
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Monitoring nucleoside metabolism in living cells with a nucleobase analogue via fluorescence lifetime imaging
Pauline
Pfeiffer
 a,
Niusha
Bagheri
b,
Chen
Qian
c,
Jerker
Widengren
b and
L. Marcus
Wilhelmsson
*a
a,
Niusha
Bagheri
b,
Chen
Qian
c,
Jerker
Widengren
b and
L. Marcus
Wilhelmsson
*a
aDepartment of Chemistry and Chemical Engineering, Chalmers University of Technology, Kemivägen 10, SE-41296 Gothenburg, Sweden. E-mail: marcus.wilhelmsson@chalmers.se
bExperimental Biomolecular Physics, Department of Applied Physics, Royal Institute of Technology (KTH), Albanova University Center, Stockholm, 10691, Sweden
cDepartment of Chemistry and Molecular Biology, Faculty of Science, University of Gothenburg, Gothenburg 412 96, Sweden
First published on 22nd August 2025
Abstract
To overcome challenges in fluorescence labelling of RNA inside living cells we have recently introduced a direct approach using the fluorescent nucleobase analogue 2CNqA. Here we demonstrate its potential for use in fluorescence lifetime imaging (FLIM) to investigate nucleoside metabolism and for metabolic RNA labelling.
Metabolic labelling of biomolecules is a beneficial approach for monitoring turnover, processes, and biochemical reactions in living cells directly coupled to the biomolecules, in their natural environment. However, major challenges are that the labels must mimic the natural biomolecular building blocks and be accepted by enzymes while carrying useful readout features.
Stable isotope tracers or radioactive analogues are frequently used as labels but are limited by sensitivity (mass spectrometry imaging) or come with special handling requirements.1 Therefore, fluorescence, with its high sensitivity, has emerged as a useful labelling and readout modality. Some biomolecules display natural fluorescence which can be used for certain metabolic studies.2–4 For most investigations, however, these signals are not specific enough, and therefore artificial fluorophores must be introduced. For metabolic RNA labelling different approaches and fluorophores have been investigated, but challenges remain.5,6 A main requirement for advanced RNA studies is to be able to perform the readout on living cells, and without applying permeabilization techniques.6 Currently, the most frequently used methodology is to attach the actual fluorophore to a chemically modified ribonucleoside, which is delivered to cells and metabolically incorporated prior to the labelling step.7,8 However, this labelling approach is based on two reaction steps and, hence, the readout is a combination of the reactions of metabolic incorporation and the attachment of the label. Furthermore, many fluorophores display low biocompatibility, limiting their reliability for labelling and readout.9 Hence, the idea of modifying RNA building blocks themselves and using cell-endogenous pathways to directly label RNA is of outmost interest. In this respect fluorescent base analogues (FBAs), which are small and closely resemble their natural counterparts, have emerged as a promising labelling strategy for imaging purposes.10–16 However, to deliver building blocks for metabolic labelling comes with the challenges of cellular uptake and intracellular processing of the analogues normally requiring engineering of the cells used.5,6 As we have shown, using our adenosine analogue 2CNqA we can image the uptake into living cells and detect cell structure changes upon addition of Actinomycin D and NaAsO2.13 By extracting the RNA from cells exposed to 2CNqATP we could detect an increase in fluorescence intensity over exposure time, which indicates metabolic incorporation of our fluorophore. However, looking only at intensity-based images using confocal microscopy we were not able to distinguish between the free (monomeric) 2CNqA derivatives (i.e. nucleotides and nucleoside) and the RNA-incorporated derivative inside living cells.
Fluorescence lifetime imaging (FLIM) enables the detection of changes in fluorescence lifetime, reflecting different molecular states, microenvironments and molecular conformations, independently of fluorophore concentrations.17 This technique is widely applied in label-free metabolic imaging by exploiting the autofluorescence of endogenous biomolecules, e.g. FLIM measurements of NADH/NADPH to monitor the redox-state of cells.18,19 Exogenous fluorophores can be used as FLIM-based sensors to monitor the microenvironment in a more specific manner, e.g. studying pH20 or diffusion.21 FLIM also enables the investigation of protein–protein interactions by detecting changes in fluorescence lifetime that arise from direct interactions or environmental alterations, including FRET-based mechanisms.22 This has allowed investigations on cellular signalling23 and other biochemical processes.24 Hence, by combining the advantages of direct labelling and fluorescence lifetime characterization FLIM is a versatile approach to study metabolic processes, by detecting subfractions of emitting species with different lifetimes, and to distinguish if a change in fluorescence intensity is due to a change in fluorophore concentration or altered fractions of its subpopulations.
