
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
NORA: non-adiabatic dynamics with force-field based representation. Application to photoisomerization in biomimetic photoswitches
Raúl
Losantos
 *ab,
Giacomo
Prampolini
c and
Antonio
Monari
*b
*ab,
Giacomo
Prampolini
c and
Antonio
Monari
*b
aDepartamento de Química, Instituto de Investigación en Química (IQUR), Universidad de La Rioja, Madre de Dios 53, 26006 Logroño, Spain
bUniversité Paris Cité and CNRS, ITODYS, F 75006 Paris, France. E-mail: raul.losantosc@unirioja.es; Antonio.monari@u-paris.fr
cIstituto di Chimica dei Composti Organo Metallici (ICCOM-CNR), Area della Ricerca, Via G. Moruzzi 1, I-56124 Pisa, Italy
First published on 29th October 2025
Abstract
Based on the JOYCE quantum-derived force field for specific electronic states, we propose a non-adiabatic dynamic procedure named Nora, to study the photochemical evolution of different chromophores at the cost of classical molecular dynamics simulations. Notably, we present its application to the E/Z photoisomerization of a biomimetic cyclocurcumin-based photoswhitch, which may have applications in oxygen-independent photodynamic therapy or photo-immunotherapy. This new methodology opens an expansive timescale in the study of photochemical processes in biological environments.
Molecular photoswitches1,2 have shown increasing interest in different scientific fields, including the development of smart materials and optoelectronic devices. Their role in photobiology3,4 can hardly be overestimated, since they lie at the very heart of crucial phenomena such as signal transduction in vision.5–7 The latter is mediated by a very fast photoisomerization of a chromophore, retinal, embedded in rhodopsin. This photo-induced process also represents one of the paradigms of the computational study of molecular photoswitches, using both static and dynamic approaches. Recently, we have shown that biomimetic photoswitches derived from cyclocurcumin (Pyro) present enhanced photochemical properties compared to the parent compound in terms of maximum absorption wavelength and photoisomerization quantum yield.8–12 Moreover, we have shown that Pyro can interact with biological membranes, perturbing their properties in a dose-dependent manner upon photoisomerization.12 This observation is attractive since it can pave the way to its use as an oxygen-independent phototherapy agent, which will allow the treatment of hypoxic solid tumours more efficiently than in conventional photodynamic therapy.9,13,14 However, the optimization of these strategies would require the use of molecular modelling and simulation approaches, which provide an atomistic-resolved vision of the photochemical pathways active in complex environments such as lipid bilayers. Yet, providing a fully resolved photochemical description of the isomerization is extremely challenging, and in some instance out of the scope of present-day simulation techniques.
As a matter of fact, the golden standard for studying excited state evolution and photochemistry relies on using non-adiabatic dynamics in which the breaking of the Born–Oppenheimer approximation is considered with different levels of theory. While quantum dynamics (QD),15 which explicitly considers the quantum nature of the nuclei, allows the propagation of relevant wave-packet, its cost is still prohibitive to treat large systems, and, furthermore, usually requires selecting only some specific degrees of freedom. On the other extreme of the spectrum, surface hopping (SH) dynamics treats the nuclei classically, propagating them on a given potential energy surface while allowing hops between them based on their non-adiabatic coupling and energy difference.16–18 While SH takes natively into account the whole vibrational degrees of freedom, it is still plagued by a high computational cost, since it requires obtaining energies, gradients and couplings for multiple electronic states at each step of the dynamics. This is even more stringent, since to simulate the probabilistic nature of the wave-packet and assure a relevant statistical sampling, multiple trajectories, usually some hundreds, should be propagated independently. Furthermore, in case strong-coupled points such as conical intersection (CI) are involved, the SH propagation should go beyond the usual density functional theory (DFT)-based approaches and eventually rely on expensive multi-configurational wave-function based methods. Advanced TD-DFT methods such as Spin–Flip (SF-TDDFT) and Multi-State (MS-DFT) can overcome some of these limitations.19 Additionally, machine learning (ML) potentials, eventually extended to the excited states, also represent a valuable alternative.20 Yet, the use of quantum mechanically derived force fields (QMD-FF), and specifically the JOYCE procedure,21 allows a detailed reproduction of the potential energy surface of a given (excited) electronic state of complex organic and organometallic systems. This high accuracy and state-specificity of QMD-FF come at the cost of transferability, since the FF is system specific. In contrast, the surroundings can be easily modelled by using a general transferable FF, which may describe both homogeneous solvents or complex biological macromolecules, such as proteins or nucleic acids. QMD-FF simulations also allow for capturing the fine solvent reorganization upon excitation and its role in tuning the optical properties.22,23 Interestingly, a double-level parameterization of the QMD-FF is also possible in which the ensemble of the vibrational degrees of freedom are treated at DFT or Time-dependent DFT (TD-DFT) level, while the coordinates involved in reaching the relevant CIs are treated at multi-configurational level.24 Recently, we have used this approach to parameterize the QMD-FF for the ground and the first excited states of the Pyro chromophore and the parent cyclocurcumin.25 The QMD-FF-obtained properties, such as vibrational frequencies, have been carefully validated against ab initio methods, showing excellent agreement and the capacity to reproduce the absorption spectrum and the main spectroscopic features. In this contribution, we aim to leverage these achievements, proposing an SH-like approach, called NORA, in which the trajectories are propagated at classical level using a QMD-FF forcing a hop between the potential surfaces in the vicinity of a conical intersection. Similar geometric approaches have been used for instance by J. Ma group.26 Our strategy, while still preliminary, is promising and allows us to recover a correct exponential decay of the Pyro excited state leading to E/Z photoisomerization, while no photochemical channel is open in the parent cyclocurcumin.
The chosen QMD-FF for the ground and the first excited state of Pyro were parametrized with the JOYCE code, following the procedure briefly described in the SI and detailed in our previous work.25 While, anharmonic degrees of freedom were sampled by relaxed scans at the DFT or TD-DFT levels of theory (TD-CAM-B3LYP/6-311++G**) using the Frozen Internal Rotation Approximation,21 the photoisomerizable coordinate, i.e. the torsion around the double bond δ4, was monitored at MS-CASPT2//CASSCF. The excellent representation of the topology of the PES near the CI, which is pivotal to the NORA approach, may be appreciated by comparing the force-field and the MS-CASPT2 energy profiles (Fig. 1 bottom). This aspect stands out as a key feature, since it allows for proper parameterization of the CI, and thus of the excited state dynamics. A more exhaustive description of the parameterization procedure is given in SI affording a set of two FF with 269 parameters and 8 LJ terms. After a first 50 ns NPT MD simulation of the chromophore embedded in a box of SPCE water in its ground state, we randomly selected 500 independent initial conditions. From each one of these starting points, S1 dynamics were realized switching to the QMD-FF representing the excited state. Note that the use of ground state geometries and velocities as the starting point mimics the Franck–Condon principle, in which the electronic excitation is considered as instantaneous. All 500 trajectories have been propagated in the S1 using the NPT ensemble for a maximum of 20 ns, systematically saving velocities and coordinates. For the aim of the present study and considering the topology of the PES in Fig. 1, we considered that excited-state Pyro reaches the CI, and thus the branching point, when the dihedral angle of the isomerizable bond reaches values of δ4 = 110°, at which one can observe a good quasi-degeneracy between the PES of the two states. In this work, we preliminarily considered that a hop to the GS happens whenever the dihedral reaches this value. Obviously, this strategy may be regarded as oversimplified compared to more suitable SH approaches based notably on the Tully minimal hops. Most notably, our strategy does neglect entirely the non-adiabatic coupling elements and considers the energy gap only implicitly. However, it provides an easy and straightforward approach to simulate the photo-decay and photochemistry of Pyro. A further discussion on how to afford that simplifications is included on the SI.
