
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct decarboxylative 18F-fluorination of benzoic acids using visible light catalysis
Lizeth Y.F.
Haveman
 *,
Cornelis A. J.
Broersen
,
Danielle J.
Vugts
and
Albert D.
Windhorst
*,
Cornelis A. J.
Broersen
,
Danielle J.
Vugts
and
Albert D.
Windhorst
Department of Radiology & Nuclear Medicine, Amsterdam UMC, Vrije Universiteit Amsterdam, De Boelelaan, 1117, Amsterdam, The Netherlands. E-mail: l.haveman1@amsterdamumc.nl
First published on 18th November 2025
Abstract
This study introduces a mild method for the direct decarboxylative 18F-fluorination of (hetero)aromatic carboxylic acids via a photoinduced ligand-to-metal charge transfer (LMCT) process. The method allows for the succesfull 18F-labeling of various benzoic acids (16–40% RCC) and the full-batch preparation of 1-fluoro-4-[18F]fluorobenzene with an overall RCY of 27 ± 2%.
The aryl halide functional group has found widespread application in organic chemistry. In particular, (hetero)aryl fluorides are of importance in drug discovery because of significant improvements in the biological properties, including membrane permeability, in vivo metabolic stability and protein binding affinities.1 This remarkable role of fluorinated molecules is aptly demonstrated by the FDA approval of thirteen aryl fluoride-containing small molecule drugs in the past three years.2 The presence of fluorine in clinically successful small molecule drugs is of great interest for positron emission tomography (PET). Upon substitution of the fluorine atom by its positron emitting fluorine-18 isotope (t1/2 = 110 minutes), a putative PET tracer is obtained without alteration of the original structure. PET enables the non-invasive visualization of physiochemical and pathological processes in vivo at a molecular level and is an essential imaging technology within patient care, clinical research and drug development.
Traditionally, simple aryl [18F]fluorides are mainly accessed via the Balz-Schiemann reaction, the nucleophilic aromatic substitution of a nitro or tri-alkyl ammonium group positioned ortho or para to an electron-withdrawing group, or the Halex process.3 However, the harsh reaction conditions generally limit the reaction scope. Structurally diverse aryl [18F]fluorides have been prepared under milder reaction conditions by Cu-mediated 18F-fluorination of aryl boronates, stannanes, iodides, iodonium ylides and sulfonium salts.3,4 However, accessing aryl [18F]fluorides from native functional groups is still a challenge. Along these lines, benzoic acids are attractive precursors because of their abundance, structural diversity and accessibility from commercial sources. Yet, no chemistry is available for the 18F-fluorination of arenes with benzoic acids as a starting point and, as such, remains an important radiosynthetic challenge (Fig. 1).
 | ||
| Fig. 1 Direct decarboxylative 18F-fluorination of (hetero)aryl carboxylic acids via Cu-LMCT catalysis as a strategy for PET tracer synthesis. | ||
In the organic chemistry fluorodecarboxylation methods rely on poorly selective electrophilic fluorine reagents or metal mediators and often necessitate ortho-substituted substrates and high reaction temperatures of >140 °C (Fig. 1).5,6 Hence, the potential transformation of such strategies into 18F-radiolabeling methods is limited. Moreover, translational difficulties are associated with the slow decarboxylation rate of benzoic acids, conflicting with the short half-life of [18F]fluoride. Recently, several research groups demonstrated that UV light-induced ligand-to-metal charge transfer (LMCT) activation is a powerful and mild approach to an oxygen-centered aryl carboxylate radical (Fig. 1).5–9 This O-centered radical can release CO2 to generate the corresponding aryl radical intermediate, which is then captured by a copper(II) salt to form a high-valent arylcopper(III) fluoride complex. Facile C(sp2)–F reductive elimination from this complex affords the desired aryl fluoride at room temperature.
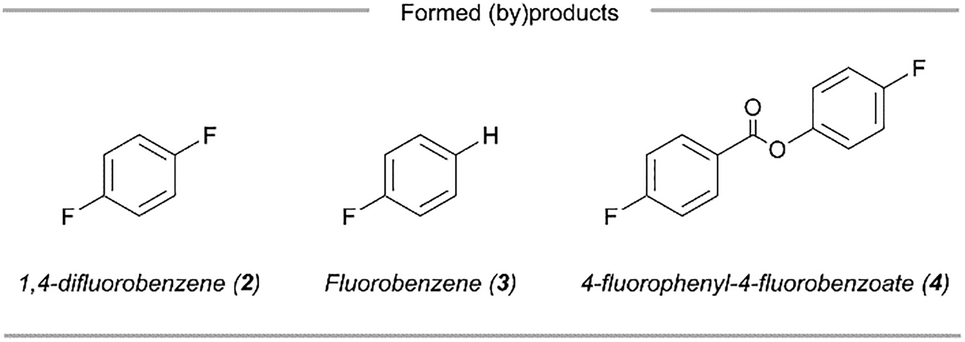
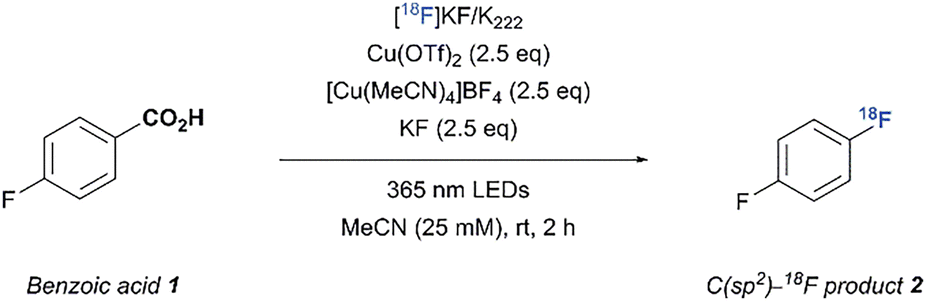
We sought to take advantage of this mild fluorodecarboxylation method to develop a strategy for the direct 18F-fluorination of benzoic acids via the combination of a copper salt, a 365 nm LED light source and a [18F]fluoride source (Fig. 1). However, transitioning from 19F− to [18F]F− changes the stoichiometric conditions of the reaction and therefore necessitated a re-examination of the fluoride source and irradiation method. With 4-fluorobenzoic acid (1) as the aryl carboxylic acid substrate, we were able to obtain 18% of fluorinated adduct ([19F]2) together with the byproducts fluorobenzene (3) and 4-fluorophenyl-4-fluorobenzoate (4) using Cu(OTf)2, [Cu(MeCN)4]BF4 and TBAF·(tBuOH)4 (5) under inert conditions and a reaction time of 6 hours. We attribute the low yield (compared to 70% as reported by Xu et al.) mainly to the absence of a glovebox in the lab, which makes careful handling of the highly air-sensitive copper and fluoride reagents difficult.5 Although the initial yields were relatively low, potassium fluoride (KF) was an effective fluorinating agent in the synthesis of [19F]2 (31 ± 4%). Replacing the 365 nm Kessil lamps with the commercially available “PhotoRedOx Box” photoreactor (365 nm LEDs) resulted in similar conversions (34 ± 1%, see SI for details). Hence, decarboxylative fluorination with KF in a photoreactor was feasible and thus we extended the method to 18F-radiofluorination.
A mixture of 1 (25 µmol), [Cu(MeCN)4]BF4 (2.5 eq), Cu(OTf)2 (2.5 eq), KF (2.5 eq) and [18F]KF/K222 in 1.0 mL MeCN was exposed to 365 nm LED irradiation for 2 h and the desired C(sp2)-18F product ([18F]2) was obtained in 24 ± 6% radiochemical conversion (RCC) (Table 1, entry 1).10,11 While control experiments revealed that the copper(II) salt and purple-light irradiation were all individually necessary (Table 1, entries 2–4), the absence of an inert atmosphere and 30 minutes pre-stirring of the reaction mixture before adding [18F]F− showed no significant effect on the RCC (Table 1, entries 5 and 6). In contrast, drying of the reaction vial equipped with a stir bar, Cu(OTf)2 and KF at 150 °C for 2 hours prior to use was necessary to obtain reliable conversions, since omission resulted in decreased product formation (Table 1, entry 7). As expected water proved to be deleterious to the reaction, as no product was observed when a 9/1 MeCN/H2O stock solution of K2CO3/K222 was used for the trapping (see SI for details). The trapping solution also required a minimum of 2 mM K2CO3 and 4 mM K222 in order to completely transform the triflyl [18F]fluoride to [18F]KF/K222 and prevent diminished conversions (see SI for details). Alternative 18F-sources, e.g. [18F]TBAF also afforded the desired product (Table 1, entry 8), but extensive drying had to be performed in order to completely remove water used in the eluents to ensure good conversions. Surprisingly, more soluble 19F-carriers such as tetramethylammonium fluoride (TMAF) and tetrabutylammonium difluorotriphenylsilicate (TBAT) were not as efficient as KF, presumably due to the strong hygroscopic character of TMAF and fluorodesilylation performed by the difluorotriphenylsilicate ion in TBAT (see SI for details).
|
|
||
|---|---|---|
| Entry | Deviations | RCC |
| a Conditions: 25 µmol scale with 2 h drying of Cu(OTf)2 and 19F-source at 150 °C, Ar sparge and 30 minutes pre-stirring. Radiochemical conversions determined by radio-HPLC, mean ± standard deviation, n = 3. See SI for experimental details. | ||
| 1 | None | 24 ± 6% |
| 2 | No Cu(OTf)2 | 0% |
| 3 | No light | 0% |
| 4 | 455 nm instead of 365 nm | 0% |
| 5 | No Ar sparge | 25 ± 3% |
| 6 | No pre-stirring | 24 ± 3% |
| 7 | No drying of chemicals and vial | 18 ± 2% |
| 8 | [18F]TBAF instead of [18F]KF/K222 | 22 ± 2% |
| 9 | 2.0 eq. Cu(OTf)2, 1.5 eq. [Cu(MeCN)4]BF4 and KF | 30 ± 2% |
| 10 | Entry 9 without [Cu(MeCN)4]BF4 | 24 ± 1% |
| 11 | Entry 9 without KF | 0% |
| 12 | Entry 9 with 4 h irradiation | 46 ± 0% |
| 13 | Entry 9 with 12.5 mM MeCN | 34 ± 3% |
|
||
A survey of the amounts of copper catalysts and KF showed that a reduction in Cu(OTf)2, [Cu(MeCN)4]BF4 and KF equivalents led to an improved RCC of 30 ± 2% (Table 1, entry 9) (see SI for details). Similarly to Xu et al., a slight decrease in RCC was observed when [Cu(MeCN)4]BF4 was removed from the reaction (Table 1, entry 10), however the effect of Cu(I) ability to capture aryl radicals and suppress the formation of the proto-decarboxylated product 3 appeared to be minimal under optimized reaction conditions.5 Hence, the presence of [Cu(MeCN)4]BF4 is not necessary for obtaining high conversions and can be excluded for the purpose of operational simplicity. We observed that the presence of exogenous fluoride anion (F−) proved critical to the overall success of the reaction (Table 1, entry 11). Since in our hypothesized mechanism KF also acts as the base to generate the carboxylate, which then coordinates to copper(II) and readily forms photoactive copper(II) carboxylates, several stoichiometric bases were investigated in order to make the transformation carrier-free. Unfortunately, neither Me4NHCO3 nor K2CO3, DBU or BTMG in the absence of KF were able to provide the desired 18F-adduct (see SI for details).
Although the 18F-fluorodecarboxylation protocol requires rather long reaction times, already after 30 minutes 16 ± 3% product could be observed (see SI for details). Whilst not interesting from a PET imaging perspective, elongating the reaction time to 4 hours resulted in a further increase of the RCC to 46 ± 0% (Table 1, entry 12). Since the yield significantly increases over time, longer reaction times in the presence of low amounts of KF can be considered in case the molar activity is an important goal, however at the dispense of loss of [18F]F− due to the decay.
Lastly, investigating the influence of the precursor concentration identified substrate 1 (12.5 µmol) with a combination of [18F]KF/K222 derived from triflyl [18F]fluoride, Cu(OTf)2 (2.0 eq), KF (1.5 eq) and 365 nm LED light in 1.0 mL MeCN as optimal. After 2 h, the 18F-fluorinated product [18F]2 was obtained in 34 ± 3% RCC (Table 1, entry 13). Notably, an increase in the concentration of precursor to 25 µmol resulted in a significant drop in RCC (24 ± 1%) (see SI for details). This observation suggests the importance of maintaining a balanced amount of substrate for a successful 18F-fluorodecarboxylation. We postulate that in the presence of higher concentrations of precursor, oxidecarboxylation via C–O reductive elimination of the aryl-copper(III) intermediate with 1 is favored over the formation of the desired 18F-adduct, resulting in the formation of 4.
After investigation of the reaction conditions, we explored the substrate scope of aryl decarboxylative 18F-fluorination (Scheme 1). This approach proved generally competent for benzoic acids with electron-donating (6–8, 16–40%), electron-neutral (2, 9 and 10, 34–40%) and electron-withdrawing (11–15, 20–36% RCC) substituents on the aryl ring. Notably, electron-deficient rings with a high oxidative potential are generally known to be problematic for photoinduced radical decarboxylation, but the oxidation-sensitive aldehyde 15 was successfully 18F-fluorinated (26 ± 2%) with this approach. As shown in Scheme 1, para- and meta-substituted aryl carboxylic acids generated 18F-adducts in good yields (2, 6, 7, 9, 11, 12 and 14, 16–40%). Moreover, the efficiency of the reaction was not hindered by ortho substitution on the aryl ring (8, 10 and 15, 25–34%), except in case of the relatively large and strongly electron-withdrawing nitro-group (13, 0%). Lastly, this copper-mediated protocol was effective for the heterocyclic substrate picolinic acid, furnishing the 18F-fluorinated derivative 16, a widely used scaffold in drug discovery, in 30 ± 5% RCC. Due to solubility issues, the pyridine-derived substrates nicotinic and isonicotinic acid were not successfully functionalized. Although we believe that the selected examples show a representative range of electronic and steric demands relevant to bioactive molecules, this method will show limitations inherent to copper-catalysis. We do notice that functionalities strongly coordinating to copper or undergoing rapid redox reactions, e.g. free amines and hydroxyl groups, will generally not be tolerated by this method and do need to be protected. In addition, polar groups like amides and esters and electrophilic groups of synthetic value, e.g. succinimidyl esters, are expected to be tolerated, but with extended reaction times and as a consequence the risk of forming more of the byproducts 3 and 4.
 | ||
Scheme 1 Scope of the direct decarboxylative 18F-fluorination of (hetero)aryl carboxylic acids via photoredox catalysis. Radiochemical conversions were determined by radio-HPLC of the crude mixtures. Reactions were performed at least three times (n = 3) and are given in the form mean ± standard deviation.a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Radiochemical yield, mean ± SD, n = 3. See SI for experimental details. Radiochemical yield, mean ± SD, n = 3. See SI for experimental details. | ||
Finally, all optimization reactions up to this point were carried out by transferring aliquots of a [18F]KF/K222 stock solution to a 4 mL vial containing the reagents. To validate the utility of this transformation on a preparative scale and to determine the overall RCY and molar activity, triflyl [18F]fluoride was synthesized from 3 GBq [18F]fluoride and trapped in a 4 mL vial containing MeCN with K2CO3/K222 and KF. After addition of Cu(OTf)2 and 1, the vial was irradiated for 2 hours at rt. To our delight, 1 was 18F-fluorinated with an overall radiochemical yield of 27 ± 2% and a molar activity of 0.21 ± 0.06 GBq/µmol (Scheme 1, n = 3). The low Am is mainly due to the presence of 1.5 eq. of KF as well as the long reaction time and is an important challenge of our method. However, the full batch application of this new 18F-fluorodecarboxylation protocol emphasizes the potential utility of this copper-mediated protocol for late-stage 18F-applications.
In conclusion, for the first time the direct decarboxylative 18F-fluorination of benzoic acids under mild conditions via photoinduced LMCT has been described. By leveraging a high-valent arylcopper(III) [18F]fluoride intermediate and subsequent C(sp2)–[18F]F reductive elimination, this strategy overcomes the harsh reaction conditions and limited substrate scope that have historically hampered aromatic decarboxylative 18F-fluorination reactions. The method demonstrates broad applicability, efficiently transforming a range of (hetero)aromatic substrates, including electron-deficient, -neutral and -rich aryl rings with para-, meta- and ortho-substituents. Its efficiency and operational simplicity were proven by the successful preparation of 1-fluoro-4-[18F]fluorobenzene on preparative scale in good isolated yield, despite a low Am. Having laid the foundation for a practical and versatile aromatic decarboxylative 18F-fluorination reaction for PET tracer synthesis, future work should focus on improving the molar activity and extending the substrate scope to bioactive compounds. Given the great diversity of benzoic acid precursors and the mildness of the reaction conditions, we expect that this transformation can become a valuable tool in the late-stage 18F-functionalization of drug-like molecules.
Conflicts of interest
There are no conflicts to declare.Data availability
All experimental procedures and data are given in the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5cc03503a.Notes and references
- N. A. Meanwell, J. Med. Chem., 2018, 61, 5822–5880 CrossRef PubMed.
- Food and Drug Administration, New Drugs at FDA: CDER's New Molecular Entities and New Therapeutic Biological Products, https://www.fda.gov/drugs/development-approval-process-drugs/new-drugs-fda-cders-new-molecular-entities-and-new-therapeutic-biological-products, (accessed 18 June 2025).
- S. Preshlock, M. Tredwell and V. Gouverneur, Chem. Rev., 2016, 116, 719–766 CrossRef PubMed.
- T. E. Spiller, K. Donabauer, A. F. Brooks, J. A. Witek, G. D. Bowden, P. J. H. Scott and M. S. Sanford, Org. Lett., 2024, 26, 6433–6437 CrossRef PubMed.
- P. Xu, P. López-Rojas and T. Ritter, J. Am. Chem. Soc., 2021, 143, 5349–5354 CrossRef PubMed.
- T. Q. Chen, P. S. Pedersen, N. W. Dow, R. Fayad, C. E. Hauke, M. C. Rosko, E. O. Danilov, D. C. Blakemore, A. M. Dechert-Schmitt, T. Knauber, F. N. Castellano and D. W. C. Macmillan, J. Am. Chem. Soc., 2022, 144, 8296–8305 CrossRef PubMed.
- Z. He and P. Dydio, Angew. Chem., Int. Ed., 2024, 63, e202410616 Search PubMed.
- N. W. Dow, P. S. Pedersen, T. Q. Chen, D. C. Blakemore, A. M. Dechert-Schmitt, T. Knauber and D. W. C. Macmillan, J. Am. Chem. Soc., 2022, 144, 6163–6172 CrossRef PubMed.
- P. S. Pedersen, D. C. Blakemore, G. M. Chinigo, T. Knauber and D. W. C. MacMillan, J. Am. Chem. Soc., 2023, 145, 21189–21196 CrossRef PubMed.
- H. H. Coenen, A. D. Gee, M. Adam, G. Antoni, C. S. Cutler, Y. Fujibayashi, J. M. Jeong, R. H. Mach, T. L. Mindt, V. W. Pike and A. D. Windhorst, Nucl. Med. Biol., 2017, 55, v–xi CrossRef PubMed.
- M. M. Herth, S. Ametamey, D. Antuganov, A. Bauman, M. Berndt, A. F. Brooks, G. Bormans, Y. S. Choe, N. Gillings, U. O. Häfeli, M. L. James, K. Kopka, V. Kramer, R. Krasikova, J. Madsen, L. Mu, B. Neumaier, M. Piel, F. Rösch, T. Ross, R. Schibli, P. J. H. Scott, V. Shalgunov, N. Vasdev, W. Wadsak and B. M. Zeglis, Nucl. Med. Biol., 2021, 93, 19–21 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2025 |