In the current study our goal was to use our fluorescent nucleobase analogue 2CNqA to develop a method to monitor nucleoside and RNA metabolism inside living cells by FLIM. In our previous work13 we concluded that 2CNqA can be applied for live-cell confocal microscopy and that it gets incorporated into RNA, but we could not conclude at what locations in the cells 2CNqA is present as a monomer and where it is found inside RNA. We know from earlier investigations that oligonucleotide-incorporated 2CNqA displays a significantly shorter average lifetime (ca. 6 ns25) than the free triphosphate (τTP = 9.9 ns11), but the determination of the fluorescence lifetime of metabolically incorporated 2CNqA in total RNA remained to be established. We used RNA from our previous study that was thoroughly purified from cell lysate to eliminate 2CNqATP monomer content in the final solutions as described in Pfeiffer et al.11 We recorded the lifetime of extracted RNA from cells that had been exposed to 2CNqATP for different time spans by time-correlated single-photon counting (TCSPC) in a cuvette (Fig. S1 and Table S1). After 2 h cellular exposure we observed a short average lifetime of the extracted RNA, similar to that of the control experiment where 2CNqA was added to cell lysate prior to RNA purification (i.e. where incorporation is impossible). At 2 h, as we have previously shown, the cellular uptake of the 2CNqA moiety is not in equilibrium, as evidenced by a continuing increase in fluorescence intensity inside the cells.13 Conversely, all RNA extracts from cells that were exposed to 2CNqATP for more than 2 h display overlapping fluorescence decays that could be fitted to a bi-exponential decay model with a longer lifetime τ1,RNA = 9.2 ± 0.2 ns (with the pre-exponential factor α1,RNA = 0.37 ± 0.02) and a shorter, dominant one τ2,RNA = 1.3 ± 0.4 ns (α2,RNA = 0.63 ± 0.02) (Table S1). While the longer lifetime of the extracted RNA (τ1,RNA) is in the same range as that of the free monomer (ca. 10 ns), the shorter lifetime τ2,RNA, is clearly distinguishable from the long-lived species. Assuming that the ratio of the amplitudes of the two lifetime components of extracted RNA is constant (α1,RNA/α2,RNA = 0.6 ± 0.04, Table S1) we aimed for the detection of changes in the amplitude contribution in the FLIM images of living cells exposed to 2CNqATP. Therefore, we exposed HEK293T cells, that were previously shown to display cellular uptake,13 to 2.5 μM 2CNqATP by addition of the compound directly to the cell medium. Since an enhancement of fluorescence intensity from extracted RNA from cells exposed to 2CNqATP with longer time was previously observed,13 we imaged cells using FLIM at two different time points. After 4 h we have previously observed a stable, maximal signal inside cells by confocal microscopy and flow cytometry, at which time the fluorescence intensity from the extracted RNA was still low.13 In the same study, after 24 h, even the intensity from the extracted RNA had increased significantly, making these two timepoints (4 h and 24 h) a good readout for our FLIM experiments. The resulting FLIM data was analysed by fitting the TCSPC data to a multiexponential decay model as well as by using the phasor approach.26
First, we fitted the TCSPC data with free parameters. All decay curves were well described by a bi-exponential function with lifetimes of around 10 ns and 3 ns in all samples. Additionally, we observed a short lifetime component when measuring 2CNqATP in water, previously reported to have a mono-exponential decay11 (details SI Section S1.6). We ascribe this component to fluorescence anisotropy decay effects, present when measuring TCSPC in the microscope setup. These effects are also present in the live cell FLIM data. To estimate the contribution of the two lifetime components we fixed the two observed lifetimes (τ1,cell = 10 ns and τ2,cell = 3 ns, Fig. S2) and investigated differences in the relative amplitudes of the two components between the two exposure times of 2CNqATP on the cells (Fig. 1 and Table 1, Fig. S2).