 | ||
| Fig. 1 Chemical structure of Pyro (left) and the PES around the isomerizable bond δ4 (right). | ||
Once a trajectory reaches a CI the QDFF is switched back to the potential of the GS and the trajectory is propagated for further 20 ns in the NPT ensemble, maintaining the velocities of the excited state to follow the evolution in the GS PES, which may lead to photoisomerization (i.e. to a dihedral angle close to 0°). All MD simulations have been performed using the Gromacs2019 package and a v-rescale thermostat coupled with a Parrinello–Rahman barostat. Additionally, solvent reorganization at the CI region was examined through a radial distribution function averaging along the trajectories to assure statistical significance. The time evolution of the process could be monitored by the population of the excited state. In this scenario, we consider that at the beginning of the MD simulation the population of the S1 state is unitary and we may retrieve the population decay by considering each enforced hop to the GS, thus considering the ensemble of the trajectories (Fig. 2a). First, we notice that the decay follows an ideal exponential decay. Furthermore, we may also appreciate that some hops to the ground state are present already during the first fs. By applying a monoexponential fit (Fig. 2) we retrieve a characteristic lifetime of the S1 state, which is of the order of 32 ps. While we do not have access to ultrafast time-resolved spectroscopic experiment, it is supposed that Pyro is isomerizing in the tens of ps regime, which is coherent with the results of our study. The solvent reorganization may play a crucial role in the first moments of the dynamics. This role is expected to be even more relevant in the branching region between S1 and GS, due to the influence on the effective isomerization QY. Different examples of the influence of solvation on photochemical processes efficiency have been reported.27,28 Therefore, we have addressed the solvent reorganization along the firsts instants after GS relaxation by computing the radial distribution function. Interestingly, we also observe that the solvent restructuring is extremely rapid and occurs in the very first moments after the relaxation, and should be considered as completed in about 2–3 ps. The most relevant solvent shakeup (SI) mainly involves the solvation of the keto oxygen (O2) involved in the n–π* and the OH of the isomerizable phenyl ring (Oh2). In both cases. A decrease on the first solvation shell is observed, flanked by a contraction of the second one. This points to a charge diminution due to the expected electronic stabilization upon GS relaxation. This solvent study is just a simple example of the possible environment analysis which could be accounted using this methodology. Our final goal is to describe and analyse the photoisomerization quantum yield (QY). To achieve this, we need to consider the fate of the GS dynamics subsequent to the decay at the CI. Fig. 2b reports the time evolution of the population of the dihedral angle around the isomerizable double bond convoluted across the ensemble of trajectories. Coherently with the presence of only E-isomer, at the beginning of the simulation only dihedral values around 180° are populated. However, and coherently with the time scale of the S1 decay, we may observe the appearance of a branch at around 100°, i.e. in the identified region of the CI. Notably, this branch is not persistent and rapidly disappears after about 50 ps. Concomitantly, we observe the population of the branch at 0°, which is indicative of the photoisomerization to the Z-Pyro. From the population at the end of the dynamics we may also propose an estimation of the photoisomerization QY which amounts at 29%, if we consider the ensemble of all the 500 trajectories. Interestingly, if we consider only the trajectories which have reached the CI the population of the Z-isomer amounts to the 45%. As a matter of fact, the values obtained for the isomerization QY are extremely closed to the experimentally observed population of the photo-stationary state, which involves 30% of the Z- and 70% of the E-isomer.
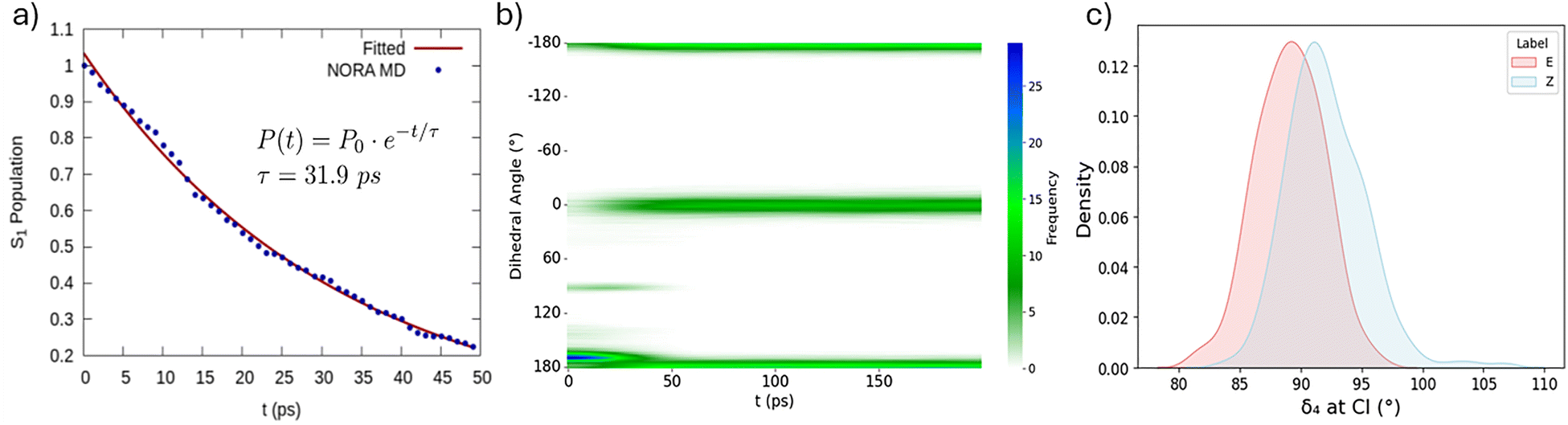 | ||
| Fig. 2 (a) Time evolution of the population of the S1 state as inferred from the NORA procedure. The exponential fitting of the population decay is also given together with the estimated lifetime τ. (b) Heat map showing the time evolution of the dihedral angle around the photoisomerizable double bond of Pyro. The branches at −180°/180° and 0° corresponds to the E- and Z-isomer, respectively. (c) Kernel Density Estimate (KDE) of the distribution of the dihedral angle of the isomerizable bond at the CI hop geometry labelled in terms of photoisomerization (Z) or photostability (E). | ||
The population of the reactant or product branch is achieved immediately after the photo-decay. This is also coherent with the topology of the CI, as it can be observed in Fig. 1. As shown in Fig. 2c it appears that photoisomerization slightly correlates with smaller values of the dihedral around the isomerizable double bond (δ4). Some less important correlations with the orientation of the nearby phenyl groups can also be observed from the correlation plot reported in SI. The inversion angle seems to play a less important role in driving photoreactivity (Fig. S5) even if a slight difference appears for the angle around the phenyl group. The impact of the variation of bond length is, instead, completely negligible. We have shown that by using a QMD-FF approach we are able to describe the photoisomerization process of the Pyro chromophore recently studied experimentally. The NORA procedure, based on the here proposed QMD-FF is inspired on non-adiabatic SH approaches and it is based on the precise and consistent parameterization of an FF for both S1 and GS states. More specifically a high-level wave-function based description of the isomerizable coordinate allows to properly model the CI and thus the branching space. Notwithstanding we have neglected specific aspects of the SH procedure such as the non-adiabatic coupling, and we enforce hops only based on geometrical parameters, the NORA approach is able to reproduce experimental results satisfactorily. This involves both the excited state lifetime and the isomerization QY, which is coherent with the experimental photostationary state. Therefore, we believe that the NORA approach represents a valuable alternative for SH dynamics in complex systems, majorly when embedded in non-homogeneous environments. Further development will be the improvement of explicitly including an FF term considering the non-adiabatic coupling or implementing the procedure into general purpose SH code, such as SHARC16 or Newton-X.29 In this scenario we plan to modify our simple geometric hopping criteria by standard algorithms,30 such as Tully fewest hopping.31 This may be realized adding a parameterization of the non-adiabatic coupling32,33 to the Joyce force field terms,34,35 which could be obtained by linear vibronic coupling30,36 or explicitly sampling the coordinates having large displacements and driving the photoreactivity. While ML based non-adiabatic dynamics,20,37,38 including its coupling with hybrid and fragment methods,39 has spurred and offered the possibility to achieve impressive timescales and detailed descriptions.
We believe that QMD-FF approaches, such as NORA, may be extremely valuable, particularly since it does not require retraining or reparameterization of the model to consider different solvents and inhomogeneous environments. The NORA approach completely integrates all interaction terms into a uniform force field expression, and it better represents the CI topology, which is challenging for ML. Thus, it represents a valuable complementary alternative to data-driven approaches.
