
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCombatting resistance: natural products as tools to drive the discovery of untapped antibiotic targets
Makayla R.
Hedges
 ,
Camden M.
Di Carlo
and
William M.
Wuest
*
,
Camden M.
Di Carlo
and
William M.
Wuest
*
Department of Chemistry, Emory University, 1515 Dickey Drive, Atlanta, GA 30322, USA. E-mail: wwuest@emory.edu
First published on 15th August 2025
Abstract
Natural products have served as a fruitful starting point for antibiotic drug development. Evolution has served both as a catalyst to optimize their structures and also as a hinderance to render them ineffective through a variety of resistance mechanisms. To combat this, there has been a significant effort to discover new antibiotics with non-conventional mechanisms of action. Toward this end, researchers have continued to leverage natural products as a key source of novel scaffolds in an effort to discover new biological targets. In this review, we provide an overview of the conventional mechanisms of action and new advances in antibiotic natural product discovery. We detail recent successes of diverted total synthesis strategies in a two-pronged approach: (1) developing potent antibiotics that overcome bacterial resistance and (2) discovering new chemical probes for biological investigation. We close by discussing affinity-based protein profiling, a chemical proteomics method that can be used in concert with resistance selection approaches to overcome some inherent limitations.
 Makayla R. Hedges | Makayla Hedges is currently pursuing her PhD at Emory University under Prof. William M. Wuest. Prior to this, Makayla attended the University of Tennessee – Knoxville where she obtained her BS in chemistry in 2024. While there, her research focused on developing isotopically labelled chemical tools for uncovering novel roles of the essential five carbon precursors of the isoprenoid pathway, under the advisement of Prof. Joshua A. Baccile. She now works on the synthesis and application of chemical probes for the discovery of novel drug targets by proteomics. |
 Camden M. Di Carlo | Camden Di Carlo is a rising second-year PhD student in Prof. William M. Wuest's lab at Emory University. Previously, he attended Colgate University, where he obtained a BA in biochemistry. Camden's undergraduate research primarily focused on the total synthesis of a Tn antigen mimic under the guidance of Dr. Ernest G. Nolen. At Emory, his current research centers around the total synthesis of complex natural products featuring antibiotic activity. |
 William M. Wuest | William M. Wuest obtained his BS degree in chemistry/business from the University of Notre Dame (Paul Helquist) in 2003 and a PhD in chemistry from the University of Pennsylvania (Amos B. Smith III) in 2008. After an NIH NRSA Postdoctoral Fellowship with Christopher T. Walsh at Harvard Medical School, he joined the department of chemistry at Temple University in 2011 before moving to Emory University in 2017. He is currently a Georgia Research Alliance Distinguished Investigator and Professor of Chemistry, and his group's research focuses on natural product-inspired approaches toward disinfectant and antibiotic development. |
1. Introduction
Antimicrobial resistance has threatened our first line treatments since the development of the first antibiotic, penicillin, in 1928.1,2 Nevertheless, the development of new antibiotics has continued, as they are vital to the healthcare industry. In 2022, seven of ten patients in outpatient settings received an antibiotic prescription; however, at least 28% of these were deemed to be unnecessary.3 The improper and over usage of antibiotics has led to a steep increase in antibiotic resistant pathogens that cannot be treated with standard medications.1–3 Thus, there is an urgent need for the development of antibiotics with novel mechanisms of action (MoAs).Bacteria utilize many mechanisms to develop resistance, which have been generally characterized into the following four categories: drug efflux, drug inactivation, limiting drug intake, and drug target modification. For additional details regarding these categories, refer to Reygaert's work in 2018, where they were outlined extensively.4 In the context of this review, we primarily focus on the consequences of the constant evolutionary changes occurring in bacteria, specifically the protein binding partners that disable drug activity. We will discuss the traditional methods used to combat common pathogens and how resistance to these methods has developed, with case studies from FDA-approved drugs. From there, we aim to give context to the efforts of the scientific community to address this healthcare emergency, specifically through efforts in academia where we share some of our stories alongside those of our peers and collaborators.
Natural products have served as an inspiration for new solutions to this growing issue by providing starting points that have been specifically evolved to kill bacteria and likely would not have been developed through rational design. Beginning at isolation, small molecules often undergo preliminary testing to determine antimicrobial activity and cytotoxicity. From there, after extensive structural analysis, compounds are either derivatized or constructed de novo via total synthesis to enable an efficient and diversifiable method to construct these small molecules and analogs thereof. In the most ideal of cases, these efforts result in a new drug candidate that is commercialized by the pharmaceutical industry. However, limitations often apply, ranging from a lack of market positioning to exceedingly demanding synthetic requirements that render translation to medicine impractical.5 The history of vancomycin, and the abundant effort put towards improving its total synthesis over 63 years, demonstrates this point well.6–8 Hence, simplified analogs are often targeted instead of the specific natural product scaffold itself.
Historically, MoA studies of these novel antibiotic scaffolds have involved resistance selection assays and genetic sequencing. However, polypharmacology or unique MoAs can confound these efforts and require novel approaches to target identification; one example is affinity-based protein profiling (AfBPP), which we have successfully employed several times in collaboration with the Sieber lab.9–12 Herein, we outline a few examples of how proteomic and proteomic-like workflows have enabled the field to identify potential drug targets as well as characterize their MoAs.
2. Conventional mechanisms of action
We and others involved in antibiotic development have focused on six primary hospital-acquired pathogens. Termed the ESKAPE pathogens, they include Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species, and are well-known for multi-drug resistance (MDR) strain development.13 Over the last century the scientific community has worked toward a comprehensive understanding of antibiotics and their corresponding MoAs (Fig. 1). This has provided a classification system and standardized methods for initializing mechanism elucidation. Through the lens of FDA-approved antibiotics, we begin this review with an outline of conventional MoAs of antibiotics and examples of how resistance can develop.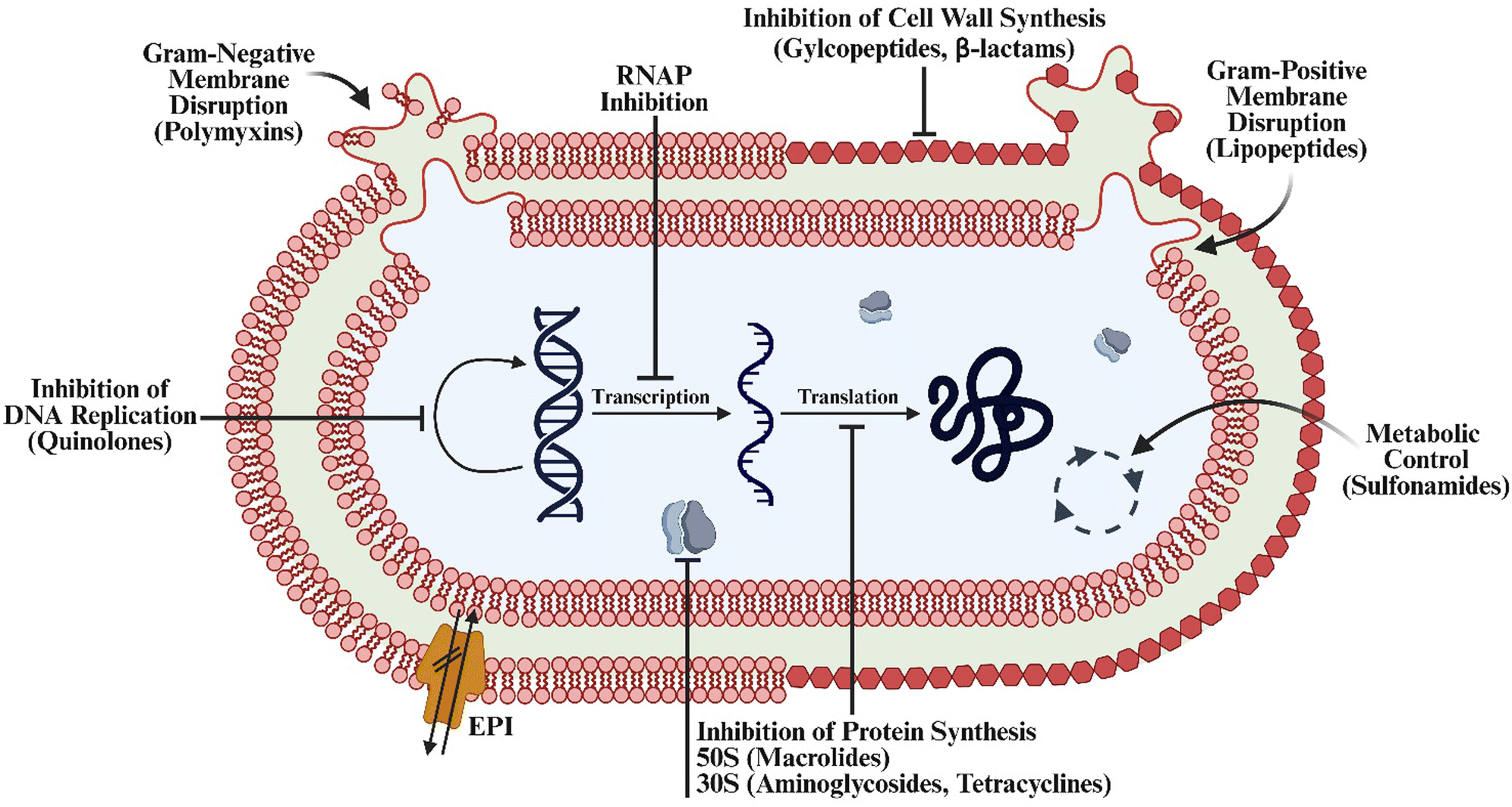 | ||
| Fig. 1 Overview of the classical antibiotic mechanisms of action. Created in BioRender. Lab, W. (2025) https://BioRender.com/uvsd811. | ||
2.1. Inhibition of cell wall synthesis: penicillins
Commonly existing right under and in our noses, S. aureus is a Gram-positive bacterium that can result in severe infections typically affecting those with weakened immune systems.13 The primary class of antibiotics used to treat this infection are β-lactams, examples including varying synthetic penicillins. Mechanistic elucidation of the penicillins was discovered in 1965 by Tipper and Strominger, where they distinguished β-lactams as bactericidal (cell death) agents which target Gram-positive bacteria effectively through the inhibition of enzymes vital in the formation of the peptidoglycan cell wall.14 In the presence of penicillin, the terminal transpeptidation reaction involving the peptide cross-linking of N-acetylglucosamine and N-acetylmuramic acid disaccharides is halted, aligning with the reported presence of uncross-linked disaccharide-peptide with a free C-terminus of D-alanine (D-ala).15 Tipper and Strominger also noted synthetic penicillin is structurally related to D-ala-D-ala.14,16 Penicillins irreversibly react with serine residues on enzymes including DD-transpeptidase and DD-carboxypeptidase, which are vital in the acylation of the peptidoglycan wall.16 Despite this, what was mainly known at the time of discovery as S. aureus, is now referred to as methicillin-susceptible S. aureus (MSSA), as the first reported case of methicillin-resistant S. aureus (MRSA) began its rise in 1961.17Resistance mechanisms can develop in many ways, typically dependent on the antibiotic MoA and the evolutionary pressures to the bacteria. Staphylococcus bacteria are known to be versatile in their evolutionary processes, developing resistance to a wide range of antibiotics. Specifically, related to penicillins, MSSA evolved to encode for both blaZ and mecA genes. blaZ encodes for the synthesis of β-lactamases which rapidly hydrolyze the β-lactam ring and mecA, encoding for PBP2a which is a modified penicillin binding partner (PBP) with reduced affinity for β-lactams. For more detailed explanations of these processes, see the work of Peacock and Paterson.18,19
2.2. Metabolic control: sulfonamides
Sulfonamides were once highly prescribed antibiotics for everyday infections like urinary tract infections; however, they have experienced a steep decline in prescription rates with both the development of more potent antibiotics and bacterial resistance mechanisms.20,21 Sulfonamides are broad-spectrum agents, active against both Gram-negative and Gram-positive bacteria including, but not limited to, E. coli and K. pneumoniae. Sulfonamides act as a competitive inhibitor for dihydropteroate synthase (DHPS), an enzyme required for the transformation of p-aminobenzoic acid into dehydroretinol acid, which is later converted into folic acid, also referred to as vitamin B9.22 As a result, these drugs are effective bacteriostatic agents, inhibiting growth while not directly killing bacteria.23,24The benefit of this drug class is that there is little effect on human hosts as folic acid can be obtained through diet and supplementation. However, any bacteria that synthesize folic acid de novo become susceptible to sulfa drugs.23,24 Bactrim (TMP-SMZ), the commonly prescribed sulfa drug, is a combination dose of trimethoprim and sulfamethoxazole. Together, they inhibit two steps of the folic acid biosynthetic pathway: sulfamethoxazole inhibiting DHPS, and trimethoprim blocking the latter conversion of dihydrofolate to tetrahydrofolate. Independently considered bacteriostatic, the synergistic mechanism which inhibits tetrahydrofolate effectively shuts down synthesis of purines, which are required for DNA and protein synthesis, making TMP-SMZ a bactericidal combination treatment.24
Like how MRSA encodes for modified enzymes with decreased affinity for competitive inhibitors, bacteria have developed resistance to sulfonamides through mutations in folP genes, which also encode for DHPS. Additionally, resistance has been identified in clinical isolates of Gram-negative species such as E. coli, A. baumannii, and K. pneumoniae; the sul (1–4) genes, which encode for sulfa-resistant DHPS, have been identified to provide plasmid-borne resistance—also known as horizontal gene transfer.25 This process involves the transfer of resistance genes through plasmids or small, circular DNA fragments to another species, expediting mutations and replication independent of the bacterial chromosome.26 For more details on this process, see Savchenko's work on plasmid-borne resistance.25
2.3. Membrane disruption: polymyxins and lipopeptides
Bacteria are classified into two primary categories, Gram-negative and Gram-positive, differentiated by the presence of an additional lipopolysaccharide (LPS) outer membrane in Gram-negative bacteria. Gram-positive and -negative bacteria both have a phospholipid-comprised cell membrane. Gram-positive bacteria solely have a thick peptidoglycan layer (30–100 nm).27Polymyxins A–E are a class of polypeptide antibiotics where B and E are the only clinically applicable of the family. Polymyxin E, more commonly known as colistin, is a leading treatment for Enterobacter species and other MDR pathogens like A. baumannii and P. aeruginosa.28 Yang and coworkers outlined the mechanism of colistin and the development of polymyxin resistance extensively in 2019.29 For context, polymyxins bind to the outer LPS through electrostatic interactions, destabilizing the membrane and allowing for the displacement of ions (Ca2+, Mg2+) and the formation of pores. This increases membrane permeability for other antibiotics to penetrate Gram-negative bacteria while simultaneously causing cellular leakage of essential components. It is important to note that the inner and outer LPS are not composed of the same sugars; polymyxins solely bind to the negatively charged lipid A component of the outer membrane, which is not housed in the inner membrane of Gram-positive bacteria.29,30
Daptomycin is the only FDA-approved and prescribed lipopeptide antibiotic which targets the integrity of the LPS membrane of Gram-positive bacteria. Interestingly, there are only a few FDA-approved usages for this drug, with other applications considered off-label. Daptomycin is approved for aggressive skin infections often caused by MSSA or MRSA and blood stream infections caused by any S. aureus strains. The MoA of daptomycin is not yet fully understood, though it is known to inhibit cell wall synthesis and cause membrane disruption.31 However, in 2020, Schneider and coworkers identified that daptomycin binds to bilayers at the septum of S. aureus, which contains lipid precursors of the undecaprenyl-coupled cell envelope and anionic phospholipid phosphatidylglycerol. This allows for the formation of a tripartite complex. In the presence of Ca2+, this complex oligomerizes and causes division of the septum, thus inhibiting cell wall synthesis through control of the biosynthetic machinery. Additionally, after prolonged exposure and cell wall discretion, daptomycin can disrupt the membrane, leading to pore formation and vital cell component leakage.32
2.4. Inhibition of protein synthesis via ribosomal subunits: aminoglycosides
The termination of protein translation operates by the targeting the 30S and 50S subunits of the ribosome. Ribosomes are vital in the translation of new proteins. Beginning with decoding at the 30S subunit, accurate base pairing of the mRNA codons and tRNA anticodons is performed. The 50S subunit then catalyses peptide bond formation.33,34 Aminoglycosides bind to the 30S subunit inhibiting interactions between the subunit and the mRNA strand. Tetracycline acts similarly by targeting the A site (residues A1492 and A1493) of the 30S subunit, inhibiting the binding of the amino acyl-tRNA and therefore halting the growing amino acid polypeptide chain. Macrolides bind to the peptidyl transferase center of the 50S subunit, leading to inhibition of the final peptide bond formation. Chloramphenicol acts like the tetracyclines but on the A site of the 50S subunit, whereas linezolid acts similarity to aminoglycosides by disruption of interactions with the 50S subunit.35Despite this wide variety of FDA-approved antibiotics, resistance against ribosomal-unit-targeting small molecules has increased significantly. This is notably due to drug efflux and active site modification (vide infra). However, ribosomal RNA methylation is the most significant and tragically compelling mechanism for how bacteria combat these antibiotics. S-Adenosyl-L-methionine (SAM)-dependent methyl transferases are enzymes responsible for this process, contributing to the methylation of intracellular substrates—including residues vital for antibiotic activity. For more details, see our recent review.36