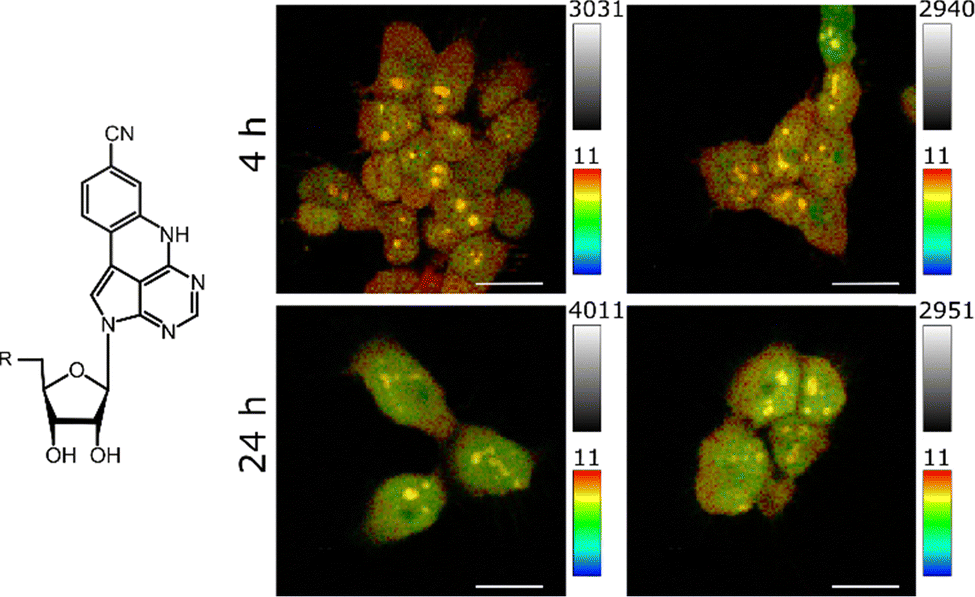 | ||
| Fig. 1 Left: Molecular structure of 2CNqATP with R indicating three phosphate groups. Right: Lifetime images of the four images summarized in Table 1 with colourized lifetime scale from 0 ns (blue) to 11 ns (red) and intensity scale, i.e. photon counting events from zero (dark) to indicated count number (bright). Exposures shown in duplicates (left/right). Scale bars = 30 μm. Details in Fig. S2. | ||
| Sample | α 1,cell | α 2,cell | τ avg,Amp [ns] | τ avg,Int [ns] |
|---|---|---|---|---|
| #1; 4 h | 0.52 | 0.48 | 6.6 | 8.5 |
| #2; 4 h | 0.47 | 0.53 | 6.3 | 8.2 |
| #3; 24 h | 0.40 | 0.60 | 5.8 | 7.8 |
| #4; 24 h | 0.40 | 0.60 | 5.8 | 7.8 |
After 4 h exposure time the amplitudes of the two lifetime components (α1,cell, α2,cell) were found to be almost equal, while we observed an increase of the shorter-lived species (α2,cell) after 24 h exposure time. This also resulted in slightly shorter average lifetimes. As described earlier, α1,cell most likely represents the abundance of a combined species with the longer lifetime of the bi-exponential 2CNqA (τ1,RNA) inside nucleic acids and the mono-exponential free monomer (τTP), whereas α2,cell originates solely from the contribution of the incorporated form (with lifetime τ2,RNA). As we suppose that the ratio of the amplitude contribution of the two lifetime species of 2CNqA incorporated into RNA is constant (α1,RNA/α2,RNA = 0.6 ± 0,04 vide supra), this shift over time indicates that the microenvironment around 2CNqA has changed and, hence, suggests that 2CNqA gets more incorporated with time. Note that due to the overlap of the two long-lived species (τTP and τ1,RNA), solely looking at α2,cell as fraction in Table 1 leads to an underestimation of the molar fraction of RNA-incorporated 2CNqA.
By inspecting the resulting images, the difference in lifetime contribution becomes visible in a spatial manner (Fig. 1): the 10 ns species τ1,cell (red) is dominant in the cytosol and comparing the two exposure times (4 h vs. 24 h), a clear difference is visible. The longer-lived species is less present in the nuclei after 24 h exposure time, which indicates that signal from the RNA-incorporated 2CNqA dominates. Interestingly, the nucleoli show longer average lifetimes compared to the nuclei, but shorter lifetime compared to the cytosol (vide infra).
