
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA fluorogenic probe for screening aldehyde dehydrogenase 1A1 (ALDH1A1) inhibitors
Shilpendu
Ghosh†
 a,
Sayar
Ghosh†
b,
Aniket
Majee†
a,
Soumee
Sengupta
b,
Sujato
Mukherjee
a,
Banshi
Roy
a,
Rahul
Das
*b and
Arindam
Mukherjee
*a
a,
Sayar
Ghosh†
b,
Aniket
Majee†
a,
Soumee
Sengupta
b,
Sujato
Mukherjee
a,
Banshi
Roy
a,
Rahul
Das
*b and
Arindam
Mukherjee
*a
aDepartment of Chemical Sciences and Centre for Advanced Functional Materials (CAFM), Indian Institute of Science Education and Research Kolkata, Mohanpur Campus, Mohanpur 741246, India. E-mail: a.mukherjee@iiserkol.ac.in
bDepartment of Biological Sciences, Indian Institute of Science Education and Research Kolkata, Mohanpur Campus, Mohanpur 741246, India. E-mail: rahul.das@iiserkol.ac.in
First published on 9th September 2025
Abstract
We report the first competitive aldehyde dehydrogenase 1A1 (ALDH1A1) inhibitor featuring a fluorogenic binding response and nanomolar affinity (KD), enabling real-time screening of non-fluorescent inhibitors with high cellular compatibility and no observed toxicity.
A fluorogenic non-covalent competitive inhibitor is often superior for direct, functional, and imaging-compatible studies of enzyme activity and inhibition, especially in live cells without genetic manipulation (e.g. unlike the nanoBRET assay). It offers key advantages in facilitating sensitive real-time measurement of enzymatic activity and is ideal for high-throughput screening to evaluate kinetic parameters of new competitive inhibitors.1–3 The fluorescence turn-on mechanism with good signal-to-noise ratio enhances the accuracy and reliability of quantification allowing screening of new candidates through kinetic assays.3
A specific fluorogenic inhibitor for ALDH1A1 (also known as retinal dehydrogenase 1) remains unavailable, although ALDH1A1 is one of 19 isoforms in the aldehyde dehydrogenase (ALDH) family, responsible for the oxidation of aldehydes in diverse metabolic pathways.4 Given the importance of ALDH1A1 in various physiological and pathological processes, including but not limited to retinoic acid biosynthesis, neuroprotection against toxic dopamine metabolites, and the maintenance of cancer stem cells (CSCs).5–7 ALDH1A1 has been identified as a CSC biomarker in multiple malignancies, including breast, lung, pancreatic, and colorectal cancers, where elevated enzymatic activity correlates with chemotherapy resistance, tumor progression, and poor clinical outcomes.4 Inhibitors targeting ALDH1A1 with nanomolar potency have drawn increasing interest due to their potential, in reducing CSC-associated phenotypes, suppressing hepatic glucose production, and attenuating triacylglycerol synthesis in the liver. These attributes position ALDH1A1 as a promising therapeutic target in oncology and in metabolic disorders (diabetes, dyslipidemia, obesity, inflammation and Sjögren–Larsson syndrome).8–12 Detailed understanding of ALDH1A1 enzyme kinetics is important for rational design of selective inhibitors for targeted therapies.10,13–17 Thus, a fluorogenic ALDH1A1 inhibitor is an important addition to this family.
Most commercially available fluorescent probes for studying ALDH activity are broadly reactive substrates that measure total isoform activity, with the exception of ‘AlDeSense,’ a fluorogenic substrate specific to ALDH1A1.18 However, such substrates are not suitable for assessing enzyme kinetics or inhibitor interactions, as they exit the catalytic site after conversion. In contrast, fluorogenic inhibitors with a good binding constant (equilibrium dissociation constant (KD) in nM range) provide a key advantage by enabling displacement-based assays to screen novel, including non-fluorescent, inhibitors and evaluate binding parameters like KD, Ki and IC50.