2.5. RNA polymerase inhibition: rifampin
The inhibition of RNA polymerase (RNAP) has become a leading MoA in targeting tuberculosis (TB) bacteria. Though not an ESKAPE pathogen, there are few antibiotics as effective as the RNAP-inhibitor classes: rifamycins, sorangicin, streptolydigin, and myxopyronin. The mechanism and structural analysis of these inhibitors and binding complexes have been detailed by Ebright and coworkers.37 Briefly put, rifamycins and sorangicins act similarly through active site binding and inhibition, while streptolydigin interacts with a proximal target that leads to steric bulk surrounding the RNAP active site. This interaction inhibits conformational cycling of the RNAP active site that is required for transcription. Myxopyronin similarly can interact with a proximal target but instead operates by interfering with opening of the RNAP cleft. This leads to the unwinding of DNA by interfering with vital interactions between RNAP and the DNA template strand.37Despite the effectiveness of drugs like rifamycin, the high frequency of resistance development has limited its usage, causing many in the health care industry to reserve its use to TB exclusively.38 Rifampin (RIF) is a semisynthetic derivative and the common TB medication, however, in 1998, Musser and Ramaswamy reported resistance mechanisms. RIF specifically binds to the β-subunit of the RNAP. Therefore, the rpoB gene of TB bacteria can modify this subunit through point mutations to survive in the presence of RNAP, justifying the conservation of this drug for TB treatment.39
2.6. Inhibition of DNA replication: quinolones
E. faecalis and E. faecium, of the enterococcal family, are high priority ESKAPE pathogens. Considered to be highly resistant to aminoglycosides and TMP-SMZ, fluoroquinolones are still a common antibiotic treatment, but resistance is reported to be on the rise. Ciprofloxacin (Cipro), the most prescribed fluoroquinolone, has experienced growing resistance and is only considered effective in Gram-negative bacteria. This, however, does include another ESKAPE pathogen, Enterobacteriaceae.40 Alternatively, levofloxacin still retains broader activity. Originating from the racemic mixture of ofloxacin, this enantiopure S-isomer shows advanced potency; nevertheless, it is still seeing drops in prescription rates.41 Fluoroquinolones inhibit DNA replication through the binding of DNA topoisomerase and DNA-gyrase, but mutations in the latter—as well as plasmid-borne resistance mechanisms—have rendered them less effective.42 Additionally, through the encoding of efflux pumps, fluoroquinolones and many other antibiotics have experienced a significant drop in potency.402.7. The role of efflux pumps in antibiotic resistance
Efflux pumps play an essential role in resistance for many MDR bacteria, however their existence as a vital cellular component of bacteria predates the usage of antibiotics. This points to alternative roles of these proteins, unlocking potentially new antibiotic targets. Acting as a trash chute for the bacteria, efflux pumps can extrude stress-inducers such as bile, toxins, metals, and by-products through a turn-over process.43 It should be noted that this mechanism of bacteria is vital for humans in terms of a healthy gut microbiome, adding additional complexity to drug development.44 Put simply, antibiotics are affected by efflux pumps by being excreted after recognition as a toxin. This leads to a low intracellular concentration of the antibiotic, making them ineffective and often at risk for increasing resistance via low concentration exposure. Recently covered extensively by Joshi and co-workers, there are many types of efflux pumps; however, understanding the specifics of each has led to new approaches for the development of antibiotic therapies, allowing for the restoration of antibiotic susceptibility.45 The development of efflux pump inhibitors (EPIs) seeks to restore the activities of FDA-approved drugs.46 Though there are currently no EPIs approved for clinical use, many FDA-approved drugs are predicted to act as EPIs, although their exact mechanism has just yet to be elucidated. Exemplified in 2022 by the Abbas lab, diclofenac sodium, glyceryl trinitrate, and domperidone were determined to have efflux pump inhibitory activities against S. aureus, and were characterized through efficient efflux assays, antibiotic minimum inhibitory concentrations (MICs), and relative gene expression analysis.47 Further mechanistic understanding of such polypharmacological applications opens the door to potential drug repurposing and optimization, exemplified by work in our lab and many others.3. Discovery of antibiotic scaffolds
The accidental discovery of penicillin has become a household tale, creating the idea that scientists simply stumble upon natural products (NPs). The reality is that the isolation and characterization of penicillin took more than a decade after first observing mold-killing bacteria.46 At this time, one can imagine the complexity of NP isolation. Therefore, some turned towards manufactured compounds, shown by the discovery of sulfonamides during investigations into azo dyes.48,49 Also fully synthetic antibiotics, fluoroquinolones are all derived from nalidixic acid, a by-product of chloroquine, a market antimalarial drug.42Nevertheless, NPs continued to prove themselves a fruitful source of antibiotic candidates. Successes include the discovery of tetracyclines from fungus and many aminoglycosides from soil bacteria. Even so, frequent discoveries were stunted by the time it took to ferment, extract, and purify NPs, many of which proved to be inactive. Hence, optimization of techniques to identify and test NPs has persisted throughout the last few decades.48
3.1. Intentional high throughput screening: qinghausu and darobactins
To efficiently identify novel scaffolds, screening of likely candidates for antibacterial compounds was undertaken by many. Tu YouYou (2015 Nobel Prize in Physiology or Medicine) spearheaded the screening of traditional Chinese medicines, knowing isolation of the active compound would lead to significant potency. After testing 2000 herbal remedies and sifting through 600 possible hits, qinghausu was isolated from Artemisia annua. This fast-acting antimalarial drug is now known as artemisinin.50,51 Through proteasome inhibition, protein damage, and production of reactive oxygen species (ROS), this drug has been the primary contributor to countries nearing malaria-free status.52,53 Screens like this have only continued to optimize. Prof. Rolf Müller, responsible for the isolation of carolacton after screening antibiotics from myxobacteria,54 has provided impactful work in the development of many antibiotic candidates. Notable also is the work on darobactins, which were first isolated by Prof. Kim Lewis through a screen of Photorhabdus isolates.55–57 Darobactins are a promising class of antibiotic NPs that inhibit BamA, which is essential in the insertion and folding of outer-membrane proteins in Gram-negative bacteria.57 Despite these advances, limitations still persist, such as an inability to isolate from environments not matched by standard laboratory conditions.3.2. iChip: teixobactin
The isolation of NPs from the rhizosphere has proven to be challenging due to its complex environment and specific growth conditions. For promysalin (vide infra), it required extensive insight into Gac regulatory system control for isolation by Prof. René De Mot.58 Epstein, however, in 2010, reported a new strategy for optimized isolation of such microbial species which cannot be grown in standard media conditions, coined isolation chip (iChip). Beginning at sample collection, serial dilution followed by incubation allows for isolation of sole microbes. Then placed independently into a single well of the iChip plate, they are provided with the appropriate nutrients and growth factors from their original environment to allow for independent growth. Preceding traditional isolation and purification, polymerase chain reaction (PCR) can be utilized for identification (Fig. 2, iChip).59,60 In 2015, Lewis leveraged this technology to discover the teixobactin class of antibiotics, which shows promising activity in S. aureus through inhibition of peptidoglycan biosynthesis.61 Unfortunately, the complexity of natural product isolation still requires alternative, complementary approaches to identify the “optimal antibiotic”. | ||
| Fig. 2 Overview of the iChip technology workflow. Created in BioRender. Lab, W. (2025) https://BioRender.com/5lofze4. | ||
4. Diverted total synthesis to access new chemical probes
Natural products (NPs) have consistently proven to be important scaffolds for drug discovery,62 with NP derivatives constituting over 20% of all new drug approvals from 1981 to 2014.63 However, the structural complexity of NPs has historically precluded thorough investigations of their structure–activity relationship (SAR) and MoAs, limiting discovery efforts. In a seminal review,64 the Danishefsky group demonstrated the power of organic synthesis in solving this problem, showcasing their success with an approach they coined diverted total synthesis (DTS). The beauty of DTS is in the design of a synthetic route that allows access to a library of hypothesis-driven NP analogs that cannot be accessed through either semi-synthesis or derivatization of the NP itself to answer specific biological questions. As Danishefsky showed, these analogs can provide helpful SAR data for the NP scaffold, allowing for the discovery of compounds with even greater potency. DTS has since gained traction in the broader synthetic community, with many examples covered in other reviews.65–67While Danishefsky's original SAR through DTS approach has clearly been effective for drug discovery, many chemists have looked to leverage it further. Indeed, there are several examples of employing DTS to make chemical probes for investigating various biological systems, as previously reviewed by Carreira.68 Some have looked to improve upon the DTS approach altogether, developing new strategies for MoA determination and drug development. For example, Romo's phamacophore-directed retrosynthesis (PDR) looks to increase efficiency by intentionally incorporating SAR opportunities throughout a synthesis. In designing the retrosynthesis around the likely pharmacophores of a NP, one can discover a less complex, but equipotent, compound earlier in the synthesis effort.69
Inspired by these promising outcomes, chemists have begun applying DTS towards combatting antimicrobial resistance (AMR). This approach has already yielded fruit, exemplified by Myers' success with the tetracyclines70 and others’ work towards arylomycin optimization.71–73 In the case of the arylomycins, this drug development effort (Phase 3 clinical trials) resulted in the discovery of a new and promising antibiotic target—the essential bacterial type I signal peptidase (SPase I)—through a novel MoA.73 Additionally, recent advances have resulted in both new antibiotics that overcome known resistance mechanisms as well as an arsenal of tool compounds that have enabled the elucidation of novel MoAs. Here, we provide an overview of some of these recent successes from our lab and others in the synthetic community.
4.1. Lincosamides
Despite the widespread success and popularity of ribosome-targeting antibiotics, bacteria have been quickly gaining resistance to this MoA, rendering crucial medicines increasingly ineffective.36 One example is the antibiotic clindamycin (Fig. 3(a)), which was derived semi-synthetically from the NP lincomycin and found to have significantly increased activity and beneficial pharmacokinetics.74 Bacteria have gained resistance to clindamycin, most often via dimethylation of the 23S ribosomal RNA subunit.75 To overcome resistance, chemists have looked to modify both the aminooctose and aminoacyl residues of clindamycin to improve upon its activity and applicability.76–88 However, most of these modifications have been realized semi-synthetically, limiting the scope of SAR studies. Seeing this gap, Myers’ group was inspired to develop a total synthesis of methylthiolincosamine 3,89 allowing future access to clindamycin analogs through DTS. | ||
| Fig. 3 (a) Semi-synthesis of clindamycin from lincomycin. (b) Myers’ retrosynthesis of lincosamide antibiotics. (c) Myers’ successful forward synthesis of novel lincosamines. | ||
Their retrosynthesis traced 3 back to glycal 4, which would be accessed via a cyclization of epoxide 5, which originates from epoxyaldehyde 6 (Fig. 3(b)). In the forward sense, TIPS protected acetylene 7 was converted to 6 with high enantiomeric excess, whereas the benzyl ether 10 was obtained in 4 steps from nitromethane 8 and acetaldehyde 9 with exquisite enantiopurity (Fig. 3(c)). A highly diastereoselective nitro-aldol reaction, followed by a serendipitous nitrone cyclization, allowed for the elaboration of 11 into isoxazolidine 12. Subsequent cyclization and epoxidation furnished glycal epoxide 13, which served as a diversification point for access to 3 as well as other aminooctose derivatives 15–17. Subsequent modifications to the aminooctoses, followed by coupling of the amines with known aminoacyl fragments produced several lincosamides (not shown), some of which had improved bioactivities over 1 and 2.89
Leveraging this new route to 3, the Myers group then synthesized hundreds of additional clindamycin analogs via DTS,80,90,91 discovering bicyclic aminoacyl scaffolds with promising activity, such as 18 (Fig. 4(a)).92 Subsequent SAR of this scaffold yielded iboxamycin (19), an antibiotic candidate they deemed worthy of further investigation, prompting the development of a gram-scale synthesis.93 Microbiological testing revealed that iboxamycin is a potent antibiotic, showing dramatically improved activity against both gram-positive and gram-negative strains. More importantly, iboxamycin retains its activity against clinically isolated strains, overcoming a variety of resistance mechanisms that have rendered clindamycin ineffective.94 Furthermore, iboxamycin did not show any major safety concerns as it is non-haemolytic and non-toxic to mammalian cells, nor does it impact membrane integrity or mitochondrial function. Subsequent mouse infection models utilizing highly resistant bacterial strains proved the clinical potential of iboxamycin, as it successfully reduced bacterial loads and rescued mice from lethal challenges.92
 | ||
| Fig. 4 (a) Development of iboxamycin via SAR analysis. (b) Selected biological activity of iboxamycin against resistant bacterial strains. | ||
Seeing the impressive activity of iboxamycin, the authors proceeded to investigate its MoA. Initial assays determined that iboxamycin is bacteriostatic and that both standard and clinical bacterial strains were very slow to develop resistance. However, a strain designed for ribosomal RNA (rRNA) mutations95 produced resistant clones with a frequency of 10−8. Sequencing these clones revealed a common A2058G or A2059G mutation within the 23S rRNA, causing a known base change present in clindamycin-resistant bacteria that occurs in the lincosamide binding site.94 Since mammalian ribosomes contain G2058 residues,96 iboxamycin selectively targets bacterial ribosomes, providing a rationale for the observed selectivity experimentally. The hypothesis that iboxamycin binds to the canonical lincosamide binding pocket was verified by an X-ray crystal structure of the antibiotic bound to the 70S ribosome of Thermus thermophilus. Finally, an additional crystal structure of iboxamycin bound to an A2058-containing ribosome was obtained, revealing how it overcomes clindamycin resistance. New hydrophobic interactions established by the unique aminoacyl side chain provide additional stability to iboxamycin's binding conformation, overcoming the methylation-mediated loss of key hydrogen bonding interactions with the aminooctose moiety that destabilize clindamycin binding.
Motivated by their success with iboxamycin, the Myers group continued their work towards developing yet more potent lincosamide antibiotics. Having already optimized the aminoacyl component with a bicyclic oxepanoprolinamide, they next looked to the aminooctose moiety. Interestingly, further examination of the crystal structures of clindamycin97–100 and iboxamycin92 bound to ribosomes revealed conformational homogeneity within the northern fragment. This observation inspired the rational design of a rigidified analog dubbed cresomycin, which employs a homoallylic sulfide linker that mimics the bound conformation of the lincosamides (Fig. 5(a)).101 Synthesis of cresomycin proceeded with the thiogalactoside 21, which was quickly elaborated to the Ellman sulfinimine 22 (Fig. 5(b)). An asymmetric crotylation efficiently set both the methyl and amino stereocenters, allowing for a ring-closing metathesis (RCM), deprotection, and amide coupling sequence to yield 20 in 16% overall yield from 21.
 | ||
| Fig. 5 (a) Design of cresomycin. (b) Forward synthesis of cresomycin from a protected thiogalactoside. (c) Selected biological activity of cresomycin compared to iboxamycin against resistant bacterial strains. | ||
Subsequent biological evaluation identified cresomycin as a broadly superior antibiotic to iboxamycin, showing drastic improvements against previously challenging strains (Fig. 5(c)).101 Similar to iboxamycin, cresomycin showed low cytotoxicity to mammalian cells and performed exceptionally in both mouse sepsis and infection models. A combination of density-functional theory (DFT) calculations, nuclear magnetic resonance (NMR) data, and single-crystal X-ray diffraction established that cresomycin resides in a single, minimum-energy conformation. Finally, crystal structures of cresomycin bound to methylated bacterial ribosomes revealed that both the antibiotic and the binding pocket exhibit slight conformational changes to accommodate their preferential binding interactions.
In sum, Myers’ work elegantly demonstrates the advantages of DTS in antibiotic discovery. Through total synthesis, hundreds of novel antibiotic scaffolds—otherwise inaccessible via semi-synthesis—were explored, resulting in the development of iboxamycin. Complementary biological investigation then inspired the rational design of cresomycin, the most potent lincosamide known to date. Thus, Myers has provided a successful blueprint for combatting AMR through DTS, further encouraging future applications of this approach.
4.2. Streptogramins
The streptogramins are a class of antibiotics that consists of two subgroups: 23-membered polyketide macrocycles (group A) and 19-membered macrocyclic depsipeptides (group B) (Fig. 6(a)).102 Historically, this class of antibiotics has been largely unexplored and limited by semi-synthetic approaches, which have resulted in a single combination therapy approved by the FDA in 1999,103 although one other combination made it to phase-II clinical trials in 2011.104,105 While several groups have completed total syntheses of group A streptogramins—virginiamycin M2106–108 and madumycin II109,110 among others111–114—the routes thus far have generally been cumbersome and low-yielding. Seeing this gap in the literature and an opportunity for drug discovery, Li and Seiple designed a highly convergent total synthesis of four group A streptogramins for future DTS and SAR studies.103 | ||
| Fig. 6 (a) The two classes of streptogramin scaffolds. (b) Seiple's total synthesis of group A streptogramins. | ||
Their synthesis begins with a highly enantio- and diastereoselective Mukaiyama aldol addition between silyl ether 23 and isobutyraldehyde 24 (Fig. 6(b)). Subsequent aminolysis and stannylation of a terminal alkyne gave alcohol 25 in high yield, which served as a key common intermediate for esters 26–28. For the fragment composing the second half of the NPs, bromoaldehyde 29 and thiazolidinethione 30 were elaborated into chiral oxazole 31via asymmetric aldol addition, silyl protection, and auxiliary substitution. A peptide coupling between 26 and 31, followed by an intramolecular Stille cross-coupling and silyl deprotection afforded madumycin I, which was easily converted to madumycin II via organoboron-directed reduction with sodium borohydride. The same sequence, employing amine coupling partners 27 and 28, allowed for the successful synthesis of virginiamycins M1 and M2, respectively; in the case of 34, an extra oxidation step was required to give the enamine functionality. In addition to the high overall yields and convergency of the reported route, another key aspect was that nearly every step in the synthesis could be performed on a multi-gram scale. Thus, Li and Seiple were the first to report a total synthesis of both virginiamycin M1 and madumycin I, as well as providing efficient routes amenable to DTS for those and two other streptogramin NPs.103
With a scalable route established, Seiple's group next looked to the design and synthesis of streptogramin analogs. They decided to begin with the scaffold of 35, first obtaining a cryo-electron microscopy (cryo-EM) structure of the NP bound to the E. coli 50S ribosome.115 Analysis of the structure suggested that C3 and C4 modifications would be tolerated, as both the methyl and isopropyl groups do not make significant binding interactions, instead projecting into pockets of empty space. Additionally, it was previously reported that these regions make crucial interactions with the active site of VatA, a protein that deactivates the streptogramins through C14 alcohol acetylation;116 hence, altering these groups may prevent a major avenue for generating resistance. Prior to Seiple's work, only one example of an analog with a modification at these positions was reported, demonstrating the dearth in SAR data for this region of the NP.117 Now with a guiding hypothesis, Seiple's group proceeded towards the DTS of 35 to produce novel analogs.
Optimizing their previous route103 in the process, nearly 60 streptogramin analogs were synthesized, featuring a variety of modifications decorating the majority of the macrocyclic scaffold.112 While many analogs showed poor biological activity, several stood out, such as 36 which features an allyl group at C4 instead of a methyl (Fig. 7(a)). This analog had improved MIC values against several resistant strains and even overcame resistance in a couple of strains compared with 35. However, it was still not as potent as the clinical candidate flopristin (37). Noting that flopristin's improved activity was a result of C16 fluorination,104 the authors designed and synthesized the analog 38. Promisingly, 38 showed broadly improved activity compared with 37, exhibiting MICs 8-fold lower in certain strains (Fig. 7(b)). While the streptogramins are usually dosed as a combination of group A and group B compounds, modifications to one of the components is not always tolerated by the other.118 Hence, the authors looked at the activity of 37 and 38 with the group B NP virginiamycin S1 (Fig. 7(c)).115 The combination of 38 + 39 proved to be superior to 37 + 39, with several MICs below the limit of detection. Furthermore, 38 alone overcame high levels of resistance, demonstrating potent MICs against strains harboring several streptogramin-resistance genes (Fig. 7(b)).
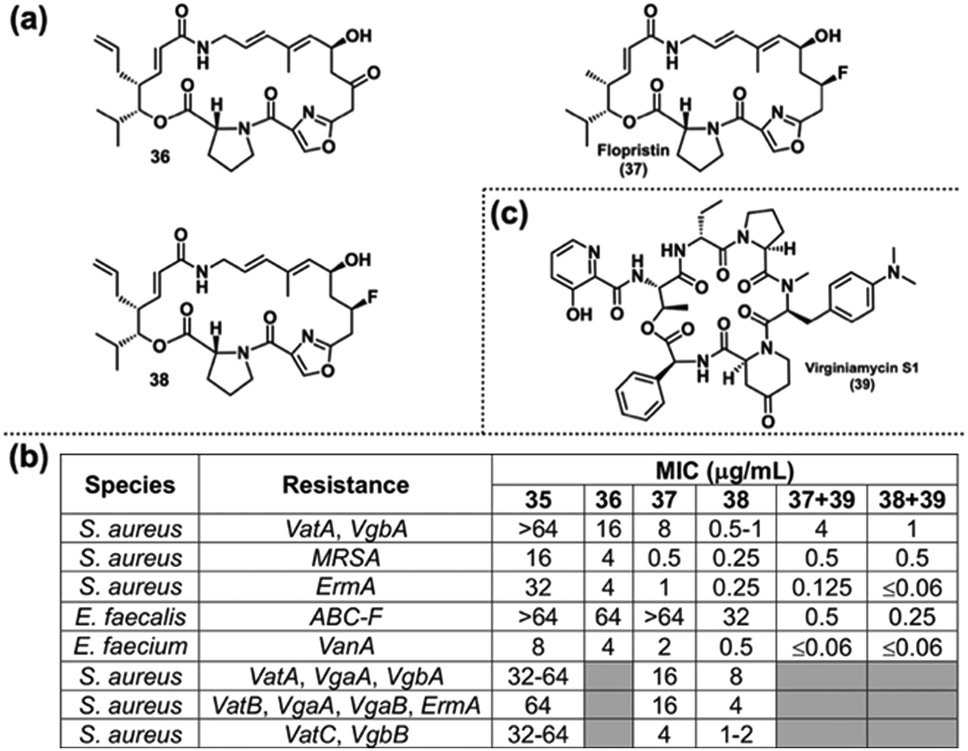 | ||
| Fig. 7 (a) Selected streptogramin analogs. (b) Selected biological activity of Seiple's streptogramin analogs against resistant bacterial strains. (c) Structure of virginiamycin S1. | ||
Following a successful mouse infection model employing 38 alone against a group A streptogramin-resistant S. aureus strain, the authors looked to elucidating the MoA of 38. Initial assays concluded that 37 and 38 have similar IC50 values in vitro (40 nM vs. 70 nM, respectively), whereas there was a 2.5-fold reduction in catalytic efficiency for VatA acetylation of 38 compared with 37.115 However, both differences do not linearly account for the extent of MIC reduction between the two compounds for all strains, prompting further investigation. An X-ray crystal structure of 38 bound to VatA supported the authors’ initial hypothesis, as the allyl substituent sterically clashed with the Leu110 residue of the enzyme. Finally, a cryo-EM structure of some analogs bound to the E. coli ribosome revealed that the allyl group of 38 projects into the streptogramin B binding site in a less strained conformation than when bound to VatA. The authors noted that this difference provides a potential explanation for the reduction in VatA acetylation rate of 38. The crystallography data obtained provide key structural information to guide the design of next generation streptogramin analogs. In preparation for this venture, Seiple has since reported a yet further-optimized synthesis of the streptogramin scaffold, replacing the Stille coupling with an RCM, which has provided higher overall yields.119
4.3. Carolacton
Initially discovered in 1998 from a myxobacterial extract, carolacton (Fig. 8(a)) was reisolated in 2010 and found to be a potent antibiotic against Streptococcus mutans,54 the primary etiological agent in dental caries.120,121 Interestingly, while S. mutans in biofilm suffered 35–66% cell death in the concentration range of 5–25 ng mL−1 of carolacton, planktonic cultures were virtually unaffected. Additionally, while several studies confirmed the disparate activity of carolacton in biofilm versus planktonic cultures of S. mutans, its MoA remained inconclusive.122–125 The combination of carolacton's structural complexity, species-specific antibiotic activity, and unknown MoA therefore prompted us to pursue its total synthesis in hopes of exploring its SAR.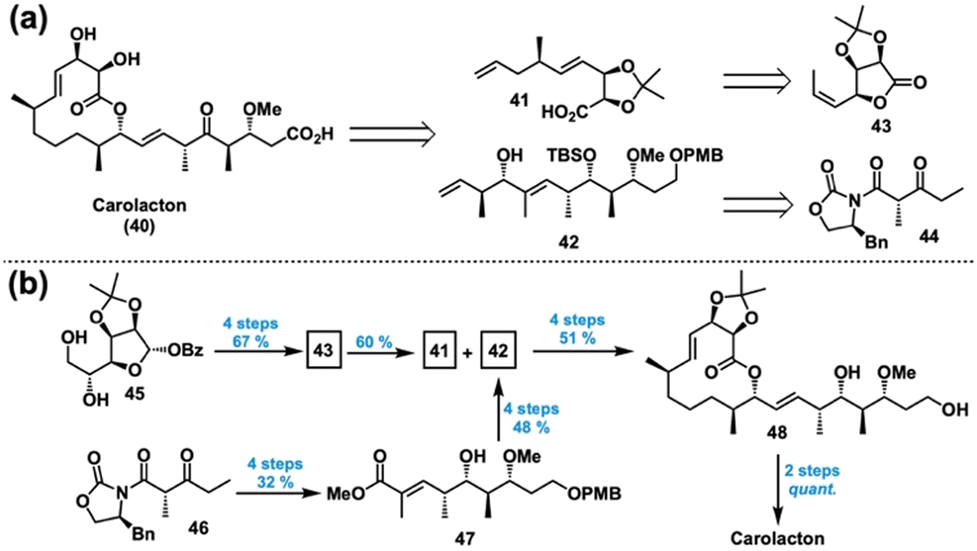 | ||
| Fig. 8 (a) Retrosynthesis of carolacton. (b) Successful forward synthesis of carolacton. | ||
Prior to our efforts, several groups had worked towards the synthesis of carolacton,126–129 with Kirschning reporting the first completed synthesis126 and Ghosh achieving a formal synthesis shortly thereafter.127 However, these previous syntheses were cumbersome in length, exemplified by Kirschning's 22 steps longest linear sequence (LLS). Hence, we looked to establish a shorter, modular synthesis that was more amenable to SAR studies. In 2014, in collaboration with the Phillips group at Yale, we reported a concise total synthesis of carolacton in 14 steps LLS and 7.8% overall yield as summarized in Fig. 8.130
We imagined that carolacton (40) could come from an esterification/RCM sequence between acid acetonide 41 and polyketide fragment 42 (Fig. 8(a)). These could trace back to lactone 43 and β-ketoimide 44, respectively. In the forward sense, transformation of the diol 45 into lactone 43 proceeded smoothly in 67% yield over 4 steps, allowing for a key SN2′ ring opening to access 41 in 60% yield (Fig. 8(b)). A series of Evans’ asymmetric aldol and redox manipulations afforded ester 47 in 4 steps and 32% yield from 46. From there, a protection, reduction/oxidation, and diastereoselective crotylation sequence accessed alcohol 42 in an additional 4 steps and 48% yield. Then, esterification of 41 and 42 was followed by an RCM, chemoselective hydrogenation, and deprotection to give lactone 48 in 51% yield over 4 steps. Lastly, oxidation of the diol to yield the ketoacid and subsequent acetonide hydrolysis yielded carolacton (40) in quantitative yield.
With our total synthesis completed, we looked to gather preliminary SAR data by testing the biological activity of advanced intermediate 48, acetonide-protected carolacton (structure not shown), and synthetic 40. We first tested the three compounds against planktonic S. mutans UA159, observing no inhibitory activity at concentrations lower than 250 μM. When incubated with S. mutans in biofilm-inducing media, carolacton (>500 nM) and 48 (>62.5 μM) significantly altered the integrity and morphology of the biofilm matrix compared to the DMSO control. However, acetonide-protected carolacton produced no observable effect at concentrations lower than 250 μM. This initial SAR data indicated that the carolacton scaffold may indeed be amenable to DTS, inspiring further investigation.
Shortly after publishing our total synthesis, Kirschning et al. reported the synthesis and biological activity of several carolacton analogs 49–54 as depicted in Fig. 9(a) and (b).131 When tested against S. mutans biofilm at 5.3 μM, compounds 49 and 50 inhibited biofilm to the same extent as carolacton but saw a dramatic decrease in activity at lower concentrations. Further investigation determined that 49 and 50 were acting as pro-drugs, as their inhibitory activity was proportional to the quantity of carolacton produced as a result of bacterial enzymatic hydrolysis. Notably, compound 51, which differed from the active 49 only in oxidation state of the C17 alcohol, showed significantly diminished biofilm inhibition at 5.3 μM. Additionally, compound 52 lost all activity, only differing from the native carolacton in stereochemistry at the C9 alcohol. Compound 53, which rearranges the allylic alcohol to produce a 14-membered lactone analog, also showed decreased activity. Therefore, the results from 51–53 suggest that major changes to the conformation and size of the 12-membered macrocycle are deleterious to biofilm inhibition. Lastly, lactone 54 was largely inactive, suggesting that either the ketone is necessary for activity or that bacterial enzymes were unable to hydrolyze 54 to its corresponding carboxylic acid to an appreciable extent.
 | ||
| Fig. 9 Kirschning's first reported active (a) and inactive (b) carolacton analogs. (c) Design of the Wuest lab's first DTS library of carolacton analogs and a summary of our major biological findings. | ||
With these results in mind, we wondered whether changes to the polyketide side chain of carolacton would be tolerated. Inspired by Chandrasekhar's design of truncated aryl pladienolide B analogs,132 we performed computational modelling and determined that a similar substitution was feasible.133 This led to the DTS proposal depicted in Fig. 9(c) (left), in which we would also probe macrocycle rigidification, saturation, and side chain oxidation state—all synthetic intermediates. With an optimized second-generation synthesis, we produced a set of 16 carolacton analogs (not shown) for biological evaluation.133 The major findings are described below as well as summarized in Fig. 9(c) (right).
At the outset, we serendipitously discovered that analog 55 inhibited biofilm formation with an MBIC50 of 63 μM without inhibiting planktonic growth, a phenotype not produced by carolacton. Interestingly, 56 was found to arrest growth at the microcolony stage as evidenced by a unique rosette phenotype visualized via confocal laser scanning microscopy. Since this phenotype was not observed for carolacton, it suggests that 56 affects biofilm growth of S. mutans by a different MoA than carolacton. Curious about our original bioisostere hypothesis, we then investigated the range of activity for 57. Excitingly, at concentrations as low as 500 nM, 57 displayed a phenotype consistent with that observed for carolacton, leading to the name “carylacton”. Notably, all analogs featuring a terminal alcohol were inactive. Shortly following our report, Kirschning published the results of a C9 lactam analog, which was found to be inactive.134
Encouraged by these results, we set out to employ our library of analogs to investigate biofilm mechanisms in hopes of gaining further insight into carolacton's MoA. Up to this point, it was known that ΔcomD,123 ΔpknB,125 and ΔcysR135 strains reduced susceptibility to carolacton; however, how those genes contributed to the biofilm response remained unknown. Additionally, carolacton had been found to inhibit folate dehydrogenase (FolD) in a TolC knockout of E. coli, but whether this interaction causes the biofilm phenotype in S. mutans was still in question.136 Despite these findings, there are still major limitations in elucidating carolacton's MoA from traditional microbiological mutant screening techniques due to its biofilm-dependent activity.137 As a result, we planned on using a chemical genetic approach instead, which has seen success in MoA identification for other natural products.138,139 Hence, an easily-accessed carolacton analog with quantifiable biofilm inhibitory activity would be required.
The biological activities of our analogs 55–57 prompted us to investigate whether a yet-more-simple analog would retain biofilm inhibitory properties and enable our desired chemical genetics approach. Toward this end, we again applied DTS to access three additional analogs featuring alkyl side chains and quickly identified (+)-58 as a promising tool compound for biological exploration (Fig. 10).137 Interestingly, (+)-58 inhibited S. mutans planktonic cells with an MIC of 250 μM, whereas carolacton and all of our other analogs showed no such activity. Under biofilm growth conditions, (+)-58 was capable of decreasing cell viability between 75% and 93% in the concentration range of 62 nM and 2 μM, whereas carolacton does so between 30% and 78% at the same concentrations, respectively. Therefore, the simplified analog (+)-58 has greater potency against S. mutans than the NP carolacton.
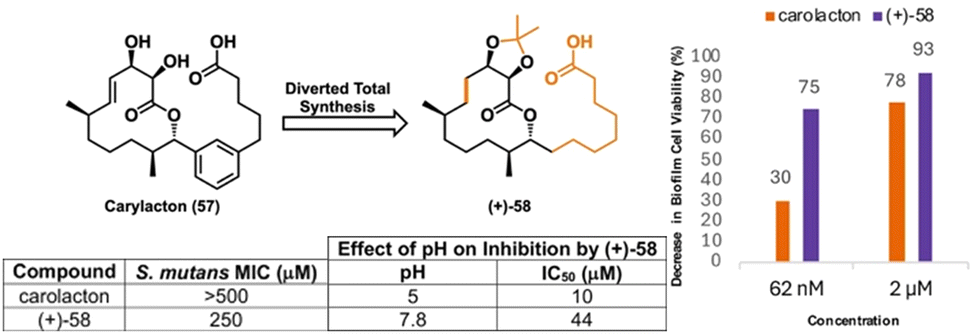 | ||
| Fig. 10 Analog (+)-58 and a summary of its biological activity. | ||
With the activity of (+)-58 verified, we next sought to determine its MoA. Since the activity of carolacton is known to be pH dependent,125 we measured the inhibition of pre-acidified planktonic cultures by (+)-58. Indeed, (+)-58 inhibited the planktonic cells with an IC50 of 10 μM at pH 5 as compared to its IC50 of 44 μM at pH 7.8, suggesting that (+)-58 acts via an acid-mediated mechanism and is not biofilm specific (Fig. 10). Due to the ability of S. mutans to survive in low pH environments via acid tolerance response (ATR) mechanisms,140,141 we suspected that (+)-58 targets the ATR pathway. With this hypothesis, we employed the S. mutans library from the Quivey lab142 to screen the susceptibility of ATR-associated knockout mutants to (+)-58, which was expedited by the unique inhibitory effect of the analog. From this, we identified two mutants with increased susceptibility, namely SMU_1276c (ΔezrA) and SMU_484 (ΔpknB); the latter was interesting to us as PknB was previously implied to be the target of carolacton.125 Nevertheless, a mutant deficient in the gene encoding carbon catabolite protein A (CcpA),142 which is associated with the ATR,143 was found to be significantly less susceptible to (+)-58.137 With this new information in hand, our lab was subsequently able to determine (+)-58's molecular target (vide infra).
Meanwhile, carolacton has continued to spark interest in the synthetic community. Goswami reported another total synthesis in 2017,144 while Kirschning evaluated several desmethyl analogs in 2023.145 Additionally, a new report of the lactone's antiviral activity146 has inspired yet more syntheses from Yu147 and Tang.148 Tang's work in particular enabled access to some of Kirschning's latest analogs. Hence, it appears that carolacton's full potential remains to be seen, motivating future work with the NP.
4.4. Promysalin
Isolated from the rhizosphere of rice plants, promysalin (59) is a metabolite with potent species-specific activity against P. aeruginosa with no bioactivity against gram-positive bacteria (Fig. 11, top left).58 Intrigued by its clinically relevant bioactivity149 and unknown MoA, we decided to pursue a total synthesis to allow for further biological investigation. While the isolation report determined the structure of promysalin, the authors did not make any stereochemical assignments nor reported an optical rotation.58 Looking to narrow this ambiguity, we started by revisiting the biosynthetic gene cluster, which hinted that the dehydroproline moiety would likely exist in the (S)-configuration.150 This analysis reduced the structural options to a set of four potential diastereomers, which we undertook synthesizing according to the representative scheme in Fig. 11 (bottom, right) for the (2R,8R) combination.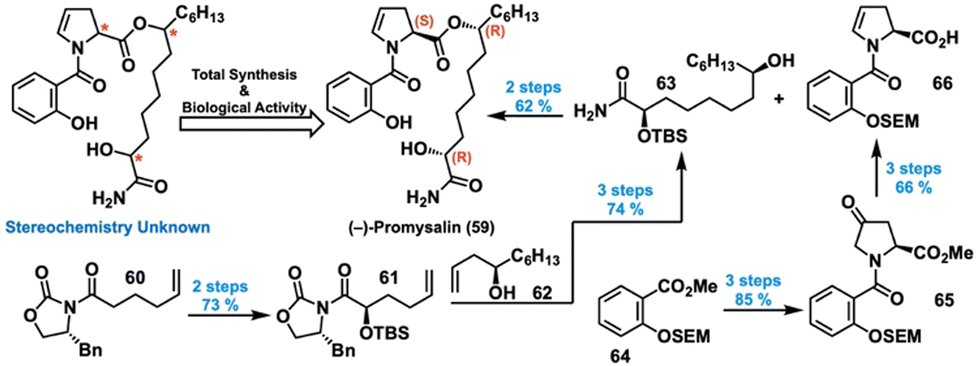 | ||
| Fig. 11 Total synthesis and structural elucidation of (−)-promysalin. | ||
For the first fragment, oxidation and protection of ketone 60 gave silyl ether 61, which was then coupled with allylic alcohol 62via olefin metathesis. Subsequent hydrogenation and ammonolysis gave alcohol 63. For the other fragment, methyl ester 64 was saponified and coupled with 4-hydroxyproline, yielding an amide alcohol that was oxidized to provide ketone 65 in high yield. Dehydration and saponification provided acid 66, which served as an esterification partner for 63. Global deprotection then afforded promysalin in 35% overall yield in eight steps, with mostly consistent yields for the other three diastereomers. We then compared the differences in NMR chemical shifts between our synthesized compounds and the literature-reported data, revealing (2R,8R,16S) as the correct relative configuration.150 Hypothesizing that only the correct enantiomer would reproduce the reported biological activity, we quantified the IC50 value for each diastereomer in the previously used PA14. Indeed, (–)-promysalin (Fig. 11, top middle) was the most potent with an IC50 of 125 nM, 10–60 times more effective than the other diastereomers. Hence, we established the absolute stereochemistry of 59 to be (2R,8R,16S).
Curious about its MoA, we decided to probe promysalin's secondary activity as a promoter of swarming and biofilm formation in other Pseudomonas species.58 Evaluating the possibility that promysalin acts as a biosurfactant to cause the swarming,151 we ran surface tension and biofilm dispersion assays, which suggested that it acts on a specific target instead.150 Furthermore, during these studies we serendipitously discovered that promysalin inhibits fluorescence in PP KT2440, implicating that it affects pyoverdine biosynthesis and/or transport.152,153 Since strains deficient in pyoverdine are known to show increased swarming,154 this pathway was now our new focus for mechanistic investigation.
We next looked to DTS to probe the moieties of promysalin's structure that are essential for its biological activity in hopes that the SAR would give hints about its MoA. Prior to analog synthesis, we were able to determine that promysalin is not an ester pro-drug, given its hydrolyzed fragments’ lack of bioactivity.155 Additionally, a preliminary set of carboxylic acid and secondary amide analogs indicated that the primary amide motif was necessary for activity.156 With this information in hand, we then synthesized a diverse set of 16 analogs probing the majority of promysalin's molecular framework (Fig. 12(a)).155 We quickly learned that the introduction of new substituents around the aromatic ring resulted in a complete loss in activity, as did an amide linkage. Probing polarity next, we observed that the α-hydroxy moiety was non-essential for activity (67), but interestingly two different methyl ester analogs 68 and 69 showed significantly reduced—but not abolished—activity (Fig. 12(b)). Furthermore, desaturation (70) and fluorine substitution (71) were tolerated, but dramatic decreases in activity were observed for methyl analog 72 and proline analog 73 (Fig. 12(c)). This data led us to the conclusion that promysalin's hydrogen bond network, which shapes its conformation, is necessary for its activity.155
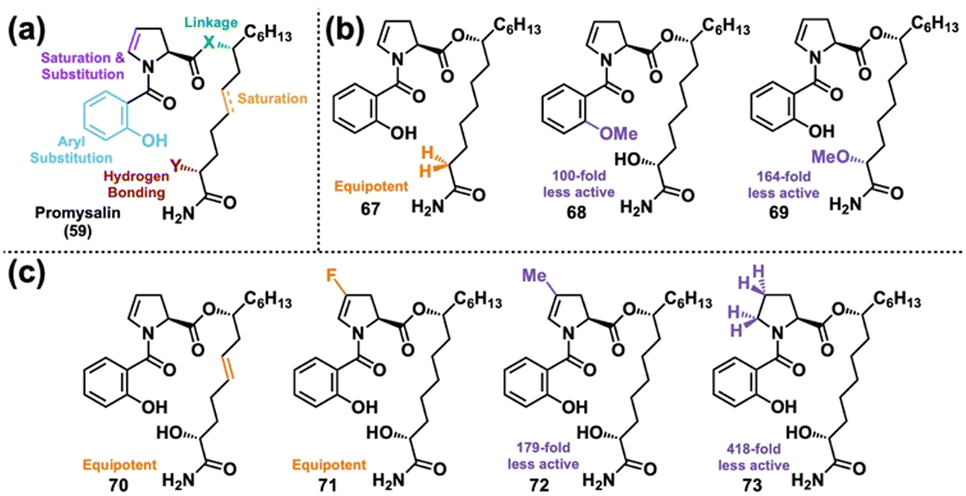 | ||
| Fig. 12 (a) Design of the Wuest lab's first DTS library of promysalin. (b) Analogs modulating hydrogen bonding capability. (c) Analogs probing conformational space. | ||
Noticing that promysalin's salicylamide moiety is reminiscent of iron-binding motifs found in the known siderophores pyochelin157,158 and pseudomonine,159–161 we questioned whether promysalin can bind iron. Indeed, a solution-based assay confirmed promysalin's ability to bind iron down to 1 mM, suggesting its potential activity as a siderophore. Siderophore activity would also explain earlier observations of its promotion of swarming and pyoverdine inhibition. However, subsequent experiments refuted the hypothesis that promysalin acts as a viable siderophore but is instead an inhibitor of succinate dehydrogenase (vide infra).162 With a molecular target now verified, we performed computational docking to aid in the rational design of additional analogs. Through this we identified promysalin's hydrogen-bond-sculpted macrocyclic conformation and that its aliphatic tail was positioned in a hydrophobic pocket.163
Looking to explore the SAR of these two features, we proceeded to synthesize ten new analogs that varied in chain length and ester positioning. Testing their biological activities, we found that ester repositioning to either C7 or C9 greatly diminished activity, indicating the sensitivity of the binding pocket to promysalin's natural macrocyclic conformation. Additionally, all truncated alkyl chain analogs showed decreased or nearly abolished activity. However, two analogs with increased alkyl chain length such as 74 showed improved activity compared with the NP, though this trend did not continue beyond two additional carbons (see 75) (Fig. 13(a)). Further computational modeling of these analogs in the binding site then revealed a previously unidentified binding cleft with a tryptophan residue within proximity to the longer alkyl chains.163 This inspired another analog 76, designed to take advantage of potential π-stacking interactions for further stabilization, but unfortunately it was less potent than the NP. Nevertheless, this new binding cleft offered yet another avenue for future analog design.
 | ||
| Fig. 13 (a) Selected alkyl side chain analogs of promysalin from the Wuest lab's second round of DTS. (b) Selected analogs from the Wuest lab's third round of DTS. (c) Terminal arene analogs designed for the newly-discovered binding cleft in succinate dehydrogenase. | ||
During our studies of promysalin and its analogs, we noticed a large difference between their IC50 values and MICs.164 Hypothesizing that this could be due to bacterial ester hydrolysis or efflux, we synthesized six new analogs to probe these mechanisms. Among these analogs were acrylamide 77, nitrile 78, and boronic acid 79 (Fig. 13(b)), which looked to take advantage of covalent target modification165,166 to circumvent the issue of efflux. We also looked to a carboxylic acid analog with an amide linkage (80) to avoid hydrolysis. Unfortunately, however, all of our analogs were less potent than the NP. We then tested our analogs in an efflux pump knockout strain, where they showed dramatic decreases in their IC50 values, confirming the impact of efflux on promysalin-based antibiotics.164 Finally, we detailed the synthesis of two more aromatic side chain analogs (81 and 82) designed to take advantage of the newly discovered binding cleft, but they were likewise not as potent as the NP (Fig. 13(c)).167 These findings demonstrate the challenge of overcoming resistance mechanisms such as efflux.
4.5. Baulamycins
Inhibitors of bacterial iron acquisition in vitro, the baulamycins (Fig. 14(a), left) were identified as competitive inhibitors of SbnE, while also showing broad-spectrum whole cell inhibitory activity.168 Noticing minimal differences in the reported IC50 values between iron-rich and -depleted media, we questioned whether the baulamycins operate according to an alternative MoA. This hypothesis, combined with ambiguity regarding their absolute and relative stereochemistry,169 prompted further investigation. We therefore looked to DTS to access baulamycin analogs for additional mechanistic insights.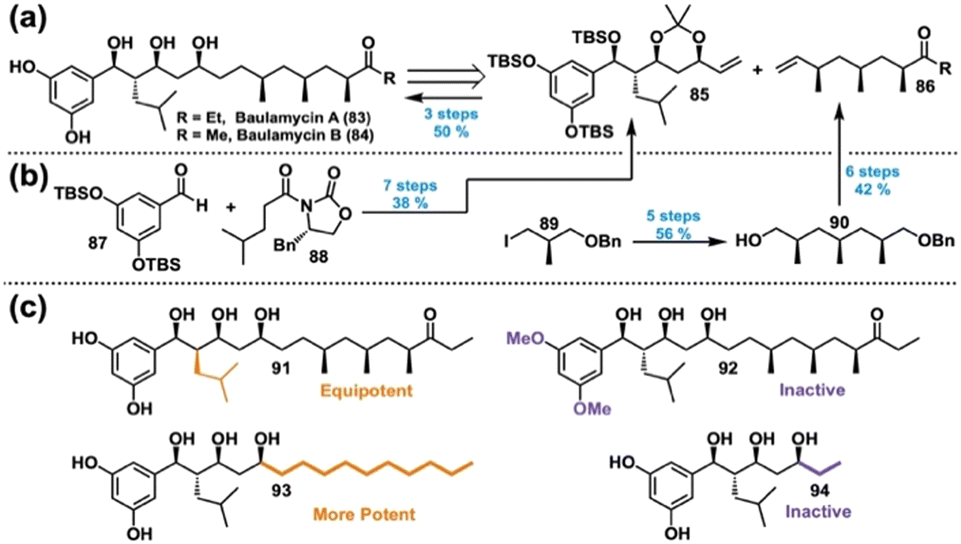 | ||
| Fig. 14 (a) Retrosynthesis of the baulamycins. (b) Successful forward synthesis of the baulamycins. Yields reported for baulamycin A. (c) Selected analogs from the Wuest lab's baulamycin DTS library. | ||
During the course of our studies, Goswami170 and Chandrasekhar171 independently reported their progress toward the total synthesis of the baulamycins. Publishing first, Goswami detailed a successful synthesis of the purported structure of baulamycin A, but unfortunately its spectroscopic data were not in agreement with that of the isolated compound.170 Subsequent syntheses of two other possible diastereomers yielded the same result. Seeing Goswami's report amidst their studies, Chandrasekhar's group was prompted to reroute, in the meantime opting to publish their completed synthesis of the reported structure's carbon framework.171 Shortly thereafter, Aggarwal et al. elucidated the correct structures of the baulamycins through a combined synthetic, computational, and NMR approach.172 However, just a couple of months later, Sim also reported a structural revision of baulamycin A determined through total synthesis, but settled on the enantiomer of Aggarwal's structure.173 While Aggarwal relied on the reported optical rotation of baulamycin A for their assignment,172 Sim argued that unidentified impurities in the original sample puts that measurement in question.173 Instead, Sim looked to verify their assignment through IC50 values against SbnE, finding that their proposed enantiomer was 3-fold less potent than the isolated natural product. An IC50 value for Aggarwal's enantiomer was not reported. Due to the incongruencies within Sim's report, we decided to reroute our synthetic efforts according to Aggarwal's enantiomer, which is the structure depicted in Fig. 14(a).
Thus, our revised retrosynthesis relied on a cross-metathesis between acetonide 85 and ketone 86 (Fig. 14(a), right). In the forward sense, aldehyde 87 and imide 88 initiated a series of asymmetric aldol reactions, protections, and reductions to yield 85 (Fig. 14(b), left).174 Likewise, iodide 89 facilitated a series of asymmetric alkylations and redox sequences to give benzyl ether 90 over 5 steps, enabling a further elaboration into ketone 86 (Fig. 14(b), right). Coupling of 85 and 86, followed by hydrogenation and global deprotection, afforded 83 in 50% yield over three steps; an analogous sequence enabled access to 84.
Considering the baulamycin structure, we hypothesized that their bioactivity in iron-rich media may be a result of nonselective membrane damage through a detergent-like MoA. We therefore designed our DTS analogs with this in mind, primarily focusing on chain length and overall polarity (Fig. 14(c)).174 In accordance with our hypothesis, an analog featuring inverted stereochemistry at C14 (91) retained activity, whereas methylation of the polyphenol (92) resulted in loss of activity. Additionally, simplifying the right half of the molecule to a simple alkyl chain (93) produced an analog with dramatically more potent activity, whereas removal of the right half (94) abolished activity. Furthermore, SYTOX uptake assays of our compounds supported our hypothesis that the active analogs were disrupting the bacterial membrane.174 Hence, our DTS campaign revealed a new, nonselective MoA of the baulamycins, informing future analog design.
Since our report, researchers have continued to show interest in the baulamycins in hopes of accessing a new potent antibiotic and/or better understand their MoAs. In 2020, Sherman and Williams reported a gram-scale synthesis of baulamycin A to enable further biological evaluation.175 Additionally, Sim and coworkers leveraged their previous route173 to produce several baulamycin analogs, focusing their efforts towards improving inhibitory potencies against SbnE. In their studies they were able to discover several cell wall disruptors that inhibit siderophore production in MRSA and even impede biofilm formation as well,176 demonstrating once again the benefits of a DTS approach. Thus, the baulamycins indeed remain a promising avenue for future investigation.
5. Resistance selection: impacts and limitations on mechanism elucidation
Traditionally, one would identify the targets of natural products through the development of resistance genes and sequencing. This can be accomplished through the gradual dosing of NPs to cells and a follow-up selection of live clones. These strains can then be regrown and sent for genome sequencing to compare to the parent strain for new mutations. Previously referenced work by the Myers group is an excellent representation of the impact this technique can have on mechanism elucidation of novel drug compounds. Found to be effective against many ESKAPE pathogens, studies were employed to understand the basis for why the specific structural moiety contained within iboxamycin led to activity against Erm-methylated MLSB-resistant ribosomes.92One of the limitations of this process is that strains are not always efficient in developing resistance. Fruitfully, the Myers lab uncovered that, in E. coli SQ110DTC, selective mutations allow for the cells to develop resistance to ribosome-targeting inhibitors through a lack of specific rRNA alleles and a multidrug efflux pump. Random selection and sequencing of resistance clones allowed for the identification of two possible single-nucleotide mutations, both of which occurred in the 23S rRNA, known to be in the canonical lincosamide binding site, also known among clindamycin-resistant bacteria.92
In almost all cases, resistance selection is a valid use of resources and time, exemplified above by uncovering the MoA by comparison with known MoAs of other antibiotics. The major limitations of this process are when the mechanism is not essential, efflux overwhelms the development of resistance to the target, or the compound has multiple targets and kills via polypharmacology.177 It is for this reason that we and others have turned to proteomic workflows to uncover non-obvious MoAs and potential drug targets that have evaded identification through traditional elucidation tactics.
6. Affinity-based protein profiling: novel target elucidation
Affinity-based protein profiling (AfBPP) allows for the determination of protein binding partners with general affinity for functionalized substrates, differing from activity-based protein profiling (ABPP) by the lack of covalent modification by the ligand. AfBPP requires a diazirine functionality to generate highly reactive carbenes which covalently crosslink the substrate to nearby affinity-based protein binding partners.178,179 This allows for the identification of cellular interactions that substrates can be participating in that have yet to be elucidated due to irreversible binding.179,180 A terminal alkyne then acts as a bioorthogonal handle, which will not be modified by any other intracellular component, to be enriched by a biotin-azide conjugate. This complex can then be “pulled down” with magnetic streptavidin beads or many other avidin-based moieties (Fig. 15). This then allows for biological analysis to determine both primary and secondary binding partners, along with initial characterization, typically by LC–MS/MS. Roles and further characterization of these proteins are then determined by biochemical and biophysical methods. Cellular assays of mutants, cellular knockouts, and overexpression are typical for target validation. However, the dynamic nature of proteins keeps this process complex and unique, and details can be found in alignment with specific projects discussed herein.179,180 Benjamin Cravatt9,10,181–183 and Matthew Bogyo11,12,184,185 are notable pioneers in proteomics that have opened the doors for groups like ours to continue to optimize this method. For details on how its development has blossomed into valuable scientific insight, see their referenced work181–185 as well as a recent review by Garber.186 | ||
| Fig. 15 Overview of the affinity-based protein profiling workflow. Created in BioRender. Lab, W. (2025) https://BioRender.com/8pczjro. | ||
Structurally built onto an 8-carbon chain in a linear fashion, the classical photo-handle contains an electrophile or nucleophile for functionalization, a diazirine for covalent linkage, and a terminal alkyne for bioorthogonal click chemistry. The order of these features is vital because—when affinity-based binding of the ligand occurs with the protein of interest—the diazirine must be within proximity for ligation to the target protein, while simultaneously preventing side reactivity. This small functionality on a linear chain leads to minimal steric hinderance, helping to retain binding affinity.187–190 Subsequently, retained binding affinity is always verified by activity comparison with the parent substrate, MICs being the standard method for antibiotic development. Often, when possible, comparison is done with multiple probes which have been functionalized at varying sites of the substrate.
Many have worked on the development and optimization of this handle. The first optimized synthesizes of the terminal carboxylic acid were completed and patented by Li and Parker in 2013 and 2015.187,188 While one resulted in higher overall yields, the other was completed in half the number of synthetic steps. Excitingly, in 2025, the Tepe group was able to report efficient access to many key functional groups from one intermediate (Fig. 16).189 In between, the Woo lab reported a shorter, 5-carbon handle that could be accessed in 8 steps. Though more steps, this handle exhibits optimized functionality. Shorter by 2.8 Å, this handle addresses the potential issues with inhibiting native interactions and off-target covalent binding.190 We look forward to further work in the optimization of these proteomic handles, not only for increased efficacy, but also in making them more accessible for academic laboratories (Fig. 16).
 | ||
| Fig. 16 Summary of synthetic routes to diazirine probes. | ||
6.1. AfBPP: promysalin
AfBPP has played an instrumental role in recent publications by our group where we leverage the technique to gain a deeper understanding of MoAs of natural products with unknown targets (Fig. 17). Touched on prior, a decade-long campaign by our group involved promysalin, whose MoA was confirmed in parallel using AfBPP and resistance selection in 2018.162,191 Originally, we hypothesized that the iron-binding ability of promysalin allowed for the use of transport channels and competitive siderophore activity to elicit an antibiotic-like response. Quickly disproven by iron-binding assays and at a crossroads, we, in collaboration with the Sieber lab, turned to proteomics. Leveraging the terminal amide of promysalin, the natural product was functionalized, and an inactive probe was also synthesized. Then, two strains of P. aeruginosa were grown and incubated with the NP probe and a control for competitive inhibition. In these experiments, promysalin was added prior to the addition of the probe, serving as a control. As promysalin will bind to the primary binding partners, only secondary interactions will remain for the handle to pull down. This process allowed for the identification of the succinate dehydrogenase C-subunit (SdhC) as the primary target of promysalin.162 | ||
| Fig. 17 Collaborative antibiotic target discoveries by AfBPP. Created in BioRender. Lab, W. (2025) https://BioRender.com/mz8cgve. | ||
Following this, computational docking and genetic knockouts were used to confirm the target; further details are thoroughly discussed in the original publication.162 Also known as succinate: ubiquinone oxidoreductase, SdhC is a crucial enzyme in both the Krebs cycle and electron transport chain, catalyzing the oxidation of succinate to fumarate.192 This is a notable target for many other NPs isolated from the rhizosphere, specifically referencing work by the Inaoka lab on siccanin, however, this did not align with the observed narrow-spectrum activity of promysalin.193
Unsatisfied, later that same year we sought to better understand the role of promysalin in the rhizosphere bacterium P. putida KT2440.191 A useful tool for mechanistic determination that has proven to be vital in the prior characterization of many natural products is global transcriptome analysis (RNASeq). Applying this technique by using promysalin-perturbation as a proxy for activity in KT2440, we discovered that of 455 affected genes, 8 down-regulated genes were involved in flagella assembly and mobility. Following further assessment of the effect promysalin had on swimming mobility—which is all outlined in the original publication—we determined that promysalin inhibits flagella mobility. The induced swarming phenotype observed is, however, flagella independent, justified rather by the lack of iron. Knowing promysalin is only a weak iron binder, we looked back at our 455 genes to find PP_4324 and PP_4325 (ccmD and ccmC, respectively) which encode part of the cytochrome C maturation (ccm) system. Additionally, we found PP_0489 (fdoG) and PP_5212, which encode iron-binding metabolic enzymes, formate dehydrogenase, and an oxidoreductase. Taken together, promysalin causes the bacteria to limit iron uptake due to the ferric-bearing enzymes it targets in primary metabolism of Pseudomonas spp.191 This work highlights the level of detail that can be achieved by leveraging proteomic and transcriptomic tools to understand natural product MoA.
6.2. ABPP vs. AfBPP: xanthocillin
Xanthocillin (Xan), an isonitrile containing NP, like many others containing this moiety, has evaded full mechanistic understanding.180,194 In collaboration with the Sieber lab, it was investigated for its MoA by leveraging a combination of both ABPP and AfBPP. In this report, a very similar workflow to that outlined above was utilized. The terminal hydroxyl was leveraged for SN2 functionalization of both a photoaffinity cross-linking alkyne handle and a sole terminal alkyne handle. The sole terminal alkyne handle allows for ABPP to identify covalent binding partners of Xan while the photoprobe allows for AfBPP to determine noncovalent binding partners. This granted us the ability to determine whether the MoA directly targets an enzyme of a vital cellular process or acts through modification of the proteome.179 When utilizing ABPP in A. baumannii, interestingly, only proteins associated with nonessential cellular processes were characterized. Specifically, through mutant development, the PbgS enzyme was identified as containing a P241S modification within the active site leading to decreased affinity for the required substrate for tetrapyrrole synthesis. However, it was determined that Xan is not a direct target of PbgS.180Looking towards whole proteome analysis, it was determined that Xan led to an upregulation of the putative proton/sodium glutamate symport protein and TonB-dependent siderophore receptors. This led to the determination that there was enhanced heme biosynthesis leading to elevated porphyrin levels in the cellular extracts. Further analysis with mutant strains pointed to the direct sequestration of the heme by Xan, therefore inhibiting heme-dependent enzymes from accessing the regulatory heme.180 This report thus demonstrates how ABPP and AfBPP can be taken together to determine the MoA of a natural product in a relatively facile way, especially in cases of polypharmalogical activity.
6.3. Biofilm AfBPP: carolacton
Collaborations between the Wuest and Sieber labs have exemplified how AfBPP can be used to discover niche MoAs. As previously mentioned, bacteria use a collection of virulence factors to become advantageous against their host and protect themselves from invaders. We have become especially interested in targeting biofilms, a community of cells at different life stages encased in a matrix, which play a major role in the protection of persister cells. These cells lie dormant to survive the presence of toxins and it happens that targeting cells that are not undergoing division and typical cellular processes remains extremely challenging.195,196Carolacton has been an ongoing story in the Wuest lab, allowing for valuable discoveries fueling antibiotic research. Recently, applications of this macrolactone natural product have set a novel precedent for the use of AfBPP to study pathways contributing to virulence factors like biofilm formation. Carolacton's simplified derivative, A2 ((+)-58), was functionalized with a bioorthogonal photoaffinity handle for biofilm-based AfBPP to determine its MoA after it was determined that it does not inhibit FolD, like carolacton.136,197 The workflow of biofilm AfBPP involves the dosage of the probe to either mature lysed biofilm cells or with probe dosage at the beginning of biofilm formation. Proteomics identified five putative targets, and by using available knockouts we were able to rule out four of the five possible options. However, without an available knockout of GbpB, its mechanistic role could not be discounted. Therefore, in collaboration with the Wu lab, who overexpressed and constructed a pVPT-GbpB plasmid for transformation into an empty vector of S. mutans, we observed that at a concentration of 150 μM (+)-58 was able to inhibit growth. This increase in IC50 strongly implicated this enzyme as the target. We next conducted binding experiments using microscale thermophoresis and observed clear binding, further validating GbpB as the bona fide target. Further studies revealed that (+)-58 binds to the conserved C-terminal CHAP (cysteine, histidine-dependent, amidohydrolase/peptidase) domain, making our derivative the first-reported CHAP binder.197
6.4. Drug repurposing: PK150 and fendiline
A major untapped source of potential antibiotics are FDA-approved drugs approved for other diseases. Through a search of human kinase inhibitors, which lack sufficient analysis into their antibacterial activity, sorafenib (SFN), an anticancer drug, was identified as a potent potential antibiotic by the Sieber lab. Further analysis and SAR of SFN resulted in 72 synthesized analogs for biological testing, of which PK150 was identified. After extensive analysis, it was determined to have enhanced activity against MRSA persister cells and biofilm targets while simultaneously lacking resistance development under laboratory conditions (SFN: 3 μM, PK150: 0.3 μM). Further mechanism elucidation was later achieved by chemical proteomics. Though it was noted that the proteomic analysis did not rule out other underlying mechanisms, it was determined that the most significant binding partners, MenG and SpsB, are contributors to the MoA. MenG catalyzes the terminal steps of menaquinone biosynthesis, making it a broad range antibiotic with activity in any strains that allow for such metabolic control.198 Menaquinone is vital in electron transport and ATP generation and is similar to folic acid in that humans can obtain the substrate from their diet.199 In terms of SpsB, this essential membrane serine endopeptidase chaperones the S. aureus protein secretion (Sec) pathway. Proven in the mechanism elucidation of the arylomycins, inhibition of SpsB leads to the accumulation of unprocessed proteins and cell death.71–73 However, here we showed how stimulation of SpsB affects the secretome by leading to the dysregulation of cellular processes. In this case, the uncontrolled secreting of proteins, including autolysins, resulted in cell death via cell-wall degradation and autolysis.198Applications of AfBPP have begun to blur the lines between chemical biology and medicinal chemistry as applications of this strategy expand. Just this year, the Wuest, Sieber, and Rather labs collaborated to identify a novel target by screening FDA-approved molecules against carbapenem-resistant A. baumannii (CRAB), β-lactams which have served as the go-to treatment for such infections. CRAB priority has risen as countries have reported 60–90% MDR strains.200,201 As previously explained, the primary MoA of antibiotic resistance to β-lactams is the encoding for β-lactamases. In CRAB, OXA-23 is responsible for the encoding of OXA β-lactamases, contributing to the resistance. Leveraging an FDA-approved drug library once again, we identified fendiline, a calcium channel blocker, as an antimicrobial reagent against cells expressing OXA-23. Initial efforts to select for a resistant mutant were ineffective and it was only through an AfBPP assay that fendiline was determined to inhibit lipoprotein trafficking pathways (Lol), by binding to LolF.200
The Lol pathway is responsible for trafficking lipoproteins to the outer membrane where they are vital in many processes including cell envelope biosynthesis and homeostasis of cellular components like the peptidoglycan layer and β-barrel proteins. Along with characterization of OXA-23 as a lipoprotein, we determined that OXA-23 was toxic to A. baumannii when contained to the periplasm, toxicity which was enhanced by the inhibition of the Lol pathway. This discovery opens a door for novel targeting of OXA-23 overproducers as we now understand that CRAB relies on the trafficking by the Lol pathway to avoid overaccumulation within the cell envelope, which can lead to cell death.200
7. Conclusions
The history of antibiotic development is rich in scope, comprising remarkable stories of both scientific breakthroughs and serendipitous discovery. Nevertheless, bacteria continue to evolve, acquiring resistance to some of medicine's greatest weapons against diseases. While these resistance mechanisms are wide-ranging and have been challenging to overcome, scientists have still managed to see success in their endeavours. Drawing inspiration from nature, we have shown here how an approach focused on natural product derivatization can result in significant advances against antimicrobial resistance to antibiotics. For example, DTS has enabled access to a variety of ways to overcome antibiotic resistance, including: (1) SAR campaigns of natural product scaffolds resulting in retained activity against even the most resistant bacterial strains, (2) the discovery of novel tool compounds to probe fundamental biological phenomena related to resistance, and (3) derivatization of natural products with photo-crosslinking probes to allow for mechanistic elucidation through strategies like AfBPP. At the same time, new isolation methodologies such as iChip are providing increasingly more natural product scaffolds for future investigation. Continued investment in this area of research is also of paramount importance as most pharmaceutical companies have divested from development. Therefore, it will be important going forward to have increased funding both from government organizations and private foundations to support these essential discoveries in the future.Author contributions
Makayla R. Hedges: writing – original draft, review & editing. Camden M. Di Carlo: writing – original draft, review & editing. William M. Wuest: writing – review & editing, supervision.Conflicts of interest
There are no conflicts to declare.Data availability
No primary research results, software or code has been included, and no new data were generated or analyzed as part of this review.Acknowledgements
The authors would like to thank Prof. Stephan Sieber (Technical University-Munich) for fruitful conversations and a decade of collaboration. Research support was provided by the National Institute of General Medical Sciences (GM119426) and the National Science Foundation (CHE2311665).Notes and references
- M. Hutchings, A. Truman and B. Wilkinson, Curr. Opin. Microbiol., 2019, 51, 72–80 CrossRef CAS PubMed.
- J. Prescott, Vet. Microbiol., 2014, 171(3–4), 273–278 CrossRef PubMed.
- Outpatient Antibiotic Prescribing in the United States CDC Antibiotic Prescribing and Use, https://www.cdc.gov/antibiotic-use/hcp/data-research/antibiotic-prescribing.html/#:˜:text=Key%20U.S.%20statistics&text=An%20estimated%2080%2D90%25%20of,in%20the%20outpatient%20setting4, (accessed June 2025).
- W. C. Reygaert, AIMS Microbiol., 2018, 4(3), 482–501 CAS.
- A. G. Atanasov, S. B. Zotchev and V. M. Dirsch, the International Natural Product Sciences Taskforce and C.T. Supuran, Nat. Rev. Drug Discovery, 2021, 20, 200–216 Search PubMed.
- M. J. Moore, S. Qu, C. Tan, Y. Moji, K. D. Jamin and D. L. Boger, J. Am. Chem. Soc., 2020, 142(37), 16039–16050 CrossRef CAS PubMed.
- E. Rubinstein and Y. Keynan, Front Public Health, 2014, 2, 217 Search PubMed.
- K. C. Nicolaou, H. J. Mitchell, N. F. Jain, N. Winssinger, R. Hughes and T. Bando, Angew. Chem., Int. Ed., 1999, 38(1–2), 240–244 CrossRef CAS.
- B. Cravatt, A. Wright and J. Kozarich, Annu. Rev. Biochem., 2008, 77, 383–414 CrossRef CAS PubMed.
- B. Cravatt, Isr. J. Chem., 2023, 63, e202300029 CrossRef CAS PubMed.
- A. B. Berger, P. M. Vitorino and M. Bogyo, Am. J. PharmacoGenomics, 2004, 4(6), 371–381 CrossRef CAS PubMed.
- L. J. Keller, B. M. Babin, M. Lakemeyer and M. Bogyo, Curr. Opin. Chem. Biol., 2020, 54, 45–53 CrossRef CAS PubMed.
- Staphylococcus aureus Basics. CDC Staphylococcus aureus. https://www.cdc.gov/staphylococcus-aureus/about/index.html#:~:text=Staphylococcus%20aureus%20(staph)%20is%20a%20bacterium%20commonly%20found%20on%20the,to%20serious%20or%20fatal%20outcomes. (accessed June 2025).
- D. J. Tipper and J. L. Strominger, Proc. Natl. Acad. Sci. U. S. A., 1965, 54(4), 1133–1141 Search PubMed.
- M. Lee, D. Hesek, M. Suvorov, W. Lee, S. Vakulenko and S. Mobashery, J. Am. Chem. Soc., 2003, 125(52), 16322–16326 Search PubMed.
- K. Bush and P. A. Bradford, Cold Spring Harbor Perspect. Med., 2016, 6(8), a025247 Search PubMed.
- About MRSA. Minnesota Department of Health. https://www.health.state.mn.us/diseases/staph/mrsa/index.html (accessed June 2025).
- F. D. Lowy, J. Clin. Invest., 2003, 111(9), 1265–1273 CrossRef CAS PubMed.
- S. J. Peacock and G. K. Paterson, Annu. Rev. Biochem., 2015, 84, 577–601 CrossRef CAS PubMed.
- A. J. Kallen, H. G. Welch and B. E. Sirovich, Arch. Intern. Med., 2006, 166(6), 635–639 CrossRef PubMed.
- G. Eliopoulos and P. Huovinen, Clin. Infect. Dis., 2001, 32(11), 1608–1614 Search PubMed.
- M. K. Yun, Y. Wu, Z. Li, Y. Zhao, M. B. Waddell, A. M. Ferreira, R. E. Lee, D. Bashford and S. W. White, Science, 2012, 335(6072), 1110–1114 CrossRef CAS PubMed.
- B. Werth, Sulfonamides, Merck Manual. https://www.merckmanuals.com/professional/infectious-diseases/bacteria-and-antibacterial-medications/sulfonamides. (accessed June 2025).
- T. R. Kemnic and M. Coleman, Trimethoprim Sulfamethoxazole, StatPearls. https://www.ncbi.nlm.nih.gov/books/NBK513232/. (accessed June 2025).
- M. Venkatesan, M. Fruci, L. A. Verellen, T. Skarina, N. Mesa, R. Flick, C. Pham, R. Mahadevan, P. Stogios and A. Savchenko, Nat. Commun., 2023, 14(1), 4031 CrossRef CAS PubMed.
- C. Wintersdorff, J. Penders, J. Niekerk, N. Mills, S. Majumder, L. Alphen, P. Savelkoul and P. Wolffs, Front. Microbiol., 2015, 7, 174871 Search PubMed.
- M. Rohde, Microbiol. Spectr., 2019, 7(3) DOI:10.1128/microbiolspec.GPP3-0044-2018.
- S. S. Mohapatra, S. K. Dwibedy and I. Padhy, J. Biosci., 2021, 46(3), 85 Search PubMed.
- Z. Li, Y. Cao, L. Yi, J. H. Liu and Q. Yang, Open Forum Infect. Dis., 2019, 6(10), 368 Search PubMed.
- S. Manioglu, S. M. Modaresi, N. Ritzmann, J. Thoma, S. Overall, A. Harms, G. Upert, A. Luther, A. Barnes, D. Obrecht, D. Müller and S. Hiller, Nat. Commun., 2022, 13(1), 6195 CrossRef CAS PubMed.
- S. Patel and S. Saw, Daptomycin, StatPearls. https://www.ncbi.nlm.nih.gov/books/NBK470407/. (accessed June 2025).
- F. Grein, A. Müller, K. M. Scherer, X. Liu, K. C. Ludwig, A. Klöckner, M. Strach, H. G. Sahl, U. Kubitscheck and T. Schneider, Nat. Commun., 2020, 11(1), 1455 CrossRef CAS PubMed.
- A. P. Carter, W. M. Clemons, D. E. Brodersen, R. J. Morgan-Warren, B. T. Wimberly and V. Ramakrishnan, Nature, 2000, 407(6802), 340–348 CrossRef CAS PubMed.
- L. J. Simpson, J. S. Reader and E. Tzima, Cells, 2020, 9(3), 650 CrossRef CAS PubMed.
- S. J. Krawczyk, M. Leśniczak-Staszak, E. Gowin and W. Szaflarski, Biomolecules, 2024, 14(10), 1263 CrossRef CAS PubMed.
- L. Jeremia, B. E. Deprez, D. Dey, G. L. Conn and W. M. Wuest, RSC Med. Chem., 2023, 14(4), 624–643 RSC.
- M. X. Ho, B. P. Hudson, K. Das, E. Arnold and R. H. Ebright, Curr. Opin. Struct. Biol., 2009, 19(6), 715–723 Search PubMed.
- E. Campbell, N. Korzheva, A. Mustaev, K. Murakami, S. Nair, A. Goldfarb and S. A. Darst, Cell, 2001, 104(6), 901–912 Search PubMed.
- S. Ramaswamy and J. M. Musser, Tuber. Lung Dis., 1998, 79(1), 3–29 CrossRef CAS PubMed.
- T. Thai, B. H. Salisbury and P. M. Zito, Ciprofloxacin, StatPearls, https://www.ncbi.nlm.nih.gov/books/NBK535454/ (accessed June 2025) Search PubMed.
- A. Sitovs, I. Sartini and M. Giorgi, Res. Vet. Sci., 2021, 137, 111–126 Search PubMed.
- D. C. Hooper and G. A. Jacoby, Ann. N. Y. Acad. Sci., 2015, 1354(1), 12–31 CrossRef CAS PubMed.
- R. Alenazy, Biology, 2022, 11(9), 1328 Search PubMed.
- R. Lathakumari, L. Vajravelu, A. Satheesan, S. Ravi and J. Thulukanam, Med. Microecol., 2024, 20, 100106 Search PubMed.
- K. M. Zack, T. Sorenson and S. G. Joshi, Pathogens, 2024, 13, 197 CrossRef CAS PubMed.
- R. Ferrer-Espada, H. Shahrour, B. Pitts, P. Stewart, S. Sánchez-Gómez and G. Martínez-de-Tejada, Nat. Sci. Rep., 2019, 9, 3452 Search PubMed.
- S. A. A. M. Abdel-Karim, A. M. A. El-Ganiny, M. A. El-Sayed and H. A. A. Abbas, PLoS One, 2022, 17(7), e0272417 CrossRef CAS PubMed.
- K. C. Nicolaou and S. Rigol, J. Antibiot., 2018, 71, 153–184 CrossRef CAS PubMed.
- H. Gaffer, Color. Technol., 2019, 135(6), 484–500 Search PubMed.
- T. YouYou. From Artemisia Annual. To Artemisinins, Chemical Industry Press, 2017 Search PubMed.
- B. Hanboonkunupakarn, J. Tarning, S. Pukrittayakamee and K. Chotivanich, Front. Pharmacol., 2022, 13, 876282 CrossRef CAS PubMed.
- J. L. Bridgford, S. C. Xie, S. A. Cobbold, C. Pafaje, S. Herrmann, T. Yang, D. Gillett, L. Dick, S. Ralph, C. Dogovski, N. Spillman and L. Tilley, Nat. Commun., 2018, 9, 3801 Search PubMed.
- X. Z. Su and L. H. Miller, Sci. China: Life Sci., 2015, 58(11), 1175–1179 CrossRef PubMed.
- R. Jansen, H. Irschik, V. Huch, D. Schummer, H. Steinmetz, M. Bock, T. Schmidt, A. Kirschning and R. Müller, Eur. J. Org. Chem., 2010, 1284–1289 Search PubMed.
- A. Kany, F. Fries, C. Seyfert, C. Porten, S. Deckarm, M. Ortiz, N. Dubarry, S. Vaddi, M. Große, S. Bernecker, B. Sandargo, A. Müller, E. Bacqué, M. Stadler, J. Herrmann and R. Müller, ACS Infect. Dis., 2024, 10(12), 4337–4346 CrossRef CAS PubMed.
- Y. Imai, K. J. Meyer, A. Iinishi, Q. Favre-Godal, R. Green, S. Manuse, M. Caboni, M. Mori, S. Niles, M. Ghiglieri, C. Honrao, X. Ma, J. J. Guo, A. Makriyannis, L. Linares-Otoya, N. Böhringer, Z. G. Wuisan, H. Kaur, R. Wu, A. Mateus, A. Typas, M. M. Savitski, J. L. Espinoza, A. O’Rourke, K. E. Nelson, S. Hiller, N. Noinaj, T. F. Schäberle, A. D’Onofrio and K. Lewis, Nature, 2019, 576(7787), 459–464 CrossRef CAS PubMed.
- H. Kaur, R. P. Jakob, J. K. Marzinek, R. Green, Y. Imai, J. R. Bolla, E. Agustoni, C. V. Robinson, P. J. Bond, K. Lewis, T. Maier and S. Hiller, Nature, 2021, 593, 125–129 Search PubMed.
- W. Li, P. Estrada-de los Santos, S. Matthijs, G. L. Xie, R. Busson, P. Cornelis, J. Rozenski and R. De Mot, Chem. Biol., 2011, 18, 1320–1330 CrossRef CAS PubMed.
- D. Nichols, N. Cahoon, E. M. Trakhtenberg, L. Pham, A. Mehta, A. Belanger, T. Kanigan, K. Lewi and S. S. Epstein, Appl. Environ. Microbiol., 2010, 76(8), 2445–2450 CrossRef CAS PubMed.
- L. Chabib, T. Rustandi, M. Fawwazi, E. Kumalasari, D. Lestari, S. Amalia and N. Normilawati, Front. Microbiol., 2025, 16 DOI:10.3389/fmicb.2025.1530273.
- L. L. Ling, T. Schneider, A. J. Peoples, A. L. Spoering, I. Engels, B. P. Conlon, A. Mueller, T. F. Schäberle, D. E. Hughes, S. Epstein, M. Jones, L. Lazarides, V. A. Steadman, D. R. Cohen, C. R. Felix, K. A. Fetterman, W. P. Millett, A. G. Nitti, A. M. Zullo, C. Chen and K. Lewis, Nature, 2015, 517, 455–459 CrossRef CAS PubMed.
- J. Szychowski, J.-F. Truchon and Y. L. Bennani, J. Med. Chem., 2014, 57(22), 9292–9308 CrossRef CAS PubMed.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2016, 79(3), 629–661 CrossRef CAS PubMed.
- R. M. Wilson and S. J. Danishefsky, J. Org. Chem., 2006, 71(22), 8329–8351 CrossRef CAS PubMed.
- J. Shimokawa, Tetrahedron Lett., 2014, 55(45), 6156–6162 CrossRef CAS.
- L. Li, Z. Chen, X. Zhang and Y. Jia, Chem. Rev., 2018, 118(7), 3752–3832 CrossRef CAS PubMed.
- R. A. Fernandes, Chem. Commun., 2023, 59, 12205–12230 RSC.
- A. M. Szpilman and E. M. Carreira, Angew. Chem., Int. Ed., 2010, 49, 9592–9628 CrossRef CAS PubMed.
- N. J. Truax and D. Romo, Nat. Prod. Rep., 2020, 37, 1436 Search PubMed.
- F. Liu and A. G. Myers, Curr. Opin. Chem. Biol., 2016, 32, 48–57 CrossRef CAS PubMed.
- T. C. Roberts, M. A. Schallenberger, J. Liu, P. A. Smith and F. E. Romesberg, J. Med. Chem., 2011, 54(14), 4954–4963 CrossRef CAS PubMed.
- Y. Zhang, D. Zhang, W. Zhao, H. Li, Z. Lu, B. Guo, X. Meng, X. Zhou and Y. Yang, J. Med. Chem., 2024, 67(8), 6585–6609 CrossRef CAS PubMed.
- P. A. Smith, M. F. T. Koehler, H. S. Girgis, D. Yan, Y. Chen, Y. Chen, J. J. Crawford, M. R. Durk, R. I. Higuchi, J. Kang, J. Murray, P. Paraselli, S. Park, W. Phung, J. G. Quinn, T. C. Roberts, L. Rougé, J. B. Schwarz, E. Skippington, J. Wai, M. Xu, Z. Yu, H. Zhang, M.-W. Tan and C. E. Heise, Nature, 2018, 561, 189–194 CrossRef CAS PubMed.
- R. D. Birkenmeyer and F. Kagan, J. Med. Chem., 1970, 13(4), 616–619 CrossRef CAS PubMed.
- R. Leclercq and P. Courvalin, Antimicrob. Agents Chemother., 1991, 35(7), 1267–1272 CrossRef CAS PubMed.
- B. Bannister, J. Chem. Soc., Perkin Trans. 1, 1972, 3025–3030 Search PubMed.
- B. Bannister, J. Chem. Soc., Perkin Trans. 1, 1972, 3031–3036 Search PubMed.
- B. Bannister, J. Chem. Soc., Perkin Trans. 1, 1973, 1676–1682 RSC.
- B. Bannister, J. Chem. Soc., Perkin Trans. 1, 1974, 360–369 Search PubMed.
- M. J. Mitcheltree, PhD Thesis, Harvard University, 2018 Search PubMed.
- H. O’Dowd, A. L. Erwin and J. G. Lewis, in Methods and Principles in Medicinal Chemistry, ed. S. Hanessian, Wiley-VCH, 2014, p. 7 Search PubMed.
- J. G. Lewis, S. K. Anandan, H. O’Dowd, M. F. Gordeev and L. Li, US Pat., 7361743B2, 2008 Search PubMed.
- A. Dondoni, S. Franco, F. Merchan, P. Merino and T. Tejero, Synlett, 1993, 78–80 CrossRef CAS.
- E. Umemura, Y. Wakiyama, K. Kumura, K. Ueda, S. Masaki, T. Watanabe, M. Yamamoto, Y. Hirai, H. Fushimi, T. Yoshida and K. Ajito, J. Antibiot., 2013, 66, 195–198 Search PubMed.
- Y. Wakiyama, K. Kumura, E. Umemura, S. Masaki, K. Ueda, Y. Sato, Y. Hirai, Y. Hayashi and K. Ajito, J. Antibiot., 2018, 71, 298–317 CrossRef CAS PubMed.
- E. Umemura, K. Kumura, S. Masaki, K. Ueda, Y. Wakiyama, Y. Sato, M. Yamamoto, K. Ajito, T. Watanabe and C. Kaji, US Pat., 7867980B2, 2008 Search PubMed.
- R. D. Birkenmeyer, S. J. Kroll, C. Lewis, K. F. Stern and G. E. Zurenko, J. Med. Chem., 1984, 27(2), 216–223 CrossRef CAS PubMed.
- Y. Hirai, K. Maebashi, K. Yamada, Y. Wakiyama, K. Kumura, E. Umemura and K. Ajito, J. Antibiot., 2021, 74(2), 124–132 CrossRef CAS PubMed.
- M. J. Mitcheltree, J. W. Stevenson, A. Pisipati and A. G. Myers, J. Am. Chem. Soc., 2021, 143(18), 6829–6835 CrossRef CAS PubMed.
- K. J. Silvestre, PhD Thesis, Harvard University, 2019 Search PubMed.
- I. Moga, PhD Thesis, Harvard University, 2019 Search PubMed.
- M. J. Mitcheltree, A. Pisipati, E. A. Syroegin, K. J. Silvestre, D. Klepacki, J. D. Mason, D. W. Terwilliger, G. Testolin, A. R. Pote, K. J. Y. Wu, R. P. Ladley, K. Chatman, A. S. Mankin, Y. S. Polikanov and A. G. Myers, Nature, 2021, 599, 507–512 CrossRef CAS PubMed.
- J. D. Mason, D. W. Terwilliger, A. R. Pote and A. G. Myers, J. Am. Chem. Soc., 2021, 143, 11019–11025 CrossRef CAS PubMed.
- B. Vester and S. Douthwaite, Antimicrob. Agents Chemother., 2001, 45(1), 1–12 CrossRef CAS PubMed.
- C. Orelle, S. Carlson, B. Kaushal, M. M. Almutairi, H. Liu, A. Ochabowicz, S. Quan, V. C. Pham, C. L. Squires, B. T. Murphy and A. S. Mankin, Antimicrob. Agents Chemother., 2013, 57(12), 5994–6004 Search PubMed.
- E. C. Böttger, B. Springer, T. Prammananan, Y. Kidan and P. Sander, EMBO Rep., 2001, 21(4), 318–323 CrossRef PubMed.
- F. Schlünzen, R. Zarivach, J. Harms, A. Bashan, A. Tocilj, R. Albrecht, A. Yonath and F. Franceschi, Nature, 2001, 413, 814–821 CrossRef PubMed.
- J. A. Dunkle, L. Xiong, A. S. Mankin and J. H. D. Cate, Proc. Natl. Acad. Sci. U. S. A., 2010, 107(40), 17152–17157 Search PubMed.
- H. Paternoga, C. Crowe-McAuliffe, L. V. Bock, T. O. Koller, M. Morici, B. Beckert, A. G. Myasnikov, H. Grubmüller, J. Nováček and D. N. Wilson, Nat. Struct. Mol. Biol., 2023, 30, 1380–1392 CrossRef CAS PubMed.
- D. Tu, G. Blaha, P. B. Moore and T. A. Steitz, Cell, 2005, 121, 257–270 CrossRef CAS PubMed.
- K. J. Y. Wu, B. I. C. Tresco, A. Ramkissoon, E. V. Aleksandrova, E. A. Syroegin, D. N. Y. See, P. Liow, G. A. Dittemore, M. Yu, G. Testolin, M. J. Mitcheltree, R. Y. Liu, M. S. Svetlov, Y. S. Polikanov and A. G. Myers, Science, 2024, 383, 721–726 CrossRef CAS PubMed.
- T. A. Mukhtar and G. D. Wright, Chem. Rev., 2005, 105, 529–542 CrossRef CAS PubMed.
- Q. Li and I. B. Seiple, J. Am. Chem. Soc., 2017, 139, 13304–13307 CrossRef CAS PubMed.
- A. D. Politano and R. G. Sawyer, Curr. Opin. Investig. Drugs., 2010, 11(2), 225–236 CAS.
- G. A. Pankuchm, G. Lin, C. Clark and P. C. Appelbaum, Antimicrob. Agents Chemother., 2011, 55(4), 1787–1791 CrossRef PubMed.
- R. H. Schlessinger and Y. J. Li, J. Am. Chem. Soc., 1996, 118(13), 3301–3302 CrossRef CAS.
- J. Wu and J. S. Panek, Angew. Chem., Int. Ed., 2010, 49(35), 6165–6168 CrossRef CAS PubMed.
- J. Wu and J. S. Panek, J. Org. Chem., 2011, 76(24), 9900–9918 CrossRef CAS PubMed.
- F. Tavares, J. P. Lawson and A. I. Meyers, J. Am. Chem. Soc., 1996, 118(13), 3303–3304 CrossRef CAS.
- A. K. Ghosh and W. Liu, J. Org. Chem., 1997, 62(23), 7908–7909 CrossRef CAS PubMed.
- P. Breuilles and D. Uguen, Tetrahedron Lett., 1998, 39(20), 3149–3152 CrossRef CAS.
- D. A. Entwistle, S. I. Jordan, J. Montgomery and G. Pattenden, J. Chem. Soc., Perkin Trans., 1996, 12, 1315–1317 RSC.
- D. A. Entwistle, S. I. Jordan, J. Montgomery and G. Pattenden, Synthesis, 1998,(S1), 603–612 CrossRef CAS.
- C. A. Dvorak, W. D. Schmitz, D. J. Poon, D. C. Pryde, J. P. Lawson, R. A. Amos and A. I. Meyers, Angew. Chem., Int. Ed., 2000, 39(9), 1664–1666 CrossRef CAS PubMed.
- Q. Li, J. Pellegrino, D. J. Lee, A. A. Tran, H. A. Chaires, R. Wang, J. E. Park, K. Ji, D. Chow, N. Zhang, A. F. Brilot, J. T. Biel, G. van Zundert, K. Borrelli, D. Shinabarger, C. Wolfe, B. Murray, M. P. Jacobson, E. Mühle, O. Chesneau, J. S. Fraser and I. B. Seiple, Nature, 2020, 586, 145–150 CrossRef CAS PubMed.
- P. J. Stogios, M. L. Kuhn, E. Evdokimova, P. Courvalin, W. F. Anderson and A. Savchenko, Antimicrob. Agents Chemother., 2014, 58(12), 7083–7092 CrossRef PubMed.
- N. H. Hoang, N. L. Huong, A. Shrestha, J. K. Sohng, Y. J. Yoon and J. W. Park, Lett. Appl. Microbiol., 2013, 57(5), 393–398 CrossRef CAS PubMed.
- C. Vidaillac, J. Parra-Ruiz, P. Winterfield and M. J. Rybak, Int. J. Antimicrob. Agents, 2011, 38(4), 301–306 CrossRef CAS PubMed.
- Q. Li and I. B. Seiple, Synlett, 2021, 647–654 CrossRef CAS PubMed.
- H. K. Kuramitsu and B. Y. Wang, Am. J. Dent., 2011, 24(3), 153–154 Search PubMed.
- W. J. Loesche, Microbiol. Rev., 1986, 50(4), 353–380 CrossRef CAS PubMed.
- C. Apel, A. Barg, A. Rheinberg, G. Conrads and I. Wagner-Döbler, Dent. Mater., 2013, 29(11), 1188–1199 CrossRef CAS PubMed.
- B. Kunze, M. Reck, A. Dötsch, A. Lemme, D. Schummer, H. Irschik, H. Steinmetz and I. Wagner-Döbler, BMC Microbiol., 2010, 10, 199 CrossRef PubMed.
- J. Li, W. Wang, Y. Wang and A. P. Zeng, Proteomics, 2013, 13, 3470–3477 CrossRef CAS PubMed.
- M. Reck, K. Rutz, B. Kunze, J. Tomasch, S. K. Surapaneni, S. Schulz and I. Wagner-Döbler, J. Bacteriol., 2011, 193(20), 5692–5706 CrossRef CAS PubMed.
- T. Schmidt and A. Kirschning, Angew. Chem., Int. Ed., 2012, 51, 1063–1066 CrossRef CAS PubMed.
- K. S. Rao and S. Ghosh, Synthesis, 2013, 2745–2751 CAS.
- G. Sabitha, K. Shankaraiah, M. N. Prasad and J. S. Yadav, Synthesis, 2013, 251–259 CAS.
- G. V. M. Sharma and S. V. Reddy, RSC Adv., 2013, 3, 21759 RSC.
- M. S. Hallside, R. S. Brzozowski, W. M. Wuest and A. J. Phillips, Org. Lett., 2014, 16, 1148–1151 CrossRef CAS PubMed.
- N. Stumpp, P. Premnath, T. Schmidt, J. Ammermann, G. Dräger, M. Reck, R. Jansen, M. Stiesch, I. Wagner-Döbler and A. Kirschning, Org. Biomol. Chem., 2015, 13, 5765 RSC.
- V. P. Kumar and S. Chandrasekhar, Org. Lett., 2013, 15(14), 3610–3613 CrossRef CAS PubMed.
- A. E. Solinski, A. B. Koval, R. Brzozowski, K. R. Morrison, A. J. Fraboni, C. E. Carson, A. R. Eshraghi, G. Zhou, R. G. Quivey Jr., V. A. Voelz, B. A. Buttaro and W. M. Wuest, J. Am. Chem. Soc., 2017, 139(21), 7188–7191 CrossRef CAS PubMed.
- J. Ammermann, T. Schmidt, J. Donner, M. Reck, M. Dalton, N. Stumpp, M. Stiesch, I. Wagner-Döbler and A. Kirschning, Org. Biomol. Chem., 2017, 15, 8553 RSC.
- P. Sudhakar, M. Reck, W. Wang, F. Q. He, I. W. Dobler and A. P. Zeng, BMC Genomics, 2014, 15, 362 CrossRef PubMed.
- C. Fu, A. Sikandar, J. Donner, N. Zaburannyi, J. Herrmann, M. Reck, I. Wagner-Döbler, J. Koehnke and R. Müller, Nat. Commun., 2017, 8, 1529 CrossRef PubMed.
- A. E. Solinski, A. M. Scharnow, A. J. Fraboni and W. M. Wuest, ACS Infect. Dis., 2019, 5(8), 1480–1486 CrossRef CAS PubMed.
- C. J. O’Connor, L. Laraia and D. R. Spring, Chem. Soc. Rev., 2011, 40, 4332–4345 RSC.
- S. M. Carlson and F. M. White, Cell Cycle, 2012, 11(10), 1903–1909 CrossRef CAS PubMed.
- J. Welin-Neilands and G. Svensäter, Appl. Environ. Microbiol., 2007, 73(17), 5633–5638 CrossRef CAS PubMed.
- J. A. Lemos and R. A. Burne, Microbiology, 2008, 154(11), 3247–3255 CrossRef CAS PubMed.
- R. G. Quivey Jr., E. J. Grayhack, R. C. Faustoferri, C. J. Hubbard, J. D. Baldeck, A. S. Wolf, M. E. MacGilvray, P. L. Rosalen, K. Scott-Anne, B. Santiago, S. Gopal, J. Payne and R. E. Marquis, Mol. Oral Microbiol., 2015, 30(6), 474–495 CrossRef CAS PubMed.
- J. Abranches, M. M. Nascimento, L. Zeng, C. M. Browngardt, Z. T. Wen, M. F. Rivera and R. A. Burne, J. Bacteriol., 2008, 190(7), 2340–2349 CrossRef CAS PubMed.
- T. K. Kuilya and R. K. Goswami, Org. Lett., 2017, 19, 2366–2369 CrossRef CAS PubMed.
- J. Ammermann, J. Meyer, J. Donner, M. Reck, I. Wagner-Döbler and A. Kirschning, Synthesis, 2023, 1961–1983 CAS.
- D. E. Anderson, J. Cui, Q. Ye, B. Huang, Y. Tan, C. Jiang, W. Zu, J. Gong, W. Liu, S. Y. Kim, B. G. Yan, K. Sigmundsson, X. F. Lim, F. Ye, P. Niu, A. T. Irving, H. Zhang, Y. Tang, X. Zhou, Y. Wang, W. Tan, L.-F. Wang and X. Tan, Proc. Natl. Acad. Sci. U. S. A., 2021, 118(39), e2104759118 CrossRef CAS PubMed.
- C.-C. Bian, Y.-Q. Li, H.-R. Yang and X.-M. Yu, Synthesis, 2023, 3349–3363 CAS.
- H. Zhang, B. Li, H. Yang, Y. Tan, X. Tan and Y. Tang, Org. Lett., 2024, 26, 370–375 CrossRef CAS PubMed.
- S. L. Gellatly and R. E. W. Hancock, Pathog. Dis., 2013, 67, 159–173 CrossRef CAS PubMed.
- A. D. Steele, K. W. Knouse, C. E. Keohane and W. M. Wuest, J. Am. Chem. Soc., 2015, 137, 7314–7317 CrossRef CAS PubMed.
- D. B. Kearns, Nat. Rev. Microbiol., 2010, 8, 634–644 CrossRef CAS PubMed.
- J. Ravel and P. Cornelis, Trends Microbiol., 2003, 11(5), 195–200 CrossRef CAS PubMed.
- I. J. Schalk and L. Guillon, Environ. Microbiol., 2012, 15(6), 1661–1673 CrossRef PubMed.
- T. C. Johnstone and E. M. Nolan, Dalton Trans., 2015, 44, 6320–6339 RSC.
- A. D. Steele, C. E. Keohane, K. W. Knouse, S. E. Rossiter, S. J. Williams and W. M. Wuest, J. Am. Chem. Soc., 2016, 138, 5833–5836 CrossRef CAS PubMed.
- K. W. Knouse and W. M. Wuest, J. Antibiot., 2016, 69, 337–339 CrossRef CAS PubMed.
- C. D. Cox, K. L. Rinehart Jr., M. L. Moore and J. C. Cook Jr., Proc. Natl. Acad. Sci. U. S. A., 1981, 78(7), 4256–4260 CrossRef CAS PubMed.
- K. Schlegel, J. Lex, K. Taraz and H. Budzikiewicz, Z. Naturforsch., 2006, 61(3–4), 263–266 CrossRef CAS PubMed.
- U. Anthoni, C. Christophersen, P. H. Nielsen, L. Gram and B. O. Petersen, J. Nat. Prod., 1995, 58(11), 1786–1789 CrossRef CAS.
- E. S. Sattely and C. T. Walsh, J. Am. Chem. Soc., 2008, 130(37), 12282–12284 CrossRef CAS PubMed.
- W. M. Wuest, E. S. Sattely and C. T. Walsh, J. Am. Chem. Soc., 2009, 131(14), 5056–5057 CrossRef CAS PubMed.
- C. E. Keohane, A. D. Steele, C. Fetzer, J. Khowsathit, D. Van Tyne, L. Moynié, M. S. Gilmore, J. Karanicolas, S. A. Sieber and W. M. Wuest, J. Am. Chem. Soc., 2018, 140, 1774–1782 CrossRef CAS PubMed.
- S. J. Post, C. E. Keohane, L. M. Rossiter, A. R. Kaplan, J. Khowsathit, K. Matuska, J. Karanicolas and W. M. Wuest, ACS Infect. Dis., 2020, 6, 1372–1377 CrossRef CAS PubMed.
- A. R. Mahoney, K. M. Storek and W. M. Wuest, ACS Omega, 2023, 8, 12558–12564 CrossRef CAS PubMed.
- M. Gehringer and S. A. Laufer, J. Med. Chem., 2019, 62(12), 5673–5724 CrossRef CAS PubMed.
- K. Messner, B. Vuong and G. K. Tranmer, Pharmaceuticals, 2022, 15(3), 264 CrossRef CAS PubMed.
- M. M. Golden, C. U. Brzezinski and W. M. Wuest, ChemBioChem, 2025, 26(10), e202401030 CrossRef CAS PubMed.
- A. Tripathi, M. M. Schofield, G. E. Chlipala, P. J. Schultz, I. Yim, S. A. Newmister, T. D. Nusca, J. B. Scaglione, P. C. Hanna, G. Tamayo-Castillo and D. H. Sherman, J. Am. Chem. Soc., 2014, 136(4), 1579–1586 CrossRef CAS PubMed.
- A. Tripathi, M. M. Schofield, G. E. Chlipala, P. J. Schultz, I. Yim, S. A. Newmister, T. D. Nusca, J. B. Scaglione, P. C. Hanna, G. Tamayo-Castillo and D. H. Sherman, J. Am. Chem. Soc., 2014, 136(29), 10541 CrossRef CAS.
- S. Guchait, S. Chatterjee, R. S. Ampapathi and R. K. Goswami, J. Org. Chem., 2017, 82, 2414–2435 CrossRef PubMed.
- S. Paladugu, P. S. Mainkar and S. Chandrasekhar, Tetrahedron Lett., 2017, 58, 2784–2787 CrossRef CAS.
- J. Wu, P. Lorenzo, S. Zhong, M. Ali, C. P. Butts, E. L. Myers and V. K. Aggarwal, Nature, 2017, 547, 436–440 CrossRef CAS PubMed.
- S. Sengupta, M. Bae, D.-C. Oh, U. Dash, H. J. Kim, W. Y. Song, I. Shin and T. Sim, J. Org. Chem., 2017, 82, 12947–12966 CrossRef CAS PubMed.
- A. D. Steele, G. Ernouf, Y. E. Lee and W. M. Wuest, Org. Lett., 2018, 20, 1126–1129 CrossRef CAS PubMed.
- J. R. Thielman, D. H. Sherman and R. M. Williams, J. Org. Chem., 2020, 85, 3812–3823 CrossRef CAS PubMed.
- N. Kim, S. Sengupta, J. Lee, U. Dash, S. Kim, H. J. Kim, C. Song and T. Sim, Eur. J. Med. Chem., 2023, 115592 CrossRef CAS PubMed.
- M. Leitão, A. Gonçalves, F. Borges, M. Simões and A. Borges, Pharmacol. Rev., 2025, 77(2), 100038 CrossRef PubMed.
- Y. Jiang, X. Zhang, H. Nie, J. Fan, S. Di, H. Fu, X. Zhang, L. Wang and C. Tang, Nat. Commun., 2024, 15, 6060 CrossRef CAS PubMed.
- J. Lehmann, M. H. Wright and S. A. Sieber, Chemistry, 2016, 22(14), 4666–4678 CrossRef CAS PubMed.
- I. Hübner, J. Shapiro, J. Hoßmann, J. Drechsel, S. Hacker, P. Rather, D. Pieper, W. M. Wuest and S. A. Sieber, ACS Cent. Sci., 2021, 7(3), 488–498 CrossRef PubMed.
- Y. Liu, M. P. Patricelli and B. Cravatt, Proc. Natl. Acad. Sci. U. S. A., 1999, 96(26), 14694–14699 CrossRef CAS PubMed.
- E. Weerapana, A. E. Speers and B. F. Cravatt, Nat. Protoc., 2007, 2, 1414–1425 CrossRef CAS PubMed.
- L. Bar-Peled, E. K. Kemper, R. M. Suciu, E. V. Vinogradova, K. M. Backus, B. D. Horning, T. A. Paul, T. Ichu, R. U. Svensson, J. Olucha, M. W. Chang, B. P. Kok, Z. Zhu, N. T. Ihle, M. M. Dix, P. Jiang, M. M. Hayward, E. Saez, R. J. Shaw and B. F. Cravatt, Cell, 2017, 171(3), 696–709 CrossRef CAS PubMed.
- D. Kato, K. M. Boatright, A. B. Berger, T. Nazif, G. Blum, C. Ryan, K. A. H. Chahade, G. S. Salcesen and M. Bogyo, Nat. Chem. Biol., 2005, 1(1), 33–38 CrossRef CAS PubMed.
- G. Blum, G. von Degenfeld, M. J. Merchant, H. M. Blau and M. Bogyo, Nat. Chem. Biol., 2007, 3(10), 668–677 CrossRef CAS PubMed.
- K. Garber, Nat. Biotechnol., 2025, 43, 1216–1220 CrossRef CAS PubMed.
- Z. Li, P. Hao, L. Li, C. Y. J. Tan, X. Cheng, G. Y. J. Chen, S. K. Sze, H. M. Shen and S. Q. Yao, Angew. Chem., Int. Ed., 2019, 52, 8551–8556 CrossRef PubMed.
- C. Parker, A. Galmozzi, Y. Wang, B. Correia, K. Sasaki, C. Joslyn, A. Kim, C. Cavallaro, R. Lawrence, S. Johnson, I. Narvaiza, E. Saez and B. Cravatt, Cell, 2017, 168(3), 527–541 CrossRef CAS PubMed.
- D. George, M. Olatunde and J. Tepe, ACS Omega, 2025, 10(4), 3622–3626 CrossRef CAS PubMed.
- C. Chang, A. Mfuh, J. Gao, H. Wu and C. Woo, Tetrahedron, 2018, 74(26), 3273–3277 CrossRef CAS.
- K. Giglio, C. Keohane, P. Stodghill, A. Steele, C. Fetzer, S. A. Sieber, M. J. Filiatrault and W. M. Wuest, ACS Infect. Dis., 2018, 4(8), 1179–1187 CrossRef CAS PubMed.
- B. Moosavi, E. A. Berry, X. L. Zhu, W. C. Yang and G. F. Yang, Cell. Mol. Life Sci., 2019, 76(20), 4023–4042 CrossRef CAS PubMed.
- K. Komatsuya, T. Sakura, K. Shiomi, S. Ōmura, K. Hikosaka, T. Nozaki, K. Kita and D. K. Inaoka, Pharmaceuticals, 2022, 15(7), 903 CrossRef CAS PubMed.
- A. D. Wright, H. Wang, M. Gurrath, G. M. König, G. Kocak, G. Neumann, P. Loria, M. Foley and L. Tille, J. Med. Chem., 2001, 44(6), 873–885 CrossRef CAS PubMed.
- T. K. Wood, S. J. Knabe and B. W. Kwan, Appl. Environ. Microbiol., 2013, 79(23), 7116–7121 CrossRef CAS PubMed.
- H. C. Flemming and J. Wingender, Nat. Rev. Microbiol., 2010, 8, 623–633 CrossRef CAS PubMed.
- A. Scharnow, A. Solinski, S. Rowe, I. Drechsel, H. Zhang, E. Shaw, J. Page, H. Wu, S. A. Sieber and W. M. Wuest, J. Am. Chem. Soc., 2024, 146(33), 23449–23456 CrossRef CAS PubMed.
- P. Le, E. Kunold, R. Macsics, K. Rox, M. C. Jennings, I. Ugur, M. Reinecke, D. Chaves-Moreno, M. W. Hackl, C. Fetzer, F. A. M. Mandl, J. Lehmann, V. S. Korotkov, S. M. Hacker, B. Kuster, I. Antes, D. H. Pieper, M. Rohde, W. M. Wuest, E. Medina and S. A. Sieber, Nat. Chem., 2020, 12(2), 145–158 CrossRef CAS PubMed.
- M. Boersch, S. Rudrawar, G. Grant and M. Zunk, RSC Adv., 2018, 8, 5099–5105 RSC.
- J. M. Colquhoun, C. U. Brzezinski, A. Ji, J. Marotta, F. A. V. Elsen, R. A. Bonomo, K. L. May, S. A. Sieber, M. Grabowicz, W. A. Wuest and P. N. Rather, Proc. Natl. Acad. Sci. U. S. A., 2025, 122(24), e2423650122 CrossRef CAS PubMed.
- C. Ma and S. McClean, Vaccines, 2021, 9(6), 570 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2025 |