The authors thank GENCI, Explor, and the Platform P3MB for computational resources. A. M. thanks the LUMA PEPR MANDALA is also acknowledged. G.P. thanks the financial support from ICSC Centro Nazionale di Ricerca in High Performance Computing, Big Data and Quantum Computing, funded by the European Union NextGenerationEU PNRR, Missione 4 Componente 2Investimento 1.4 and funding from the Italian Ministry of University and Research (PRIN grant 2022K3AY2K). The Joyce code can be downloaded free of charges at https://www.iccom.cnr.it/en/joyce-2/.
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI).Supplementary information: Extended methodology, hopping and final geometries, CI geometrical analysis. See DOI: https://doi.org/10.1039/d5cc04585a.
References
- M. M. Russew and S. Hecht, Adv. Mater., 2010, 22, 3348–3360 Search PubMed.
- J. Volarić, W. Szymanski, N. A. Simeth and B. L. Feringa, Chem. Soc. Rev., 2021, 50, 12377–12449 Search PubMed.
- W. Szymański, J. M. Beierle, H. A. V. Kistemaker, W. A. Velema and B. L. Feringa, Chem. Rev., 2013, 113, 6114–6178 Search PubMed.
- N. K. Singer, L. González and A. Monari, J. Phys. Chem. Lett., 2023, 14, 10333–10339 Search PubMed.
- D. Polli, P. Altoè, O. Weingart, K. M. Spillane, C. Manzoni, D. Brida, G. Tomasello, G. Orlandi, P. Kukura, R. A. Mathies, M. Garavelli and G. Cerullo, Nature, 2010, 467, 440–443 Search PubMed.
- P. Kukura, Science, 2005, 310, 1006–1009 Search PubMed.
- S. Hayashi, E. Tajkhorshid and K. Schulten, Biophys J., 2009, 96, 403–416 Search PubMed.
- M. Girardon, S. Parant, A. Monari, F. Dehez, C. Chipot, E. Rogalska, N. Canilho and A. Pasc, ChemPhotoChem, 2019, 3, 1034–1041 Search PubMed.
- J. Pecourneau, R. R. Losantos, A. Delova, Y. Bernhard, S. S. Parant, M. Mourer, A. Monari and A. Pasc, Langmuir, 2022, 38, 15642–15655 Search PubMed.
- J. Pecourneau, R. Losantos, A. Gansmuller, S. Parant, Y. Bernhard, M. Mourer, A. Monari and A. Pasc, J. Photochem. Photobiol., A, 2023, 439, 114583 Search PubMed.
- M. Marazzi, A. Francés-Monerris, M. Mourer, A. Pasc and A. Monari, Phys. Chem. Chem. Phys., 2020, 22, 4749–4757 Search PubMed.
- A. Delova, R. Losantos, J. Pecourneau, Y. Bernhard, M. Mourer, A. Pasc and A. Monari, J. Chem. Inf. Model., 2023, 63, 299–307 Search PubMed.
- X. Yang, M. Li, X. Qin, S. Tan, L. Du, C. Ma and M. Li, J. Am. Chem. Soc., 2022, 144, 3863–3874 Search PubMed.
- M. Das, C. Zhu and V. K. Kuchroo, Immunol. Rev., 2017, 276, 97–111 Search PubMed.
- B. F. E. Curchod and T. J. Martínez, Chem. Rev., 2018, 118, 3305–3336 Search PubMed.
- S. Mai, P. Marquetand and L. González, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1370 Search PubMed.
- M. Barbatti, M. Ruckenbauer, F. Plasser, J. Pittner, G. Granucci, M. Persico and H. Lischka, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 26–33 Search PubMed.
- M. Barbatti, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 620–633 Search PubMed.
- D. Casanova and A. I. Krylov, Phys. Chem. Chem. Phys., 2020, 22, 4326–4342 Search PubMed.
- C. Müller, Š. Sršeň, B. Bachmair, R. Crespo-Otero, J. Li, S. Mausenberger, M. Pinheiro, G. Worth, S. A. Lopez and J. Westermayr, Chem. Sci., 2025, 16, 17542–17567 Search PubMed.
- S. Giannini, P. M. Martinez, A. Semmeq, J. P. Galvez, A. Piras, A. Landi, D. Padula, J. G. Vilhena, J. Cerezo and G. Prampolini, J. Chem. Theory Comput., 2025, 21, 3156–3175 Search PubMed.
- G. Prampolini, A. Andersen, B. I. Poulter, M. Khalil, N. Govind, E. Biasin and M. Pastore, J. Chem. Theory Comput., 2024, 20, 1306–1323 Search PubMed.
- G. Prampolini, F. Ingrosso, J. Cerezo, A. Iagatti, P. Foggi and M. Pastore, J. Phys. Chem. Lett., 2019, 10, 2885–2891 Search PubMed.
- H. Moumene, G. Prampolini, C. García-Iriepa, J. Cerezo, I. Navizet and F. Santoro, J. Phys. Chem. B, 2025, 129, 2829–2844 Search PubMed.
- R. Losantos, G. Prampolini and A. Monari, Molecules, 2024, 29, 1752 Search PubMed.
- Y. Lei, Z. Zheng, L. Vasquez, J. Zhao, J. Ma and H. Ma, J. Phys. Chem. Lett., 2022, 13, 4840–4848 Search PubMed.
- G. Prampolini, F. Ingrosso, A. Segalina, S. Caramori, P. Foggi and M. Pastore, J. Chem. Theory Comput., 2019, 15, 529–545 Search PubMed.
- J. Cerezo, S. Gao, N. Armaroli, F. Ingrosso, G. Prampolini, F. Santoro, B. Ventura and M. Pastore, Molecules, 2023, 28, 3910 Search PubMed.
- M. Barbatti, M. Bondanza, R. Crespo-Otero, B. Demoulin, P. O. Dral, G. Granucci, F. Kossoski, H. Lischka, B. Mennucci, S. Mukherjee, M. Pederzoli, M. Persico, M. Pinheiro, J. Pittner, F. Plasser, E. Sangiogo Gil and L. Stojanovic, J. Chem. Theory Comput., 2022, 18, 6851–6865 Search PubMed.
- E. X. Salazar, M. F. S. J. Menger and S. Faraji, J. Chem. Theory Comput., 2024, 20, 5796–5806 Search PubMed.
- J. C. Tully, J. Chem. Phys., 1990, 93, 1061–1071 Search PubMed.
- X. Zhao, I. C. D. Merritt, R. Lei, Y. Shu, D. Jacquemin, L. Zhang, X. Xu, M. Vacher and D. G. Truhlar, J. Chem. Theory Comput., 2023, 19, 6577–6588 Search PubMed.
- A. K. Belyaev, W. Domcke, C. Lasser and G. Trigila, J. Chem. Phys., 2015, 142, 104307 Search PubMed.
- A. Segalina, D. Aranda, J. A. Green, V. Cristino, S. Caramori, G. Prampolini, M. Pastore and F. Santoro, J. Chem. Theory Comput., 2022, 18, 3718–3736 Search PubMed.
- L. L. E. Cigrang, J. A. Green, S. Gómez, J. Cerezo, R. Improta, G. Prampolini, F. Santoro and G. A. Worth, J. Chem. Phys., 2024, 160, 174120 Search PubMed.
- S. Polonius, O. Zhuravel, B. Bachmair and S. Mai, J. Chem. Theory Comput., 2023, 19, 7171–7186 Search PubMed.
- B. Cao, J. Dong, Z. Wang and L. Wang, J. Phys. Chem. Lett., 2025, 16, 4907–4920 Search PubMed.
- C. Zhang, Y. Zhong, Z.-G. Tao, X. Qin, H. Shang, Z. Lan, O. V. Prezhdo, X.-G. Gong, W. Chu and H. Xiang, Nat. Commun., 2025, 16, 2033 Search PubMed.
- S. K. Chandy and K. Raghavachari, J. Chem. Theory Comput., 2023, 19(19), 6632–6642 Search PubMed.
| This journal is © The Royal Society of Chemistry 2025 |