Cell-permeable fluorogenic inhibitors uniquely support both novel inhibitor screening, and live-cell imaging apart from mechanistic dissemination for pathway analysis upon inhibition of the target protein. Hurley et al. identified isatin derivatives as potent nanomolar inhibitors of ALDH enzymes and enhanced selectivity for ALDH1A1 over ALDH3A1 by incorporating an N-alkyl spacer to accommodate additional functional groups.14
We report the development of a fluorogenic ALDH1A1 inhibitor synthesized by conjugating a green-emitting naphthalimide fluorophore with an isatin scaffold. The resulting isatin–naphthalimide hybrid shows strong and competitive binding to the ALDH1A1 substrate-binding site. Owing to its competitive and reversible binding, the compound can be displaced by other inhibitors, enabling its use in displacement-based assays for screening binding constants of novel competitive ALDH1A1 inhibitors (Scheme 1).
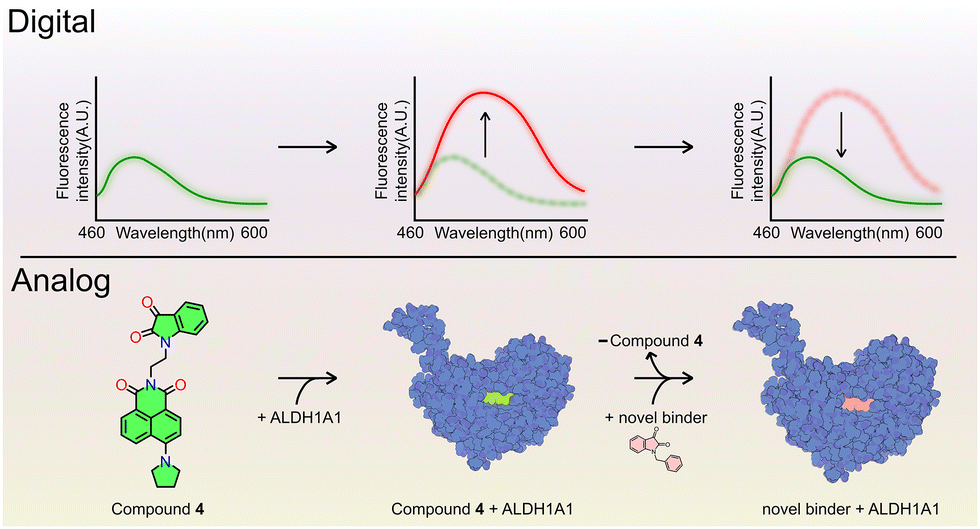 | ||
| Scheme 1 Illustration of the fluorogenic mechanism where compound 4 binds competitively to ALDH1A1, enabling displacement-based inhibitor screening. | ||
This study initiated with in silico design and investigation of a potential fluorogenic ALDH1A1 inhibitor by linking a green-emitting naphthalimide derivative to an isatin via an ethyl linker. Given the possibility that the naphthalimide conjugation might impair binding affinity, we investigated docking of 4 against the substrate-binding site of ALDH1A1 (PDB ID: 7JWU).
The docking studies show that 4 favors the substrate-binding pocket over the NAD+ site, indicating higher affinity and competitive binding (Fig. 1(A)).