Measurements in aqueous solution do not straightforwardly provide a benchmark to FLIM data from 2CNqA acquired in the complex microenvironments inside cells. The interpretation of differences in parameter values between the image decays (τ1,cell = 10 ns and τ2,cell = 3 ns, Table 1) and the measured values in solution (τ1,RNA = 9.2 ns and τ2,RNA = 1.3 ns, Table S1) is complex. Hence, a direct comparison between the lifetimes and amplitudes of extracted 2CNqA-labelled RNA and 2CNqA inside cells might not be entirely valid. While it cannot include all features contributing to the different microenvironments affecting the 2CNqA fluorescence, it nonetheless provides a basis for a simplified comparison. Given the complex influence on the 2CNqA fluorescence decay, by the microenvironment but also by the nearest neighbour nucleotides as we observed in previous studies,25 we also evaluated the FLIM data in a model-independent manner, separating the fluorophore populations based on differences in their overall fluorescence decay features. For this, we analysed the images in the frequency domain by the phasor approach (Fig. 2).26 Pixels (phasors) with mono-exponential decay profiles are located on the semicircle, whereas phasors with combinations of several decay profiles are linear combinations of the phasors of these decay components and are, thus, located inside the semicircle. Phasors of a mixture of two species therefore lie on a straight line connecting the phasors of the component species. The indicated photon weighted centre of mass distribution (Fig. 2A, crosses and circles) of the four FLIM images correspond well to the intensity-weighted average lifetimes observed from the fitting (Table 1), i.e. shorter exposure time (4 h, crosses) results in a longer average lifetime than exposure for 24 h (circles, overlapped). The contribution of the long-lifetime species decreases with incubation time as we also found above (Table 1). Again, because the ratio of the amplitudes of the two lifetimes of 2CNqA inside RNA (α1,RNA/α2,RNA) is supposedly constant (see above) this shift in the phasor indicates 2CNqA RNA-incorporation. By inspecting the resulting images, where the colours represent the relative lifetime along the fraction line (Fig. 2A) it is evident that cells exposed for only 4 h show a larger contribution of the longer lifetime component compared to cells exposed for 24 h (Fig. 2B).
 | ||
| Fig. 2 (A) Phasor plot sector presenting the phasors of the four FLIM images in B. The photon-weighted centre of mass for each image is shown as a circle for 24 h exposure time and as a cross for 4 h exposure time. Black line shows the intensity fraction profile through the four phasor centres. Mono-exponential lifetime of 8 ns indicated on the semi-circle, shown as dashed line. (B) FLIM images of HEK293T cells exposed to 2CNqATP for 4 h or for 24 h, respectively (from same raw image data as in Fig. 1). Exposures shown in duplicates (left/right). Colouring is based on the intensity fraction line in A (black line), details in SI Section S1.4. Scale bar = 30 μm. | ||
Examining the localization of the different 2CNqA lifetime species inside the cells, we observe the same inhomogeneous distribution as in the decay-fitted images: the longer lifetime (red to green) contributes most in the cytosol and least in the nuclei. Moreover, the nucleoli show a high fraction of the long-lived species after both exposure time points (4 h and 24 h), but the contribution of the long-lived species inside the nucleoli decreases (from red after 4 h to yellow after 24 h, Fig. 2B arrows). Previously, we have observed stronger fluorescence intensity regions in confocal microscopy images,13 indicating that 2CNqA accumulates inside these nuclear sub-structures. From the FLIM data we can now further resolve the underlying reason for the stronger fluorescence in these regions, getting information on the fluorescent species of 2CNqA inside the nucleoli. The contribution of the long-lived species is higher in the nucleoli than in the nuclei, which suggests accumulation of the free monomer. By pulse-chase experiments with compounds affecting the nucleoli (e.g. Actinomycin D or NaAsO2) or possibly by isolating nucleoli, additional analyses of their 2CNqA content may be possible to further confirm this conclusion.
To our knowledge, this is the first study in which an FBA is used in FLIM measurements in live cells and, specifically, to monitor nucleoside and RNA metabolism. Unlike other metabolic labelling strategies, via spontaneous uptake FBAs enable one-step labelling, supporting diverse fluorescence-based readouts in live cells. Building on our recent report of 2CNqA's spontaneous uptake and accumulation in the cytosol, nuclei, and nucleoli,13 we now present a method to distinguish and track its species in living cells with spatiotemporal resolution. Hence, our FLIM approach enables direct studies of metabolic labelling of RNA and nucleoside metabolism in live cells. By co-exposure of cells to 2CNqATP and small molecules like inhibitors or drugs in a pulse-chase manner, this imaging method, together with other readout techniques promises to give novel, detailed information on inhibitor/drug mechanisms of action. With FLIM instrumentation and software currently becoming more common and easily available we envision that the methodology we present here has a large potential in the basic research on RNA, but also in pharmaceutical research, e.g. drug development and screening. Our previous finding that 2CNqA also is a good two-photon label for antisense oligonucleotide gapmers11 suggests that the imaging approaches presented herein for single cells could also be feasible in organoids/spheroids/tissue by two-photon excitation. Future FBA development will enhance spectral properties (e.g. redshift, brightness) for greater labelling flexibility, and our findings suggest continued method development will broaden specificity (e.g. organelles, RNA types).
P. P. and N. B. performed FLIM experiments. N. B. fitted decays and prepared lifetimes images. C. Q. performed phasor plot analysis and prepared corresponding images. P. P. performed and analysed TCSPC in solution, designed figures and drafted the manuscript. J. W. and L. M. W. aided in interpreting results and worked on the manuscript. P. P. and L. M. W. conceived the project. All authors have read and given feedback on the manuscript.
We thank Mr A. Kulkarni and Dr C. V. Srambickal at KTH for help in the laboratory, Dr A. Kenesei at KTH for providing cells, and Dr J. R. Nilsson at LanteRNA, Prof. M. Ericson at Gothenburg University as well as Profs. B. Albinsson and J. Andréasson at Chalmers for valuable input on the interpretation on the results, and for manuscript feedback, respectively. Funding from Area of Advance Nano at Chalmers University of Technology to P. P. and L. M. W. and conducted as part of the FoRmulaEx research center for nucleotide delivery and with associated financial support to L. M. W. from the Swedish Foundation for Strategic Research (SSF) [IRC15-0065] and the Swedish Research Council [VR, 2021-04409]. Financial support to J. W. from the Swedish Foundation for Strategic Research (SSF) [BENVAC RMX 18-0041] and the Swedish Research Council [VR, 2021-04556]. Financial support the C. Q. from Horizon Europe MSCA Postdoctoral Fellowship [101103885] and the Swedish Research Council [VR, 2023-03787].
Conflicts of interest
L. M. W. and P. P. own shares in LanteRNA.Data availability
Experimental details and data analysis is in the SI. Raw data available upon reasonable request. See Doi: https://doi.org/10.1039/d5cc03959bNotes and references
- M. Schwaiger-Haber, et al. , Nat. Commun., 2023, 14, 2876 CrossRef PubMed.
- B. Chance, et al. , J. Biol. Chem., 1979, 254, 4764–4771 CrossRef.
- I. Georgakoudi, et al. , Cancer Res., 2002, 62, 682–687 Search PubMed.
- A. B. T. Ghisaidoobe and S. J. Chung, Int. J. Mol. Sci., 2014, 15, 22518–22538 CrossRef PubMed.
- R. E. Kleiner, Mol. Omics, 2021, 17, 833–841 RSC.
- J. I. H. Knaack and C. Meier, ChemMedChem, 2024, 19, e202400160 CrossRef PubMed.
- N. Z. Fantoni, et al. , Chem. Rev., 2021, 121, 7122–7154 CrossRef PubMed.
- N. Klöcker, et al. , Chem. Soc. Rev., 2020, 49, 8749–8773 RSC.
- S. T. Steiner, et al. , Photochem. Photobiol. Sci., 2023, 22, 2093–2104 CrossRef PubMed.
- T. Baladi, et al. , J. Am. Chem. Soc., 2021, 143, 5413–5424 CrossRef PubMed.
- J. R. Nilsson, et al. , Phys. Chem. Chem. Phys., 2023, 25, 20218–20224 RSC.
- J. R. Nilsson, et al. , Sci. Rep., 2021, 11, 11365 CrossRef CAS PubMed.
- P. Pfeiffer, et al. , Nucleic Acids Res., 2024, 52, 10102–10118 CrossRef CAS PubMed.
- D. Wang, et al. , J. Am. Chem. Soc., 2022, 144, 14647–14656 CrossRef CAS.
- Y. Zhang and R. E. Kleiner, J. Am. Chem. Soc., 2019, 141, 3347–3351 CrossRef CAS.
- K. Hadidi, et al. , Angew. Chem., Int. Ed., 2023, 62, e202216784 CrossRef CAS PubMed.
- R. Datta, et al. , JBO, 2020, 25, 071203 CAS.
- T. S. Blacker, et al. , Nat. Commun., 2014, 5, 3936 CrossRef CAS PubMed.
- J. Vergen, et al. , Microsc. Microanal., 2012, 18, 761–770 CrossRef CAS.
- S. Ogikubo, et al. , J. Phys. Chem. B, 2011, 115, 10385–10390 CrossRef CAS PubMed.
- K. Suhling, et al., Multiphoton Microscopy in the Biomedical Sciences XIX, SPIE, 2019, 10882, pp. 115–123 Search PubMed.
- W. R. Algar, et al. , Nat. Methods, 2019, 16, 815–829 CrossRef CAS.
- F. S. Wouters and P. I. H. Bastiaens, Curr. Biol., 1999, 9, 1127-S1 CrossRef.
- P. I. H. Bastiaens and A. Squire, Trends Cell Biol., 1999, 9, 48–52 CrossRef CAS.
- A. Wypijewska del Nogal, et al. , Nucleic Acids Res., 2020, 48, 7640–7652 CrossRef.
- M. A. Digman, et al. , Biophys. J., 2008, 94, L14–L16 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |