heteronuclear correlation.
To this aim, we enriched B2(OH)4 with 17O to prepare an 17O-enriched BO material. Enrichment of tetrahydroxydiboron by direct 17O exchange with water proved ineffective due to its poor solubility and slow decomposition to form boric acid, a non-isolable impurity. We therefore instead produced tetramethoxydiboron as previously described,22 which that then hydrolyzed with an excess of 17O-enriched water (39.9%) to yield pure B2(17OH)4. From liquid-state 17O NMR, we estimate the enrichment level to be ∼3.9% (Fig. S5). Synthesis of B17O was then carried out in the usual fashion by the self-condensation of B2(17OH)4 at 200 °C, see SI for further details.
We acquired 11B and 17O multiple-quantum (MQ) magic-angle spinning (MAS) NMR spectra of the resulting material (Fig. 2).23–25 As expected,10 we observed a single 11B resonance with an isotropic (δiso) chemical shift of 35.1 ppm, a quadrupolar coupling constant (CQ) of 3.5 MHz, and a quadrupolar asymmetry parameter (η) of 0.5. The 17O MQMAS spectrum was equally simple, being described with a single site (δiso = 178 ppm; CQ = 3.8 MHz; η = 0.7, see Table 1). These parameters were obtained by fitting the MAS lineshape and the MQMAS shift simultaneously using dmfit.26
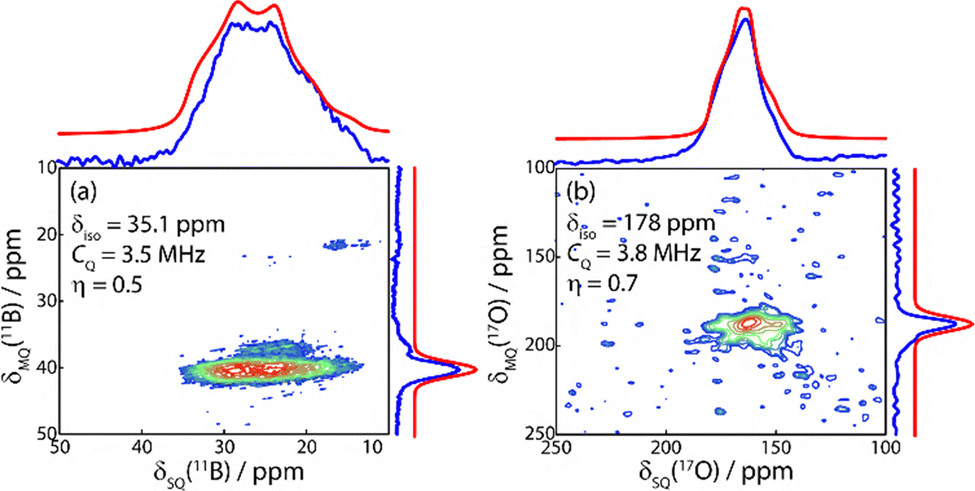 | ||
| Fig. 2 11B (a) and 17O (b) 2D MQMAS NMR spectra. There is one well-defined site present for both 11B and 17O, suggesting a structure containing only a single crystallographically unique site for each element. Sum projections (blue) are overlaid with a fit to a single site with the listed chemical shift and EFG tensor parameters (red). | ||
| ρ/g cm−3 | Site | δ iso/ppm | |C Q|/MHz | η | |
|---|---|---|---|---|---|
| a Calculation performed on the Immm polymorph.9 | |||||
| Expt. | 2.08 ± 0.13 | B | 35 ± 1 | 3.5 ± 0.1 | 0.5 ± 0.2 |
| O | 178 ± 2 | 3.8 ± 0.1 | 0.7 ± 0.1 | ||
| A | 2.05 | B | 33.8 | 3.90 | 0.32 |
| O | 203.9 | 4.73 | 0.52 | ||
| B | 1.53 | B | 28.2 | 3.86 | 0.44 |
| Ointra | 223.1 | 5.16 | 0.35 | ||
| Ointer | 149.8 | 6.87 | 0.51 | ||
| C | 1.44 | B | 25.5 | 3.65 | 0.50 |
| B | 31.0 | 3.95 | 0.38 | ||
| Ointra | 217.0 | 4.84 | 0.50 | ||
| Ointra | 219.4 | 4.48 | 0.66 | ||
| Ointer | 161.3 | 5.03 | 0.81 | ||
| D | 0.84 | B | 31.7 | 3.97 | 0.38 |
| B | 25.5 | 5.66 | 0.95 | ||
| Ointra | 215.9 | 4.48 | 0.70 | ||
| Ointra | 205.4 | 4.31 | 0.40 | ||
| Ointer | 163.2 | 4.79 | 0.30 | ||
| Ointer | 162.2 | 4.84 | 0.36 | ||
| E | 1.35 | B | 30.9 | 4.28 | 0.64 |
| B | 27.7 | 6.18 | 0.94 | ||
| Ointra | 213.6 | 4.60 | 0.63 | ||
| Ointra | 218.8 | 4.67 | 0.26 | ||
| Ointer | 153.6 | 4.81 | 0.95 | ||
Immediately, these results strongly suggest the formation of structure A (see Fig. 1), which uniquely features only one type of chemically distinct oxygen site. We then performed plane-wave density functional theory (DFT) calculations to predict the 11B and 17O chemical shifts and electric field gradient (EFG) tensor27,28 so that they may be compared with experiment. We found that the 17O chemical shifts of sites within ring structures were highly sensitive to the method used, and as such a monomer correction29 was applied using the resolution of identity second-order Møller–Plesset perturbation theory method (RI-MP2) for all calculated 17O chemical shifts. The results are listed in Table 1. There is a clear distinction between the intraring (δiso = 200–225 ppm) and interring oxygen species (δiso = 150–165 ppm) suggesting that structure B–F are inconsistent with the 17O NMR observations.
The differences between the various model are perhaps most clearly depicted by comparing experimental and computed 17O{11B} dipolar heteronuclear multiple-quantum correlation (D-HMQC)30 spectra (Fig. 3) that depend on the 11B and 17O EFG tensors (including their relative orientations) in addition to the chemical shifts. We acquired such a spectrum using rotational-echo double-resonance (REDOR) recoupling,31 applied at the 11B frequency, and 17O detection.32,33 Owing to the strong 11B–11B homonuclear dipolar interactions and the low 17O concentration, 11B-detection was not feasible. DFT-predicted 17O{11B} correlation spectra were calculated using SIMPSON34,35 with the parameters listed in Table 1, Euler angles calculated using MagresView ver. 1.6.2 (Table S2),36 and assuming a 100% efficient magnetization transfer. Clearly only structure A yields a correlation spectrum in close agreement with that measured experimentally, with the others having clearly defined intraring and interring correlations. Three-dimensional structures D and E feature ring distortions that disagree with prior J coupling measurements.10,11 These distortions further dramatically increase the magnitude of the 11B quadrupolar interactions beyond that observed experimentally. Comparisons between the 1D Hahn echo NMR spectra and the five models are also shown in Fig. S2.
 | ||
| Fig. 3 Experimental (a) and calculated (b)–(g) 17O{11B} D-HMQC 2D correlation spectra. Simulations were performed using the periodic DFT-calculated chemical shifts and EFG tensor parameters for the structures indicated on the Figure. For the simulation shown in (c), the Euler angles from the DFT calculations were used together with the experimentally-determined EFG tensor parameters and chemical shifts. | ||
To further confirm that structure A is the correct polymorph for the BO materials produced by the condensation of B2(OH)4, we performed pycnometry measurements to determine the density of the material. As described by Claeyssens,9 and as listed in Table 1, the polymorphs differ greatly in their densities, with structure A being the densest at a predicted 2.05 g cm−3. We measured a density of 2.08 ± 0.13, which is within error of the predicted value for A, and far denser than structures B–E.
It thus seems that structure A, a one-dimensional polymer, is indeed the correct model for BO, however, this does not explain the peculiar X-ray reflections that pointed to a two-dimensional material. Claeyssens predicted four different packing arrangements for the polymers in A,9 which lead to very different diffraction patterns (Fig. 4), none of which agree with the experimental result. If we randomize the stacking arrangement in a 1 × 1 × 10 P1 supercell,10 however, we do reproduce a similar powder pattern as that predicted for structure B. There are still differences with the experiment, however, and a broader study of the stacking faults may be required to fully understand the diffraction pattern.
 | ||
| Fig. 4 Comparison of the experimentally-measured PXRD pattern to those predicted using different polymorphs of model A, as indicated on the Figure, including turbostratic models of A and B. None of the predicted patterns agree with the experimental measurement, suggesting that some degree of random stacking is common. | ||
To conclude, density measurements and 17O{11B} solid-state NMR experiments were used to narrow the potential structural candidates for boron monoxide (BO), which was earlier shown to be made of interconnected B4O2 rings. We discovered that the material was, in fact, not a two-dimensional nanomaterial but instead was composed of one-dimensional polymers composed of fused B4O2 rings. Interestingly, this exact model was initially suggested by Wartick and Apple in 1955 when the material was first prepared.3 We were nevertheless unable to narrow the possibilities for a space group, with the material seeming to lack long-range order. Learning about the long-range order of the material will require methods that are sensitive to such length scales, such as scanning tunneling microscopy.37
Prof. Aaron J. Rossini is thanked for many fruitful discussions. Qasim Q. Kashif and Alex Kopeny are thanked for help with the DFT analysis. This work was supported by the US. Department of Energy, Office of Science, Basic Energy Sciences, Materials Science and Engineering Division. The Ames National Laboratory is operated for the U.S. DOE by Iowa State University under contract no. DE-AC02-07CH11358.
Conflicts of interest
There are no conflicts to declare.Data availability
Raw data for this study are available at https://doi.org/10.5281/zenodo.16884299.Supplementary information (SI): synthesis details, solution-phase NMR data, differential scanning calorimetry, 11B J-resolved NMR data, thermogravimetric analysis, infrared spectroscopy, and further calculation details. See DOI: https://doi.org/10.1039/d5cc04723d.
References
- V. E. Zintl, W. Morawietz and E. Gastinger, Z. Anorg. Allg. Chem., 1940, 245, 8–11 CrossRef.
- F. A. Kanda, A. J. King, V. A. Russell and W. Katz, J. Am. Chem. Soc., 1956, 78, 1509–1510 CrossRef CAS.
- T. Wartik and E. F. Apple, J. Am. Chem. Soc., 1955, 77, 6400–6401 CrossRef CAS.
- A. L. McCloskey, J. L. Boone and R. J. Brotherton, J. Am. Chem. Soc., 1961, 83, 1766–1767 CrossRef CAS.
- A. K. Holliday and A. G. Massey, Chem. Rev., 1962, 62, 303–318 CrossRef CAS.
- D.-Z. Li, H. Bai, Q. Chen, H. Lu, H.-J. Zhai and S.-D. Li, J. Chem. Phys., 2013, 138, 244304 CrossRef PubMed.
- Z. Zhang, L. Pu, Q.-S. Li and R. B. King, Inorg. Chem., 2015, 54, 2910–2915 CrossRef CAS.
- Y. Liu, C. Liu, Z. Zhang and R. B. King, Chem. Commun., 2017, 53, 3239–3241 RSC.
- F. Claeyssens, N. L. Allan, N. C. Norman and C. A. Russell, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 094119 CrossRef.
- F. A. Perras, H. Thomas, P. Heintz, R. Behera, J. Yu, G. Viswanathan, D. Jing, S. A. Southern, K. Kovnir, L. Stanley and W. Huang, J. Am. Chem. Soc., 2023, 145, 14660–14669 CrossRef CAS.
- F. A. Perras and D. L. Bryce, J. Am. Chem. Soc., 2013, 135, 12596–12599 CrossRef CAS PubMed.
- F. A. Perras and D. L. Bryce, J. Magn. Reson., 2014, 242, 23–32 CrossRef CAS PubMed.
- F. A. Perras and D. L. Bryce, Chem. Sci., 2014, 5, 2428–2437 RSC.
- C. J. Carmalt, W. Clegg, A. H. Cowley, F. J. Lawlor, T. B. Marder, N. C. Norman, C. R. Rice, O. J. Sandoval and A. J. Scott, Polyhedron, 1997, 16, 2325–2328 CrossRef CAS.
- H. Yamauchi, A. Asano and S. Hayashi, Heterocycles, 2022, 104, 979–986 CrossRef CAS.
- R. Rahimi and M. Solimannejad, J. Mol. Liq., 2022, 354, 118855 CrossRef CAS.
- B. Mortazavi, F. Shojaei, F. Ding and X. Zhuang, FlatChem, 2023, 42, 100575 CrossRef CAS.
- W. Othman, W. Alfalasi, T. Hussain and N. Tit, J. Energy Storage, 2024, 98A, 113014 CrossRef.
- M. M. Kadhim, N. Sadoon, H. A. Gheni, S. K. Hachim, A. Majdi, S. A. H. Abdullaha and A. M. Rheima, Comput. Theor. Chem., 2023, 1219, 113941 CrossRef CAS.
- M. Sotudeh, Z. Rastipour, F. Shojaei, A. Mohajeri and H. S. Kang, ACS Appl. Electron. Mater., 2025, 7, 2696–2708 CrossRef CAS.
- R. Mardanian, A. Kokab and S. B. Touski, Phys. Scr., 2025, 100, 065409 CrossRef CAS.
- R. Fornwald, A. Yadav, J. Montero Bastidas, M. SmithIII and R. MaleczkaJr, J. Org. Chem., 2024, 89, 6048–6052 CrossRef CAS PubMed.
- L. Frydman and J. S. Harwood, J. Am. Chem. Soc., 1995, 117, 5367–5368 CrossRef CAS.
- J.-P. Amoureux, C. Fernandez and S. Steuernagel, J. Magn. Reson., Ser. A, 1996, 123, 116–118 CrossRef CAS PubMed.
- Z. Gan and H.-T. Kwak, J. Magn. Reson., 2004, 168, 346–351 CrossRef CAS PubMed.
- D. Massiot, F. Fayon, M. Capron, I. King, S. Le Calvé, B. Alonso, J.-O. Durand, B. Bujoli, Z. Gan and G. Hoatson, Magn. Reson. Chem., 2002, 40, 70–76 CrossRef CAS.
- C. J. Pickard and F. Mauri, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 245101 CrossRef.
- M. Profeta, F. Mauri and C. J. Pickard, J. Am. Chem. Soc., 2003, 125, 541–548 CrossRef CAS PubMed.
- J. Hartman and J. K. Harper, Solid State Nucl. Magn. Reson., 2022, 122, 101832 CrossRef CAS PubMed.
- Z. Gan, J. Magn. Reson., 2007, 184, 39–43 CrossRef CAS PubMed.
- T. Gullion and J. Schaefer, J. Magn. Reson., 1989, 81, 196–200 CAS.
- R. W. Dorn, L. O. Mark, I. Hung, M. C. Cendejas, Y. Xu, P. L. Gor’kov, W. Mao, F. Ibrahim, Z. Gan, I. Hermans and A. J. Rossini, J. Am. Chem. Soc., 2022, 144, 18766–18771 CrossRef CAS.
- R. W. Dorn, A. L. Paterson, I. Hung, P. L. Gor’kov, A. J. Thompson, A. D. Sadow, Z. Gan and A. J. Rossini, J. Phys. Chem. C, 2022, 126, 11652–11666 CrossRef CAS.
- M. Bak, J. T. Rasmussen and N. C. Nielsen, J. Magn. Reson., 2000, 147, 296–330 CrossRef CAS.
- Z. Tošner, R. Andersen, B. Stevensson, M. Edén, N. C. Nielsen and T. Vosegaard, J. Magn. Reson., 2014, 246, 79–93 CrossRef PubMed.
- S. Sturniolo, T. F. G. Green, R. M. Hanson, M. Zilka, K. Refson, P. Hodgkinson, S. P. Brown and J. R. Yates, Solid State Nucl. Magn. Reson., 2016, 78, 64–70 CrossRef CAS.
- D. Cui, J. M. Macleod, M. Ebrahimi and F. Rosei, CrystEngComm, 2017, 19, 4927–4932 RSC.
| This journal is © The Royal Society of Chemistry 2025 |