 | ||
| Fig. 1 (A) Structural depiction of the interaction between 4 and the catalytic site of ALDH1A1 (PDB: 7JWU). (B) Evaluation of the contributions of individual residues to the stabilization or destabilization of the ALDH1A1-4-NAD complex. (C) RMSD analysis of the system for 100 ns. (D) Comparison of binding energies (mean ± SD) of 4, NBI, and VMA (the native ligand of 7JWU). (E) 4 shifts deeper into the catalytic pocket over 100 ns, enhancing binding energy. (F) Change in the fluorescence intensity of 4 when it binds to the active site of ALDH1A1 (n = 3). (G) and (H) are the plots of time-dependent association and dissociation binding kinetics of ALDH1A1 and 4, respectively. The inset represents the single exponential fitting in one-phase association and one-phase decay. The error bar represents the SD from three independent experiments (n = 3). | ||
Furthermore, docking against other ALDH isoforms (ALDH1A3, ALDH2, and ALDH3A1) suggested potential pan-ALDH inhibition, with strong affinity for ALDH1A1, comparable to the selective inhibitor N-benzylisatin (NBI)14 (Fig. S1). Notably, 4 yielded a higher docking score of 93 ± 6 for ALDH1A1 in comparison to 67 ± 3 for NBI. Interestingly, in the most favorable docking conformation, the naphthalimide moiety of 4 occupied the ALDH1A1 active site rather than the isatin moiety. This is contrary to the well-documented affinity of isatin for the substrate-binding pocket.14
To validate the docking conformation, we performed 100 ns molecular dynamics (MD) simulations. The trajectory supported the docking pose, with the ligand gradually migrating deeper into the binding cavity (Fig. 1(B)–(E)). RMSD analysis indicated system equilibration after ∼20 ns. Binding free energy calculations using the molecular mechanics Poisson–Boltzmann surface area (MMPBSA) method (100 snapshots, final 10 ns) predicted −29.68 ± 2.48 kcal mol−1 (Table S1). Stabilizing interactions included π-stacking of the isatin moiety with Y297 (−2.21 ± 0.05 kcal mol−1) and van der Waals interactions between the naphthalimide core and V460 (−2.13 ± 0.09 kcal mol−1), along with additional π-stacking involving F290, F171, and W178. Destabilizing contributions arose from D122, E289, and G458. Pose validation compared docking scores of isatin and naphthalimide with their alkyl chain derivatives (5 and 6), where naphthalimide–alkyl derivatives scored higher than isatin but lower than 4 (Fig. S3 and S4). Further docking of 4 into ALDH1A1 (PDB: 7JWU) with its isatin motif placed in the active site (Fig. S5) yielded a lower binding free energy (−26.87 ± 2.14 kcal mol−1, Table S2) than when the naphthalimide moiety occupied the catalytic site.
Encouraged by these results, we synthesized 4 in a few steps (Scheme S1 and Fig. S6–S19) and isolated it in pure form (>95%, Fig. S20). The compound exhibited green fluorescence ((λabsmax/λemmax = 450/535 nm in methanol, quantum yield (Φ) = 0.0056); Fig. S21), high aqueous stability over 72 h (HPLC, single peak at 19 min; Fig. S22), and a lipophilicity of log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Po/w = 3.7 (Fig. S23 and Table S2), consistent with Lipinski's rule of five and suggestive of favorable drug-like properties.
Po/w = 3.7 (Fig. S23 and Table S2), consistent with Lipinski's rule of five and suggestive of favorable drug-like properties.
Upon binding to ALDH1A1 (purified from E. coli BL21 (pLysS) cells, Fig. S24 and S25) compound 4 exhibited a fluorogenic response characterized by a 3-fold increase in fluorescence intensity, Φ = 0.0264 (ca. 4.7-fold higher than the free state), and a redshift of ∼20 nm, consistent with its interaction within the hydrophobic active site of the enzyme (Fig. 1(F)). This pronounced fluorescence shift enabled the characterization of pre-steady-state kinetics for 4 (1 μM) binding to ALDH1A1 (100 nM) via stopped-flow spectroscopy with a cutoff filter of 515 nm. The absolute binding affinity of 4 yielded KabsD = 102 ± 4 nM, derived from the ratio koff/kon where koff = (2.885 ± 0.201) × 10−2 s−1 and kon = (2.84 ± 0.32) × 10−4 nM−1 s−1 (Fig. 1(G) and (H)). The inhibitory potency of pure, stable 4 against ALDH1A1 was evaluated by monitoring NAD+ reduction to NADH. Assays were conducted in a buffer containing 20 mM HEPES (pH 7.4), 150 mM NaCl, 1 mM NAD+, and 1 mM DTT, with 1 mM benzaldehyde as a substrate and 3.5 μM ALDH1A1 enzyme. NADH formation was measured via absorbance at 340 nm over 90 minutes at 25 °C. Compound 4 exhibited potent inhibition with an IC50 of 370 ± 8 nM (Ki = 63 ± 14 nM) (Fig. 2(A)). Considering isatin's previously reported mild inhibitory activity against ALDH1A1,14 we investigated the inhibitory effect of the naphthalimide moiety alone (compound 2). We further evaluated the inhibitory effects of compounds 5 and 6, in which the naphthalimide moiety is linked via a propyl or ethyl spacer containing a nitrogen atom (Fig. S26–S30). Compounds 2, 6 and 7 showed no inhibition, confirming that both the isatin and naphthalimide components contribute synergistically to the inhibitory effect (Fig. 2(B)). Under identical conditions, the ALDH1A1-selective inhibitor NBI displayed an IC50 of 220 ± 20 nM (Ki = 38 ± 5 nM) (Fig. S31). Using propionaldehyde as the enzymatic substrate, we assessed ALDH1A1 inhibition by 4 and NBI at 1 μM.
 | ||
| Fig. 2 (A) Enzyme inhibition profile of 4 targeting ALDH1A1 (n = 3). (B) Percentage of enzyme activity in the presence of 1 μM 2 and 4 showing that 2 is not able to inhibit ALDH1A1 (n = 3, ns = non-significant). (C) Comparative Michaelis–Menten kinetics between ALDH1A1 and ALDH1A1 with 4 and NBI using benzaldehyde as a substrate (n = 3). (D) The table represents the kinetic parameters of ALDH1A1 in the presence of 4 and NBI at their IC50 concentrations, implying that 4 serves as a competitive inhibitor. (E) Fluorescence titration to determine the equilibrium binding constant (KeqD) of 4 and apparent binding constants KappD of NBI, Etphisa, and Br-Bzisa by fluorescence displacement assay (n = 3). (F) KappD values of NBI, Etphisa, and Br-Bzisa. Statistical significance was determined as follows: p < 0.05 (*), p < 0.01 (**), p < 0.001 (***), p < 0.0001 (****); ns denotes not significant. | ||
Both agents potently suppressed catalytic activity, evidenced by a marked reduction (>80%) in specific activity relative to controls (Fig. S32). Michaelis–Menten kinetics were assessed by measuring NADH production at varying benzaldehyde concentrations in the presence of 4 and NBI (Fig. S33). The kinetics observed with compound 4 (IC50 concentration) indicate a threefold Km increase, an unchanged Vmax, and a fourfold decrease in kcat/Km (Fig. 2(C) and (D)), confirming competitive inhibition of ALDH1A1. In contrast, NBI is identified as a noncompetitive inhibitor using a Line–Weaver Burk plot (Fig. S34). 4 increases Km without affecting Vmax, showing classical competitive inhibition at the active site. In contrast, NBI reduces Vmax with little change in Km, consistent with non-competitive inhibition. This distinction explains that displacement assays are suitable for compound 4. Leveraging this mechanism, the equilibrium dissociation constant (KeqD) of 4 was determined by fluorescence titration to be 127 ± 16.3 nM (Fig. 2(E)). Thus, the approximate equivalence of KeqD to the KabsD confirms the validity of our assay setup. Furthermore, as a fluorescent competitive inhibitor, 4 enables measurement of apparent dissociation constants (KappD) via fluorescence quenching, providing a robust platform for screening new inhibitors and guiding inhibitor design. The compound's fluorescence intensity, sensitivity, and emission wavelength are compatible with standard fluorimeters, facilitating assay development. To demonstrate the use of 4 in evaluating new inhibitors using its fluorogenic properties, we tested three ALDH1A1 inhibitors with known IC50 values and evaluated their KD, including NBI, 1-phenethylindoline-2,3-dione (EtPhisa), and 1-benzyl-5-bromoindoline-2,3-dione (Br-Bzisa). ALDH1A1 (500 nM) was pre-incubated with 4 (1 μM) at 4 °C to achieve active-site saturation. Competitive binding of candidate inhibitors was then assessed via titration against the pre-formed ALDH1A1:4 complex (Fig. S35). The fluorescence intensity of 4 is negligible at 510 nm unless it binds to ALDH1A1, so titration with an inhibitor can be tracked by monitoring the decrease in fluorescence at this wavelength until it plateaus. This allowed determination of the KappD for each inhibitor (Fig. 2(F) and Table S4). These KappD values correlated well with their reported IC50 values (Fig. S36),14 validating 4 as an effective probe for screening and ranking the binding affinities of ALDH1A1 inhibitors.
Given the presence of a naphthalimide fluorophore, the aggregation behavior of 4 in aqueous media was further evaluated using dynamic light scattering (DLS) in 1× PBS (pH 7.4) across concentrations. Aggregation was observed at concentrations ≥3 μM. At 1 μM, the average particle size remained ∼50 nm (Fig. S37 and S38). Field emission scanning electron microscopy (FESEM) corroborated the DLS data, revealing small, dispersed aggregates. Thus, the aggregation occurs at concentrations ca. 7-fold higher than the KD value (127 ± 16 nM), suggesting that the measured KD is genuine and not an artifact of aggregation-induced enzyme inactivity.
ALDH1A1 is primarily cytoplasmic, and co-localization studies confirmed that 4 similarly distributes within cells (Fig. 3(A) and (B)). Fixed-cell immunofluorescence and spinning disk confocal microscopy showed widespread cytoplasmic localization of both 4 and ALDH1A1 (Fig. S39). Live-cell imaging in MDA-MB-231 cells revealed that 1 μM 4 exhibited minimal nuclear presence and was equally partitioned between mitochondria and lysosomes (PCC ∼ 0.5; Fig. 3(A) and (B)), supporting its cytoplasmic accumulation essential for probing ALDH1A1 inhibition and downstream mechanisms.
 | ||
| Fig. 3 (A) Co-localization analysis of lysosomal, mitochondrial, and nuclear markers in MDA-MB-231 cells treated with 1 μM of 4 (scale bar = 10μm). (B) Pearson's correlation coefficient (PCC) values quantify the degree of co-localization with 4 across cellular compartments (n > 100). (C) Aldefluor assay representing ALDH activity reduction (n = 3) in MIA PaCa-2 in the presence of 1 μM 4, 15 μM DEAB and 1 μM NBI. Statistical significance was determined as follows: p < 0.05 (*), p < 0.01 (**), p < 0.001 (***), p < 0.0001 (****); ns denotes not significant. | ||
To study the effect of 4 at the cellular level, we selected three cancer cell lines (MIA PaCa-2, PANC-1, and MDA-MB-231) expressing ALDHA1A1 (Fig. S40). 4 exhibited minimal cytotoxicity (IC50 > 100 μM) in all of them (Fig. S41 and Table S5), in contrast to the ALDH1A1 inhibitor NBI, which showed a cellular IC50 of 18.7 ± 1.8 μM in MIA PaCa-2 cells. MIA PaCa-2 has the highest expression of ALDH1A1 among the three cell lines (Fig. S40). The minimal cytotoxicity of 4 (>100 μM in MTT assays) suggests reduced off-target effects compared to NBI, apart from the stronger binding affinity toward ALDH1A1. ALDH1A1 inhibition in cells was confirmed using the AldeRed assay, where red fluorescence increases upon ALDH-mediated aldehyde oxidation and retention in ALDH1A1high MIA PaCa-2 cells. Treatment with 1 μM of compound 4 markedly reduced fluorescence (Fig. 3(C) and Fig. S42), indicating effective intracellular ALDH inhibition.
ALDH1A1 regulates cancer stem cell (CSC) traits,19–21 and we examined the effect of treatment of MIA PaCa-2 cells, known for high CSC activity,19 with 4 or NBI on a few key stemness-related genes (Table S5). ALDH1A1 and c-MYC mRNA levels reduced upon treatment with 4 or NBI (Fig. S43 and S44), consistent with ALDH1A1's role in c-MYC regulation via the Wnt/β-catenin pathway.22 Although increase in compensatory stemness-related genes NOTCH1, SOX2, SOX9, PI3K, and OCT4, highlighted the multifactorial regulation of stemness beyond ALDH1A1 alone.
In summary, 4 is a potent fluorogenic ALDH1A1 inhibitor with nanomolar KD and low cytotoxicity. MD simulations reveal cooperative binding of its isatin and naphthalimide groups, consistent with competitive inhibition and fluorogenic response. The displacement-based assay, the first of its kind, enables precise evaluation of non-fluorescent ALDH1A1 inhibitors, with KD values aligning with the reported IC50. Beyond inhibition, 4 suggests that ALDH1A1 inhibition may trigger compensatory stemness mechanisms, highlighting the complexity of ALDH1A1 biology. These results support context-specific therapeutic strategies and provide a foundation for next-generation fluorogenic inhibitors with optimized emission and reduced aggregation. AM thanks SERB (CRG/2021/001118) and IISER Kolkata for support. S. G., B. R., A. M., and S. M. from AM Lab acknowledge CSIR and KVPY; S. S. and S. G. (R.D. Lab) thank UGC and IISER Kolkata for fellowships.
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the SI which contains synthetic schemes, NMR and mass spectra, docking and MD simulation data, kinetic plots, additional figures, and tables. See DOI: https://doi.org/10.1039/d5cc03947a..
References
- W. Chyan and R. T. Raines, ACS Chem. Biol., 2018, 13, 1810–1823 CrossRef CAS PubMed.
- X. Wu, W. Shi, X. Li and H. Ma, Acc. Chem. Res., 2019, 52, 1892–1904 CrossRef CAS PubMed.
- S. M. Usama, B. Zhao and K. Burgess, Chem. Soc. Rev., 2021, 50, 9794–9816 RSC.
- M. Wang, T. Wang, J. Wang, Y. Yang, X. Li, H. Chen and J. Liao, Cell Death Dis., 2024, 15, 568 CrossRef CAS PubMed.
- S. A. Marchitti, R. A. Deitrich and V. Vasiliou, Pharmacol. Rev., 2007, 59, 125–150 CrossRef CAS PubMed.
- V. Ciccone, E. Terzuoli, S. Donnini, A. Giachetti, L. Morbidelli and M. Ziche, J. Exp. Clin. Cancer Res., 2018, 37, 311 CrossRef CAS PubMed.
- S. Nie, B. Li, M. Wang, Z. Chen, J. Ren, Z. Li, X. Xu, Z. Qian, Z. Xie, J. Han, Z. Zhang, Z. Zhang, Y. Zhu, Z. Chen, X. Yang and K. Ye, Adv. Sci., 2025, 12, e2409477 CrossRef PubMed.
- Y. C. Chang, H. L. Lee, W. Yang, M. L. Hsieh, C. C. Liu, T. Y. Lee, J. Y. Huang, J. Y. Nong, F. A. Li, H. L. Chuang, Z. Z. Ding, W. L. Su, L. Y. Chueh, Y. T. Tsai, C. H. Chen, D. Mochly-Rosen and L. M. Chuang, Nat. Commun., 2023, 14, 5971 CrossRef CAS PubMed.
- J. Xia, S. Li, S. Liu and L. Zhang, MedComm, 2023, 4, e195 CrossRef CAS PubMed.
- B. C. Huddle, E. Grimley, C. D. Buchman, M. Chtcherbinine, B. Debnath, P. Mehta, K. Yang, C. A. Morgan, S. Li, J. Felton, D. Sun, G. Mehta, N. Neamati, R. J. Buckanovich, T. D. Hurley and S. D. Larsen, J. Med. Chem., 2018, 61, 8754–8773 CrossRef CAS PubMed.
