
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ammonia reactivity with ferriotetrylenes: a structural snapshot of a highly dynamic process
Phuong Anh
Cao
a,
Alice C.
Phung
a,
Ella L.
Schwirzke
a,
James C.
Fettinger
 a,
Theo A. H.
Rusmore
a,
Kristian L.
Mears
*a,
Petra
Vasko
*b and
Philip P.
Power
*a
a,
Theo A. H.
Rusmore
a,
Kristian L.
Mears
*a,
Petra
Vasko
*b and
Philip P.
Power
*a
aDepartment of Chemistry, University of California, 95616, USA. E-mail: klmears@ucdavis.edu; pppower@ucdavis.edu
bDepartment of Chemistry, University of Helsinki, (A. I. Virtasen aukio 1), P.O. Box 55, 00014, Finland. E-mail: petra.vasko@helsinki.fi
First published on 24th October 2025
Abstract
The reaction of ammonia with the ferriotetrylenes Ar′EFeCp(CO)2 (Cp = η5-C5H5; E = Sn(1), Pb(2); Ar′ = −C6H3-{C6H3-2,6-iPr2}2) afforded a product interpreted as either a tin amide hydride (3) or a tin ammonia complex, which has a very long Sn–N bond of 2.4289(3), and which readily regenerates 1 and NH3 under ambient conditions.
Ammonia (NH3) is a highly valuable chemical feedstock, with 176 million tons being produced via the Haber–Bosch process in 2024.1 Given its relatively low cost and its vital role in plastics and fertilizer production, considerable research has been directed towards the conversion of NH3 into value-added chemicals (e.g., organoamines).2–4 Various methods for NH3 derivatization have been reported,5,6 among which the use of homogeneous molecular catalysts have shown promise. For example, several transition metal complexes are known to react with NH3 to generate dihydrogen and transition metal amides.7–9 However, in some transition metal systems, oxidative addition of NH3 leads to the formation of a metal amido hydride complex.10,11 In contrast, main group compounds, especially those incorporating group 14 elements in low-oxidation states have also displayed similar reactivity to transition metals.12–19 However, until recently, none of these processes were shown to be reversible, but in 2021, Goicoechea and coworkers reported a reversible activation of NH3 by a geometrically constrained phosphine. This feat relied on a ligand imposed strained coordination at the phosphorus to increase reactivity, and facilitate the addition of NH3 across a P–N bond.20 Aldridge and coworkers also showed reversible coordination of NH3 is possible with the use of a lithium xanthene-linked diphosphine dihydroborate oligomer (based on frustrated P–B Lewis pair type reactivity).21 In 2023, Long and coworkers characterized a three-dimensional metal–organic framework species that could reversibly bind ammonia to form a one-dimensional coordination polymer.22
Despite these findings, relatively few simple compounds are known to provide a direct insertion of an element into an N–H bond under mild conditions. Hadlington and coworkers reported that a germylene-Ni0 complex undergoes a metathesis reaction involving a Ge–Cl bond with NH3 to form a Ge–NH2 complex and HCl.23 The reverse reaction can be achieved by treating the Ge–NH2 species with ammonium chloride to regenerate the germylene-Ni0 starting compound. Breher reported the reversible N–H activation of NH3 under mild conditions with the use of a so-called hidden frustrated Lewis pair, which consisted of a phosphorus ylide and an aluminium Lewis acid moiety.24 In addition, we reported previously that a ferriogermylene species displays an irreversible insertion reaction with NH3 to form a stable germanium amido hydride species (Fig. 1).25
 | ||
| Fig. 1 Overview of the work presented in this study. | ||
In this report, we describe the synthesis and reactivity of the heavier congeners of this ferriotetrylene system, of the formula Ar′EFeCp(CO)2 (E = Sn, 1; Pb, 2; Ar′ = −C6H3-{C6H3-2,6-iPr2}2) (Fig. 1) with NH3. Interestingly, we observe highly dynamic behaviour of NH3 with Ar′SnFeCp(CO)2, 1, room temperature, leading to the formation of either a ferriostannylene ammonia complex or an inserted tin amido hydride species, Ar′Sn(H)(NH2)FeCp(CO)23 (Fig. 1 and 3) which proved difficult to characterise due to spontaneous release of NH3 from the complex and the reformation of 1. Compound 3 (Fig. 3) initially appeared to be the first direct experimental illustration of reversible dynamic ammonia activation under near ambient conditions by an unstrained tin species, but due to the extremely lengthened Sn–N bond distance (2.4289(3) Å) and unstable solution-state behaviour, a conclusive assignment proved difficult.
Initially, we synthesized the ferriostannylene 1 by a procedure similar to that described in the literature.26 In addition, we prepared its lead(II) analogue (compound 2) in a similar manner (Scheme 1).
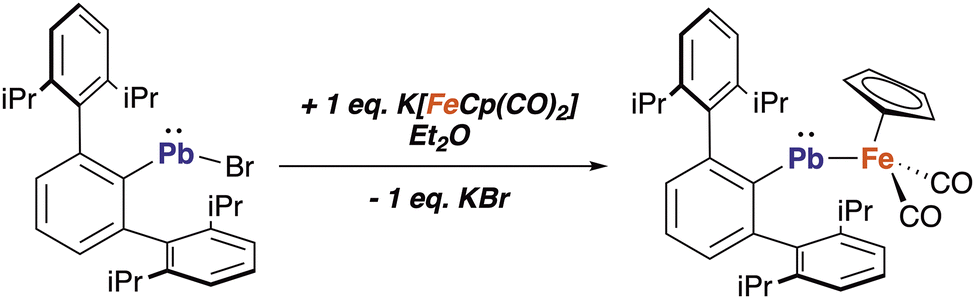 | ||
| Scheme 1 Synthetic route to compound 2. | ||
Investigation of the reactivity of solutions of 1 or 2 with NH3 afforded a dramatic color change from green to orange/red for 1 but no change was observed for 2. We further examined this process for the tin species 1 to determine whether the reaction occurring with NH3 was a simple coordination of ammonia to Sn or an N–H insertion reaction.
It is worth noting that our previous work on the reactivity of NH3 with the ferriogermylene analogue of 1 (Fig. 1) revealed that an irreversible insertion into the N–H bond occurs at the tetrel, as it also does for the related diarylstannylene  and diarylgermylene
and diarylgermylene  . In the case of the diarylstannylene, reaction with NH3 afforded the thermodynamically stable tetrel-amido species [Ar′Sn(μ-NH2)]2via elimination of Ar′H. The Ge(IV) amido hydride
. In the case of the diarylstannylene, reaction with NH3 afforded the thermodynamically stable tetrel-amido species [Ar′Sn(μ-NH2)]2via elimination of Ar′H. The Ge(IV) amido hydride  was formed from the diarylgermylene, which did not release NH3, even on heating.13
was formed from the diarylgermylene, which did not release NH3, even on heating.13
Evidence of N–H bond insertion was attempted via the growth of X-ray quality single crystals of 3 (Fig. 2), which were grown by the condensation of pure, anhydrous NH3 into a forest green solution of 1 in toluene (condensed at ca. −78 °C). The resulting orange crystals were initially assigned as the insertion product 3 (Fig. 2). The X-ray data could also be modelled as the NH3 complex Ar′Sn(NH3)FeCp(CO)2 to a similar residual value, but in this case, all the ammonia hydrogens could not be located on a difference map. At room temperature, under ambient conditions, it was observed that the orange crystals returned to their initial green color (i.e.1) within ca. 10 minutes, with liberation of NH3.
 | ||
| Fig. 2 Molecular structure of compound 3 (thermal ellipsoids are shown at 50% probability). The 2,6-diisopropylphenyl groups are shown in wireframe format and the H-atoms (except those of the amido group and the tin hydride moieties) are not shown for clarity. Color code: carbon = grey, iron = orange, oxygen = red and tin = blue, nitrogen = lilac, hydrogen = white. | ||
Compound 3 has a distorted tetrahedral coordination at the tin atom. The most striking structural feature is the lengthened Sn1–N1 bond which is 2.4289(3) Å. This is longer than any Sn–N bond distance previously reported for either a Sn–(NH3) complex or a Sn–NH2 amido species and may suggest an assignment of an ammonia complex, over an inserted product.27 This may be contrasted with the related germanium amido hydride compound Ar′Ge(NH2)(H)FeCp(CO)2 and Ar#Ge(NH2)(H)FeCp(CO)2 (Ar# = C6H3-2,6-{C6H2-2,4,6-Me3}2), whose bonds show only a small dependence on the type of terphenyl substituent employed (1.8505(2) Å for the Ge–N bond in Ar#Ge(NH2)(H)FeCp(CO)2 and 1.757(4) Å in Ar′Ge(NH2)(H)FeCp(CO)2).12 These variations are obviously far smaller than those seen for 3, which further highlights its unique structure. However, it could be that the structure obtained is indicative of the weakening Sn–NH2 bond within a ammonia-inserted ferriostannylene 3 and the incipient formation of a complexed NH3 moiety, which rapidly dissociates. As crystal selection was occurring at room temperature, rapid NH3 loss from the sample was clear, particularly in the colour change from orange back to green, Thus, the X-ray data for 3 could be considered as a structural snapshot of a reversible NH3 insertion and regeneration of 1 at ambient temperature (as shown in Fig. 2).
The Sn1–C1 bond length 2.2921(3) Å of 3 is comparable to that of the Sn1–C1 bonds (2.240(3) Å) in compound 1 (2.208(3) Å), as well as those in the diarylstannylene bridged amide (2.240(3) Å) [Ar′Sn(μ-NH2)]2.13 The Sn1–Fe1 distance in 3 (2.6703(4) Å) is far longer than the Sn1–Fe1 distance observed in the starting material 1 (2.6040(16) Å). The C1–Sn1–Fe1 angle (109.13(5)°) in 3 is slightly narrower than the corresponding angle in compound 1 (112.65(9)°), due to the increased coordination number.
To further pin down the identity of compound 3, we attempted variable temperature NMR experiments by the condensation of dried NH3 into a J. Young NMR tube containing a solution of 1 in C7D8. This experiment yielded a change in the chemical shift of the 119Sn NMR signal, from δSn = 2958.6, which corresponds to the parent ferriostannylene, to a broadened δSn = 752.2 at 293 K, which we assign to a dynamic coordination of NH3 to 1.28 The signal shifted further upfield to 557.9 ppm when the temperature was decreased to 193 K, which resembles more of a Sn(IV) signal, and could allude to the insertion pathway. A 1H-coupled 119Sn NMR spectrum of 3 did not show clear coupling (cf. Fig. S9). A coupling consistent with a four-coordinate Sn(IV) complex could be expected to be in the range of 1J(119Sn–1H) = 103.13 Hz.28,29 The 1H NMR spectrum also revealed a broad signal at −0.05 ppm, which is caused by the dynamic coordination of free and complexed NH3. The 1H VT NMR studies further showed a change of the broad singlet to a sharper signal at −0.09 ppm at 213 K and then finally to a narrow signal with a chemical shift at −0.12 ppm at 193 K. The upfield shift at lower temperatures again made an assignment of either the inserted amide product or an ammonia complex difficult. Additionally, a broad signal detected at 2.97 ppm could potentially be assigned as the Sn–H group, though a signal that could reasonably be assigned to the Sn–NH2 resonance was not observed.
Despite the VT-NMR experiments, the results of a van’t Hoff analysis proved insufficiently definitive to determine very accurate energy values, as the concentration of ammonia was difficult to measure with our equipment, and the highly unstable nature of the coordination rendered FTIR experiments with our setup impossible. Instead, the feasibility of an insertion mechanism was probed using density function theory (DFT). The DFT calculations indicated that indeed, the first step of an activation process is the complexation of an NH3 molecule to the unoccupied, virtually pure 5p orbital of the tin atom giving the ammonia adduct (Int1, Fig. 3). This is followed by the association of three NH3 molecules via N–H interactions with the complexed NH3, which facilitates Sn–H and Sn–NH2 formation via a concerted process (TS1, Fig. 3).
 | ||
| Fig. 3 DFT-calculated reaction profile between Ar′SnFeCp(CO)2 (1) and 3 molecules of NH3 to give 3 at 193 K (red) in 1 atm ([Fe] = FeCp(CO)2 and Ar′ = −C6H3-{C6H3-2,6-iPr2}2). Calculated at the SMD-PBE0-GD3BJ/Def2-TZVP//PBE0-GD3BJ/Def2-SVP/Def2 TZVP(Sn,Fe) level of theory. Energies represent Gibbs free energies (kcal mol−1) in 1 atm at the given temperature. | ||
These results indicate that at room temperature, the reaction is essentially thermoneutral, with reaction energy barriers consistent with the observed X-ray and spectroscopic data. At lower temperatures, the insertion becomes slightly exergonic despite the lower entropy value. A similar mechanism has been calculated for a diarylgermylene complex reacting with hydrazines as reported earlier.30
For the ferrioplumblyene species (compound 2), dark green crystals of 2 (60% yield) were characterized by single crystal X-ray diffraction (Fig. 4) which showed the expected, bent two coordinate geometry at lead with a Fe1–Pb1–C1 angle of 111.09(16)°.31 The Fe1–Pb1 bond length in 2 is 2.6388(11) Å, which is shorter than the bond length (2.7367(5) Å) of a plumbylone complex reported by Driess and coworkers, which has the formula {[SiII(Xant)SiII]Pb0Fe(CO)4} (SiII(Xant)SiII = PhC(NtBu)2Si-(Xant)Si(NtBu)2CPh).32 In Driess’ compound, the lead atom also coordinates datively to the tetracarbonyl iron. The Pb1–C1 bond length (2.321(6) Å) in 2 is comparable to those (2.313(13) Å and 2.307(13) Å) in the ferrioplumbylene species of formula Fe(CO)4(PbAr*)2 (Ar* = −C6H3-2,6-{C6H2-2,4,6-iPr3}2) reported earlier by our group.33
 | ||
| Fig. 4 Molecular structure of compound 2 (thermal ellipsoids are shown at 50% probability). The 2,6-diisopropylphenyl groups are shown in wireframe format and H-atoms are not shown for clarity. Color code: carbon = grey, iron = orange, oxygen = red and lead = and lead = purple. | ||
The 207Pb NMR spectrum of 2 at 298 K shows a signal at 11724.1 ppm, which lies in the expected region for a two-coordinate Pb(II) complex.34 In addition, the 1H NMR spectrum displayed the expected signals, from the η5-C5H5 Cp protons (4.18 ppm) and 2,6-diisopropyl methyl protons (1.12 ppm and 1.39 ppm). UV-Vis spectroscopy of 2 showed λmax absorbances at 337 nm (π → π* transition) and 634 nm (n → π* transition). FTIR spectroscopic analysis showed CO stretches at nCO = 1961 and 1916 cm−1.
In summary, we report the synthesis and characterisation of the heavier congeners of a ferriotetrylene system of the formula Ar′EFeCp(CO)2 (Cp = η5-C5H5; E = Sn(1), Pb(2); Ar′ = −C6H3-{C6H3-2,6-iPr2}2) and probed their reactivity with ammonia. The ferriostannylene 1 afforded, at the least, coordination of ammonia to the Sn atom in 1 as determined using single crystal X-ray diffraction and multinuclear VT-NMR experiments, but the extent as to which an insertion into the N–H bond of NH3 proved difficult to determine due to low energy barriers for both mechanisms, as calculated using DFT. However, the reactivity of 1 with NH3 lies between the profiles of its lighter and heavier congeners. The analogous ferriogermylene irreversibly inserts into the N–H bond of ammonia, and the ferrioplumbylene 2 is unreactive with NH3. Based on the crystallographic evidence for 3, the Sn–N bond distance is elongated due low energy barriers for both the coordination, insertion and liberation of NH3 at ambient temperature and pressure. This structure provides an insight into NH3 coordination at 1 and may provide a unique view into a dynamic process of reversible N–H bond scission and subsequent NH3 regeneration at a main group metal. This study further provides a complete picture of ferriotetrylene reactivity with ammonia, which could inform future catalyst design for small molecule activation. The activation of other industrially relevant small molecules using this ferriostannylene complex is currently in hand.
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information is available. The SI includes synthetic procedures and analyses of the compounds. See DOI: https://doi.org/10.1039/d5cc04597e.CCDC 2248740 (2) and 2468055 (3) contain the supplementary crystallographic data for this paper.35a,b
Notes and references
- N. Salmon and R. Bañares-Alcántara, Sustainable Energy Fuels, 2021, 5, 2814–2839 RSC
.
- J. Hoover, Science, 2016, 354, 707–708 CrossRef PubMed
- A. S. Guram, R. A. Rennels and S. L. Buchwald, Angew. Chem., Int. Ed. Engl., 1995, 34, 1348–1350 CrossRef
- J. Louie and J. F. Hartwig, Tetrahedron Lett., 1995, 36, 3609–3612 CrossRef
- J. I. van der Vlugt, Chem. Soc. Rev., 2010, 39, 2302 RSC
- S. Streiff and F. Jérôme, Chem. Soc. Rev., 2021, 50, 1512–1521 RSC
- J. L. Klinkenberg and J. F. Hartwig, J. Am. Chem. Soc., 2010, 132, 11830–11833 CrossRef PubMed
- M. J. Bezdek, S. Guo and P. J. Chirik, Science, 2016, 354, 730–733 CrossRef PubMed
- Y. Nakajima, H. Kameo and H. Suzuki, Angew. Chem., Int. Ed., 2006, 45, 950–952 CrossRef PubMed
- J. Zhao, A. S. Goldman and J. F. Hartwig, Science, 2005, 307, 1080–1082 CrossRef PubMed
- E. Morgan, D. F. MacLean, R. McDonald and L. Turculet, J. Am. Chem. Soc., 2009, 131, 14234–14236 CrossRef PubMed
- Y. Peng, J.-D. Guo, B. D. Ellis, Z. Zhu, J. C. Fettinger, S. Nagase and P. P. Power, J. Am. Chem. Soc., 2009, 131, 16272–16282 CrossRef PubMed
- Y. Peng, B. D. Ellis, X. Wang and P. P. Power, J. Am. Chem. Soc., 2008, 130, 12268–12269 CrossRef PubMed
- S. L. McOnie, G. A. Özpınar, J. L. Bourque, T. Müller and K. M. Baines, Dalton Trans., 2021, 50, 17734–17750 RSC
- D. Sarkar, P. Vasko, L. Ying, J. J. C. Struijs, L. P. Griffin and S. Aldridge, Angew. Chem., Int. Ed., 2025, 64, e202502326 CrossRef PubMed
- Z. Zhu, X. Wang, Y. Peng, H. Lei, J. C. Fettinger, E. Rivard and P. P. Power, Angew. Chem., Int. Ed., 2009, 48, 2031–2034 Search PubMed
- D. M. J. Krengel, N. Graw, R. Herbst-Irmer, D. Stalke, O. P. E. Townrow and M. Fischer, Inorg. Chem. Front., 2024, 11, 8649–8659 RSC
- D. C. H. Do, A. V. Protchenko, M. Á. Fuentes, J. Hicks, P. Vasko and S. Aldridge, Chem. Commun., 2020, 56, 4684–4687 RSC
- J. Cui, Y. Li, R. Ganguly, A. Inthirarajah, H. Hirao and R. Kinjo, J. Am. Chem. Soc., 2014, 136, 16764–16767 CrossRef PubMed
- J. Abbenseth, O. P. E. Townrow and J. M. Goicoechea, Angew. Chem., Int. Ed., 2021, 60, 23625–23629 CrossRef PubMed
- A. E. Crumpton, A. Heilmann and S. Aldridge, Angew. Chem., Int. Ed., 2024, 63 Search PubMed
- B. E. R. Snyder, A. B. Turkiewicz, H. Furukawa, M. V. Paley, E. O. Velasquez, M. N. Dods and J. R. Long, Nature, 2023, 613, 287–291 CrossRef PubMed
- P. M. Keil, T. Szilvási and T. J. Hadlington, Chem. Sci., 2021, 12, 5582–5590 RSC
- F. Krämer, J. Paradies, I. Fernández and F. Breher, Nat. Chem., 2024, 16, 63–69 CrossRef PubMed
- A. C. Phung, J. C. Fettinger and P. P. Power, Organometallics, 2021, 40, 3472–3479 CrossRef
- H. Lei, J.-D. Guo, J. C. Fettinger, S. Nagase and P. P. Power, Organometallics, 2011, 30, 6316–6322 CrossRef
- A. V. Protchenko, J. I. Bates, L. M. A. Saleh, M. P. Blake, A. D. Schwarz, E. L. Kolychev, A. L. Thompson, C. Jones, P. Mountford and S. Aldridge, J. Am. Chem. Soc., 2016, 138, 4555–4564 CrossRef CAS PubMed
-
B. Wrackmeyer, 119Sn-NMR Parameters, Annual Reports on NMR Spectroscopy, Elsevier, 1985, vol. 16, pp 73–186 DOI:10.1016/S0066-4103(08)60226-4
-
A. G. Davies, Organotin Chemistry, 2nd edn, Wiley-VCH, 2004, p. 18 Search PubMed
- Z. D. Brown, J.-D. Guo, S. Nagase and P. P. Power, Organometallics, 2012, 31, 3768–3772 CrossRef CAS
- C. Stanciu, S. S. Hino, M. Stender, A. F. Richards, M. M. Olmstead and P. P. Power, Inorg. Chem., 2005, 44, 2774–2780 CrossRef CAS PubMed
- J. Xu, S. Pan, S. Yao, G. Frenking and M. Driess, Angew. Chem., Int. Ed., 2022, 61, e202209442 CrossRef CAS PubMed
- Q. Zhu, J. C. Fettinger, P. Vasko and P. P. Power, Organometallics, 2020, 39, 4629–4636 CrossRef CAS
-
B. Wrackmeyer, 207Pb-NMR Parameters, Annual Reports on NMR Spectroscopy, Elsevier, 1990, vol. 22, pp 249–306 DOI:10.1016/S0066-4103(08)60257-4
-
(a)
CCDC 2248740: Experimental Crystal Structure Determination, 2025 DOI:10.5517/ccdc.csd.cc2fh00z
| This journal is © The Royal Society of Chemistry 2025 |