- F. W. Kiefer, C. Vernochet, P. O'Brien, S. Spoerl, J. D. Brown, S. Nallamshetty, M. Zeyda, T. M. Stulnig, D. E. Cohen, C. R. Kahn and J. Plutzky, Nat. Med., 2012, 18, 918–925 CrossRef CAS PubMed.
- C. Grim, R. Noble, G. Uribe, K. Khanipov, P. Johnson, W. A. Koltun, T. Watts, Y. Fofanov, G. S. Yochum, D. W. Powell, E. J. Beswick and I. V. Pinchuk, J. Crohns Colitis, 2021, 15, 1362–1375 Search PubMed.
- S. M. Yang, N. J. Martinez, A. Yasgar, C. Danchik, C. Johansson, Y. Wang, B. Baljinnyam, A. Q. Wang, X. Xu, P. Shah, D. Cheff, X. S. Wang, J. Roth, M. Lal-Nag, J. E. Dunford, U. Oppermann, V. Vasiliou, A. Simeonov, A. Jadhav and D. J. Maloney, J. Med. Chem., 2018, 61, 4883–4903 CrossRef CAS PubMed.
- A. C. Kimble-Hill, B. Parajuli, C. H. Chen, D. Mochly-Rosen and T. D. Hurley, J. Med. Chem., 2014, 57, 714–722 CrossRef CAS PubMed.
- S. M. Yang, A. Yasgar, B. Miller, M. Lal-Nag, K. Brimacombe, X. Hu, H. Sun, A. Wang, X. Xu, K. Nguyen, U. Oppermann, M. Ferrer, V. Vasiliou, A. Simeonov, A. Jadhav and D. J. Maloney, J. Med. Chem., 2015, 58, 5967–5978 CrossRef CAS PubMed.
- L. Quattrini, E. L. M. Gelardi, V. Coviello, S. Sartini, D. M. Ferraris, M. Mori, I. Nakano, S. Garavaglia and C. La Motta, J. Med. Chem., 2020, 63, 4603–4616 CrossRef CAS PubMed.
- A. I. M. Ibrahim, E. Batlle, S. Sneha, R. Jimenez, R. Pequerul, X. Pares, T. Rungeler, V. Jha, T. Tuccinardi, M. Sadiq, F. Frame, N. J. Maitland, J. Farres and K. Pors, J. Med. Chem., 2022, 65, 3833–3848 CrossRef CAS PubMed.
- C. Anorma, J. Hedhli, T. E. Bearrood, N. W. Pino, S. H. Gardner, H. Inaba, P. Zhang, Y. Li, D. Feng, S. E. Dibrell, K. A. Kilian, L. W. Dobrucki, T. M. Fan and J. Chan, ACS Cent. Sci., 2018, 4, 1045–1055 CrossRef CAS PubMed.
- I. Chefetz, E. Grimley, K. Yang, L. Hong, E. V. Vinogradova, R. Suciu, I. Kovalenko, D. Karnak, C. A. Morgan, M. Chtcherbinine, C. Buchman, B. Huddle, S. Barraza, M. Morgan, K. A. Bernstein, E. Yoon, D. B. Lombard, A. Bild, G. Mehta, I. Romero, C. Y. Chiang, C. Landen, B. Cravatt, T. D. Hurley, S. D. Larsen and R. J. Buckanovich, Cell Rep., 2019, 26, 3061–3075 Search PubMed.
- D. Zhao, Y. Mo, M.-T. Li, S.-W. Zou, Z.-L. Cheng, Y.-P. Sun, Y. Xiong, K.-L. Guan and Q.-Y. Lei, J. Clin. Invest., 2014, 124, 5453–5465 Search PubMed.
- P. Kumari, S. Ghosh, S. Acharya, P. Mitra, S. Roy, S. Ghosh, M. Maji, S. Singh and A. Mukherjee, J. Med. Chem., 2023, 66, 14061–14079 CrossRef CAS PubMed.
- P. Ordonez-Moran, C. Dafflon, M. Imajo, E. Nishida and J. Huelsken, Cancer Cell, 2015, 28, 815–829 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |