
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and electronic structure elucidation of bioinspired heterobimetallic nickel complexes†
Claire R.
Patterson
 a,
Paul H.
Oyala
b and
Joshua A.
Buss
*a
a,
Paul H.
Oyala
b and
Joshua A.
Buss
*a
aWillard Henry Dow Laboratory, Department of Chemistry, University of Michigan, Ann Arbor, MI 48109, USA. E-mail: jbuss@umich.edu
bDivision of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, CA 91125, USA. E-mail: phoyala@caltech.edu
First published on 7th July 2025
Abstract
Despite relevance to metalloenzyme active-sites, the selective synthesis of heterobimetallic compounds featuring unbiased, biologically relevant coordination environments remains a challenge. Herein, we disclose a stepwise synthetic strategy, leveraging kinetic stabilization by an alkali metal cation, as a means of accessing spectroscopically pure M(II)/Ni(II) (M = Co & Cu) bimetallics. Electron paramagnetic resonance spectroscopy, variable temperature magnetometry, and DFT calculations support perturbation of the electronic structure as a function of the heterometal within these closely related complexes.
A critical design element underpinning biological catalysis is metallocofactor active site dissymmetry.1 Enzymes leverage subtly distinct coordination pockets,2 electronic asymmetry,3 and disparate metal pairings4 to modulate the functionalization chemistry of inert substrates (Fig. 1A). Whereas so-called “cambialistic” enzymes have evolved to maintain function irrespective of their metallic composition,5 significant differences in reactivity are observed for select metal pairings, even within identical protein environments.6 These observations highlight the importance of elucidating the fundamental interplay between metal identity, electronic structure, and subsequent reactivity; however, general synthetic routes to analytically pure mixed-metal model complexes remain rare.7 Small molecule systems commonly suffer from mixed metalation,8 metallic exchange,9 or rely on biased coordination environments10 that convolute the influence of the ligand versus the metal on the physicochemical properties of the complex.
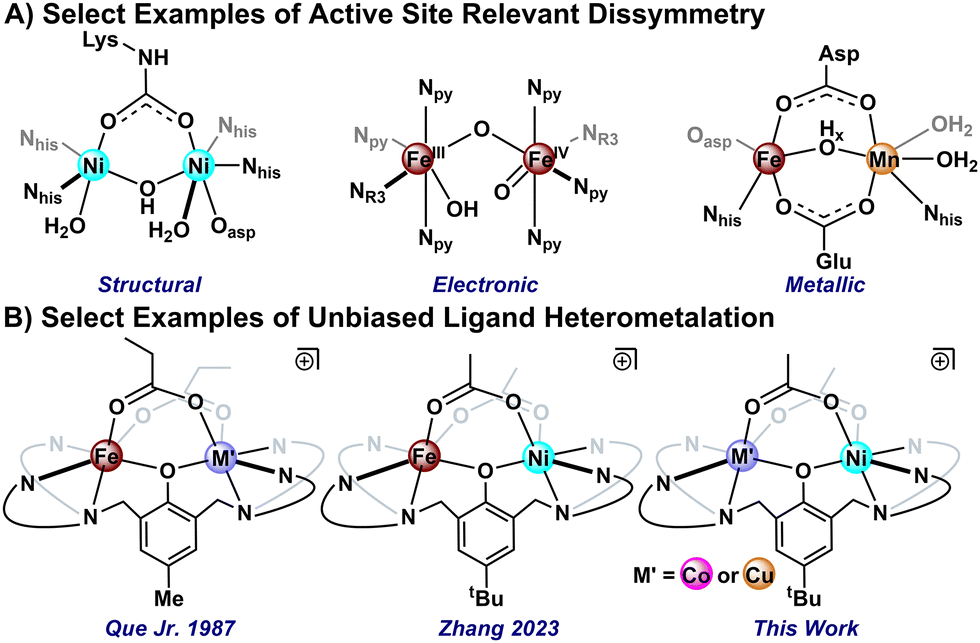 | ||
| Fig. 1 Examples of (A) dissymmetric metallocofactor active sites or models thereof and (B) previously reported M(II)/Fe(III) complexes featuring symmetric ligation. | ||
As an extension of our interest exploring the dinickel chemistry of the symmetric bicompartmental 2,6-bis[(bis(2-pyridylmethyl)amino)methyl]-4-tert-butylphenol (BPtBuP, HL) ligand,11 we sought to access analogues in which Ni(II) is paired in close contact with divalent first-row heterometals. Seminal work by the Que group on the related BPMeP scaffold leverages the Lewis acidity of Fe(III) to sequentially heterometalate the unbiased binding pockets (Fig. 1B).9b,12 More recently, Zhang and coworkers disclosed a route to related BPtBuP-supported Fe(III)/Ni(II) complexes, wherein the metalation order is inverted, installing the nickelous centre prior to deprotonation of the ligand phenol.13 Herein, we disclose a complementary strategy in which stepwise metalation is facilitated by lithium cation binding in a monometalated reaction intermediate, 1. The isolation of 1 facilitates high yielding—and generalizable—routes to analytically pure heterobimetallic complexes both in accord (Co) and contrary (Cu) to the Irving–Williams series.14,15 Notably, these are unprecedented metal pairings in the well-vetted chemistry of the BPRP ligand family.16 Detailed spectroscopic studies reveal disparate physical properties as a function of metal identity, including ferromagnetic exchange coupling in the Co(II) congener.
Recent examples of successful monometalation of symmetric dinucleating ligands13,17 prompted the treatment of LiL with substoichiometric Ni(OTf)2. The resulting pale-blue paramagnet displayed a distinct NMR spectroscopic signature to that of the related dinickel complex and was tentatively assigned as the desired mononickel intermediate. Single crystal X-ray diffraction (SCXRD) studies corroborated the Ni:L stoichiometry, but likewise revealed a five-coordinate lithium cation adjacent to the Ni(II) centre, which fills its coordination sphere via acetonitrile binding (1; Fig. 2). We postulated that the salt-metathesis byproduct formed upon the initial metalation, LiOTf, binds to the adjacent pocket of the BPtBuP ancillary ligand and kinetically inhibits a second nickel binding event. Accordingly, either rapid addition or addition of stoichiometric Ni(OTf)2 predominantly forms the undesired dinickel product (Fig. S12, ESI†).11 These results suggest that substitution of Li+ in 1 with a divalent metal is facile.
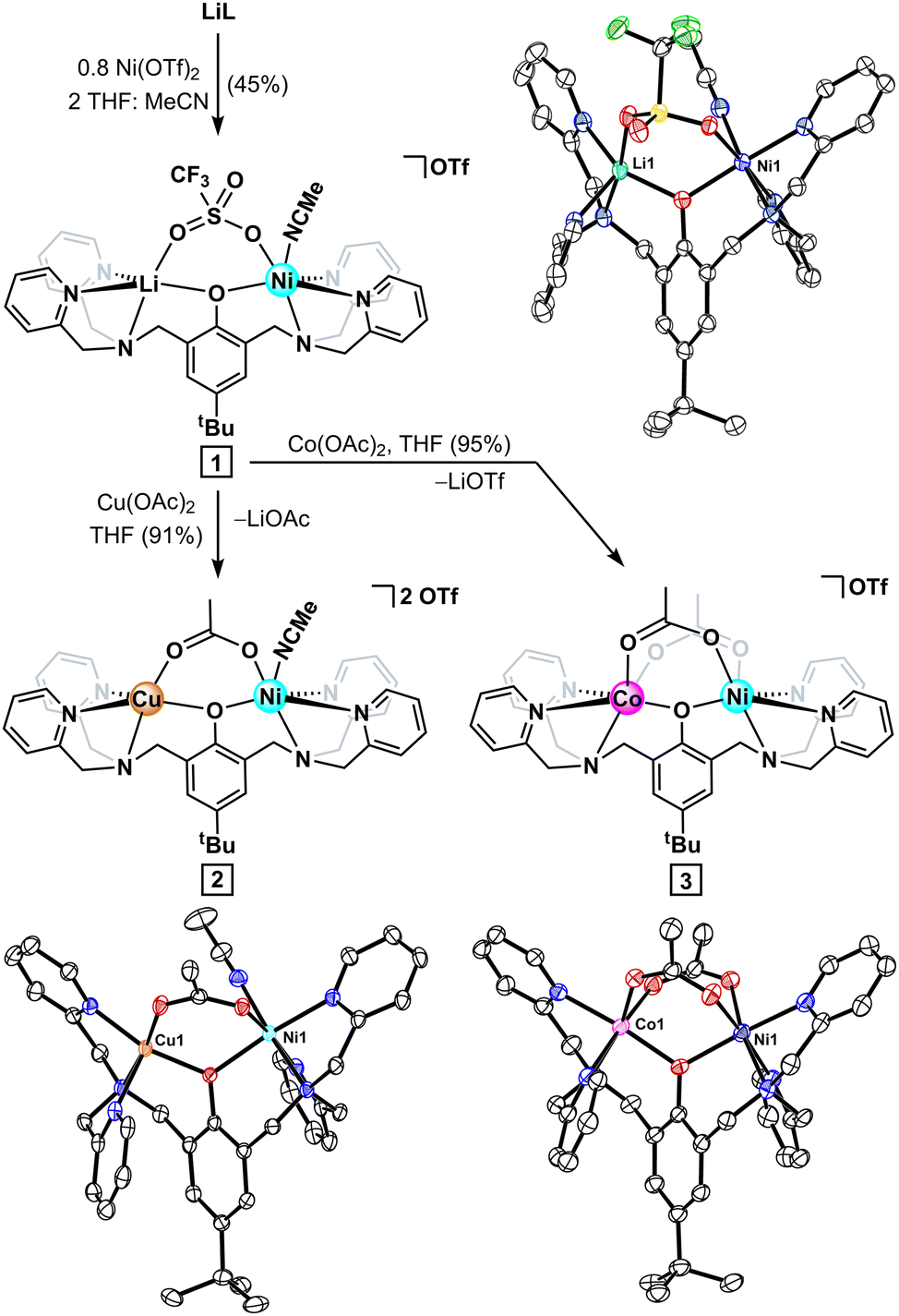 | ||
| Fig. 2 Synthesis and solid-state structures of well-defined LiNi intermediate (1) and heterometallic CuNi (2) and CoNi (3) complexes. Thermal anisotropic displacement ellipsoids are displayed at a 50% probability and both hydrogen atoms and triflate counterions are omitted for clarity. | ||
Leveraging this observation en route to the desired heterometallics, intermediate 1 was converted to either a CuNi (2) or CoNi (3) complex, in excellent yield, via addition of the corresponding M(OAc)2 salt. SCXRD analysis of 2 reveals a doubly bridged dinuclear core with a pseudo square-pyramidal Cu(II) centre (τ5 = 0.28)18 adjacent to a six-coordinate Ni(II). Complex 3 crystallizes as a triply bridged dinuclear core, reminiscent of other BPtBuP-supported bimetallics.13 The metrical parameters of both 2 and 3 are in agreement with related structures supported by this ligand class—the phenolate rings are canted relative to the M–O–Ni plane (ca. 50°) and the M–Oacetate bonds trans to the tertiary amine donor are slightly contracted.9b,12,19 However, the M⋯Ni contacts for 2 and 3 (3.453(1) and 3.429(1) Å, respectively) oppose the trend seen for the heterometal ionic radii (Cu – 0.65 Å, Co – 0.74 Å),20 a consequence of the shorter M–Ophenol bond of the five-coordinate Cu(II). Whereas the similarity of the Co(II) and Ni(II) ions prohibits distinguishing the metals by SCXRD, 3 features a well-resolved 1H NMR spectrum with 29 signals, ranging from −72 to 142 ppm, corroborating the C1 solution symmetry anticipated for the mixed-metal complex. Furthermore, the formulations of both 2 and 3 were corroborated by ESI-MS, which shows no evidence of homobimetallic impurities.
To elucidate the impact of heterometal identity on electronic structure, we first prepared the dinickel(II) homobimetallic control complex, 4. Closely related dinickel species display weak antiferromagnetic coupling.19,21 Accordingly, variable temperature magnetic susceptibility data for 4 corroborate weak antiferromagnetic exchange, with a μeff of 4.49μB at 300 K that attenuates to 3.58μB at 5 K. Fitting the susceptibility data to an isotropic Heisenberg model (H = −2JS1S2) affords a small negative exchange coupling (J = −0.66 cm−1). A low-temperature parallel mode EPR spectrum, with a geff of 7.5 (Fig. S13, ESI†) supports a total spin of ST = 2 at 5 K, consistent with a low-lying quintet spin-coupled excited state.19 No significant signal is observed for 4 in the perpendicular mode spectrum, across a wide range of temperatures (5 to 298 K).
As Kramer's systems, glassed propionitrile solutions of both 2 and 3 were readily probed by CW X-band EPR spectroscopy. For 2, an axial signal with g⊥ = 2.224 and g‖ = 2.140 is observed at 5 K (Fig. 3A), substantiating both a ST = 1/2 spin state (arising from antiferromagnetic coupling between the S = 1/2 Cu(II) and S = 1 Ni(II) centres) and the five-coordinate geometry at Cu observed in the SCXRD structure.22 The spectrum displays an axial hyperfine coupling to 63/65Cu (A = [20, 20, 170] MHz), consistent with significant Cu-centred spin. The most resolved spectra for 3 were obtained at lower temperature (5 K) and display an axial spectrum with effective g-values of g⊥ = 5.29 and g‖ = 2.06 (Fig. 3A), attributable to ferromagnetic coupling between the S = 1 Ni(II) and high-spin (HS) S = 3/2 Co(II) (ST = 5/2). This broad spectrum features a large axial zero-field splitting (D > 1 cm−1) that was well modelled in the simulation with a Gaussian distribution of E/D centred at 0.035 and an applied FWHM linewidth of 0.7. The spectral breadth further suggests weak exchange coupling, a common feature of μ-phenoxy bridged bimetallic cores. For 2 and 3, parallel mode EPR spectroscopy reveals no meaningful integer spin contribution.
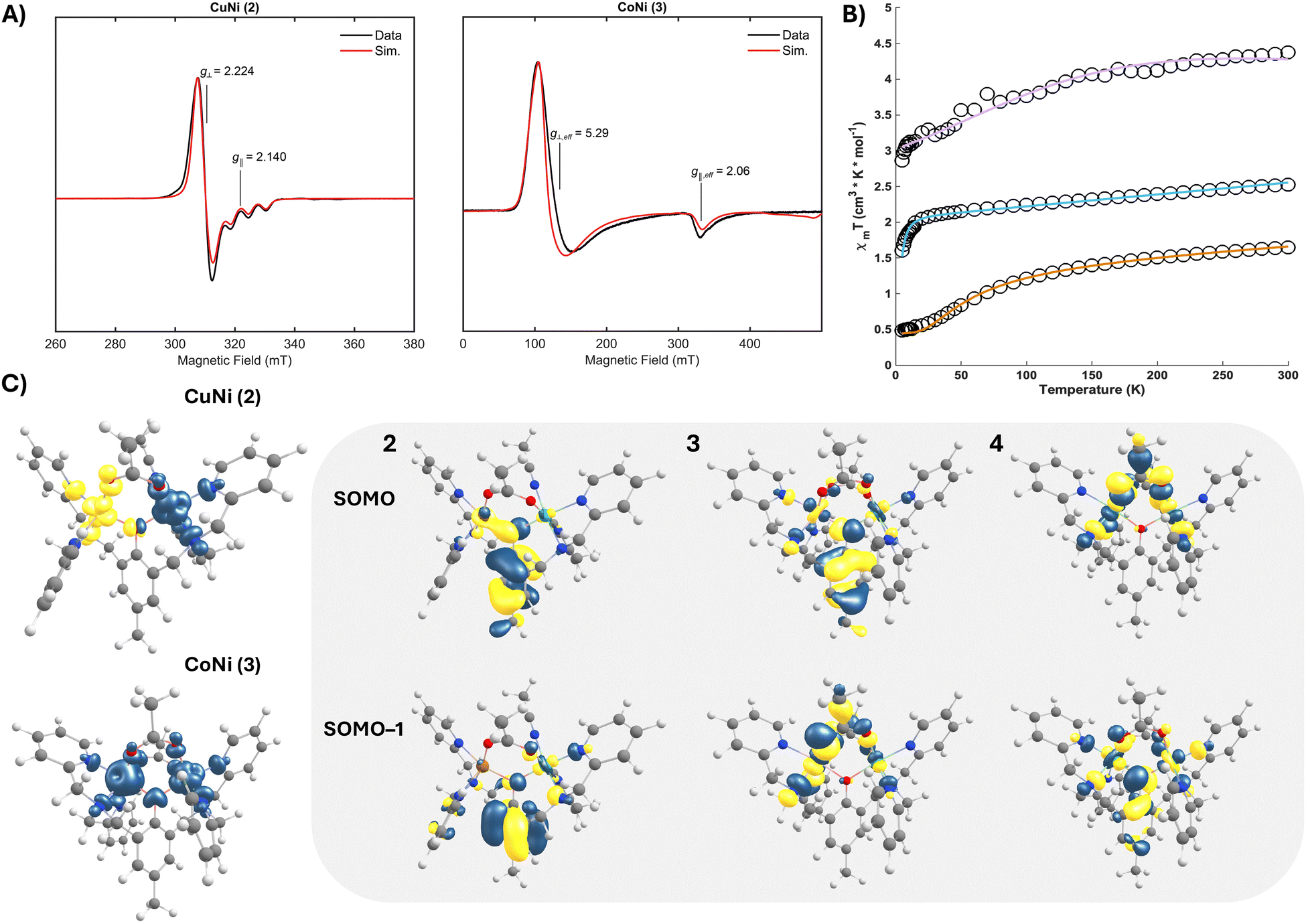 | ||
| Fig. 3 (A) X-band CW-EPR spectra of 2 and 3 (5 K). (B) Temperature-dependent magnetic susceptibility measurements (open circles) and simulated data (coloured lines) for 2 (orange), 3 (purple), and 4 (blue) from 5 to 300 K at B = 1 T. (C) Calculated spin density iso-surfaces (0.004 e− Å−3) and select SOMOs (0.04 e− Å−3) for 2–4. | ||
To corroborate the magnetic exchange coupling properties inferred from the EPR studies, temperature dependant magnetic susceptibility data were collected for polycrystalline samples of both 2 and 3 (Fig. 3B).23 The 5 K μeff value of 1.97μB agrees with an antiferromagnetically coupled system with ST = 1/2 ground state for 2; a higher μeff at 300 K (3.63μB) indicates thermal population of higher spin excited states. In comparison to a closely related dicopper variant,21a exchanging one cuprous centre for Ni(II) inverts the sign of the coupling, reflecting the importance of controlling the magnetic orbitals available for exchange. The measured μeff for 3 increases from 4.78μB at 5 K to 5.92μB at 300 K, which is again consistent with the theoretical spin-only μeff value for a ferromagnetically coupled system with ST = 5/2. The strength of the isotropic magnetic exchange coupling was extracted from fits of the magnetometry data (see ESI†). For 2, the data (J = −21.78 cm−1 and g = 2.2) are in excellent agreement with the EPR spectroscopy and a moderate antiferromagnetic superexchange. The susceptibility data for complex 3 displayed axial ZFS, and the fit parameters (J = 0.1 cm−1, g = 2.09, and D = 4.23 cm−1) corroborate the properties inferred from EPR spectroscopy (Table 1). The comparable exchange couplings observed for 3 and 4 (both near zero) are consistent with both complexes featuring similar magnetic orbital configurations, a phenomenon we sought to explore via broken-symmetry DFT.24
| Complex | g iso(EPR) | g iso(SQUID) | J squid (cm−1) | J comp (cm−1) | ρ M | ρ Ni | D (cm−1) | S T |
|---|---|---|---|---|---|---|---|---|
| 2 | 2.20 | 2.20 | −21.78 | −35.01 | −0.61 | 1.63 | — | 1/2 |
| 3 | 2.09 | 2.09 | 0.1 | 2.27 | 2.71 | 1.66 | 4.23 | 5/2 |
The calculated frontier molecular orbitals from optimized structures of 2–4 (B3LYP(D4)|def2-TZVP(–f)|def2-SVPD) afforded additional insight into both the electronic structures and magnetic properties of these complexes. The spin density iso-surface for 2 indicates unpaired electrons of opposite sign in Cu dx2−y2 and Ni dx2−y2/dz2 parentage orbitals. Both the μ-phenoxy and μ-acetoxy bridges participate in the superexchange pathway, yet the primary contributor is the phenolate (Fig. 3C). Intuitively, the orbital configuration at Ni(II) remains unperturbed across the series; however, both 3 and 4 introduce a second μ-acetate and additional unpaired spin(s) on the adjacent metal. In 3, Co dx2−y2 and dx2−y2/dz2 parentage orbitals dominate the SOMOs, engaging in exchange through both the phenolate and acetate linkages. A near-analogous bonding situation is observed for 4, but with the relative orbital energies inverted. The remaining unpaired spin on the Co centre of 3 appears in the dxy orbital, orthogonal to the phenolate but well-suited for overlap with both acetates. Computed exchange couplings mirror the signs and relative magnitudes of the experimental values for 2 and 3 (Table 1). The mixed-ligand bridges result in counter-complimentarity of the magnetic orbitals,25 decreasing the antiferromagnetic contribution to the total exchange parameter and ultimately affording the weak ferromagnetic coupling observed for 3. In this way, metal pairings can tune the spin state of the bimetallic, a property correlated to reactivity in related systems.4b
The foregoing results demonstrate a rational synthetic route to analytically pure mixed-metal complexes supported by an unbiased ancillary ligand environment. Through a Lewis-acid stabilized intermediate (1), selective heterometalations both contra (Cu – 2) and pro (Co – 3) the Irving–Williams series have been achieved in high-yield (>90%).15 These metal pairings represent novel members of a growing family of mixed-metal BPRP-type bimetallics. These complexes provide a platform to investigate structure/property relationships as an exclusive function of metal identity. Here, EPR spectroscopy, magnetometry, and computation were employed to probe electronic differences stemming from metal differentiation. Ongoing work is expanding this series of heterobimetallics and further evaluating heterometal effects on electronic structure, redox properties, and small molecule reactivity.
This work was supported by the University of Michigan and the NSF (XRD Instrumentation – CHE-0840456). The Caltech EPR facility is supported by the Beckman Institute and the Dow Next Generation Educator Fund. We thank Dr Eugenio Alvarado for NMR spectroscopy expertise, Dr Fengrui Qu for assistance with SCXRD, and Dr Hayley L. Lillo for XRD data of 4. Dr Reza Loloee (MSU) and Prof. John Berry (UW Madison) aided with acquisition and analysis of the magnetism data, respectively. Dr Mukunda Mandal is gratefully acknowledged for fruitful DFT discussions. Roy Wentz aided in the design and fabrication of custom glassware that made this work possible.
Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data have been deposited in the CCDC (2455269–2455272)—https://www.ccdc.cam.ac.uk/structures.Conflicts of interest
There are no conflicts to declare.Notes and references
- C. Belle and J.-L. Pierre, Eur. J. Inorg. Chem., 2003, 4137 CrossRef CAS.
- (a) L. Mazzei, F. Musiani and S. Ciurli, J. Biol. Inorg. Chem., 2020, 25, 829 CrossRef CAS PubMed; (b) D. E. Wilcox, Chem. Rev., 1996, 96, 2435 CrossRef CAS PubMed; (c) R. H. Holm, P. Kennepohl and E. I. Solomon, Chem. Rev., 1996, 96, 2239 CrossRef CAS PubMed.
- (a) R. Than, A. A. Feldmann and B. Krebs, Coord. Chem. Rev., 1999, 182, 211 CrossRef; (b) B. E. Sturgeon, D. Burdi, S. Chen, B.-H. Huynh, D. E. Edmondson, J. Stubbe and B. M. Hoffman, J. Am. Chem. Soc., 1996, 118, 7551 CrossRef CAS.
- (a) S. Yoshikawa and A. Shimada, Chem. Rev., 2015, 115, 1936 CrossRef CAS PubMed; (b) G. Xue, R. De Hont, E. Münck and L. Que, Nat. Chem., 2010, 2, 400 CrossRef CAS PubMed; (c) N. Sträter, T. Klabunde, P. Tucker, H. Witzel and B. Krebs, Science, 1995, 268, 1489 CrossRef PubMed; (d) J. A. Tainer, E. D. Getzoff, J. S. Richardson and D. C. Richardson, Nature, 1983, 306, 284 CrossRef CAS PubMed.
- (a) R. Valenti, J. Jabłońska and D. S. Tawfik, Protein Sci., 2022, 31, e4423 CrossRef CAS PubMed; (b) A. Barwinska-Sendra, Y. M. Garcia, K. M. Sendra, A. Baslé, E. S. Mackenzie, E. Tarrant, P. Card, L. C. Tabares, C. Bicep, S. Un, T. E. Kehl-Fie and K. J. Waldron, Nat. Commun., 2020, 11, 2738 CrossRef CAS PubMed; (c) M. Schmidt, B. Meier and F. Parak, J. Biol. Inorg. Chem., 1996, 1, 532 CrossRef CAS.
- (a) C. Liu, G. Rao, J. Nguyen, R. D. Britt and J. Rittle, J. Am. Chem. Soc., 2025, 147, 2148 CrossRef CAS PubMed; (b) C. Liu, M. M. Powell, G. Rao, R. D. Britt and J. Rittle, J. Am. Chem. Soc., 2024, 146, 1783 CrossRef CAS PubMed; (c) Y. Kutin, R. Kositzki, R. M. M. Branca, V. Srinivas, D. Lundin, M. Haumann, M. Högbom, N. Cox and J. J. Griese, J. Biol. Chem., 2019, 294, 18372 CrossRef CAS PubMed.
- M. Jarenmark, H. Carlsson and E. Nordlander, C. R. Chim., 2007, 10, 433 CrossRef CAS.
- (a) A. L. Poptic, Y.-P. Chen, T. Chang, Y.-S. Chen, C. E. Moore and S. Zhang, J. Am. Chem. Soc., 2023, 145, 3491 CrossRef CAS PubMed; (b) S. J. Tereniak, R. K. Carlson, L. J. Clouston, V. G. Young, Jr, E. Bill, R. Maurice, Y.-S. Chen, H. J. Kim, L. Gagliardi and C. C. Lu, J. Am. Chem. Soc., 2014, 136, 1842 CrossRef CAS PubMed.
- (a) W. D. Kerber, J. T. Goheen, K. A. Perez and M. A. Siegler, Inorg. Chem., 2016, 55, 848 CrossRef CAS PubMed; (b) T. R. Holman, Z. Wang, M. P. Hendrich and L. Que, Inorg. Chem., 1995, 34, 134 CrossRef CAS.
- For select examples, see: (a) A. Nicolay and T. D. Tilley, Chem. – Eur. J., 2018, 24, 10329 CrossRef CAS PubMed; (b) C. Pathak, S. K. Gupta, M. K. Gangwar, A. P. Prakasham and P. Ghosh, ACS Omega, 2017, 2, 4737 CrossRef CAS PubMed; (c) Y. Sano, N. Lau, A. C. Weitz, J. W. Ziller, M. P. Hendrich and A. S. Borovik, Inorg. Chem., 2017, 56, 14118 CrossRef CAS PubMed; (d) R. J. Eisenhart, P. A. Rudd, N. Planas, D. W. Boyce, R. K. Carlson, W. B. Tolman, E. Bill, L. Gagliardi and C. C. Lu, Inorg. Chem., 2015, 54, 7579 CrossRef CAS PubMed; (e) L. J. Clouston, R. B. Siedschlag, P. A. Rudd, N. Planas, S. Hu, A. D. Miller, L. Gagliardi and C. C. Lu, J. Am. Chem. Soc., 2013, 135, 13142 CrossRef CAS PubMed; (f) A. R. Paital, J. Ribas, L. A. Barrios, G. Aromí and D. Ray, Dalton Trans., 2009, 256 RSC.
- H. L. Lillo and J. A. Buss, Chem. Commun., 2024, 60, 8549 RSC.
- (a) T. R. Holman, C. Juarez-Garcia, M. P. Hendrich, L. Que, Jr and E. Munck, J. Am. Chem. Soc., 1990, 112, 7611 CrossRef CAS; (b) A. S. Borovik, L. Que, V. Papaefthymiou, E. Muenck, L. F. Taylor and O. P. Anderson, J. Am. Chem. Soc., 1988, 110, 1986 CrossRef CAS.
- H.-T. Zhang, Y.-H. Guo, Y. Xiao, H.-Y. Du and M.-T. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202218859 CrossRef CAS PubMed.
- H. Irving and R. J. P. Williams, J. Chem. Soc., 1953, 3192 RSC.
- For a more detailed discussion, refer to the ESI†.
- To our knowledge, the only ligand-symmetric M(II)/Ni(II) (M = Co or Cu) complexes are: (a) R. A. Lal, K. Arvind, S. Ibanphylla and S. D. Kurbah, J. Coord. Chem., 2017, 70, 2722 CrossRef CAS; (b) U. Turpeinen, R. Hämäläinen and J. Reedijk, Polyhedron, 1987, 6, 1603 CrossRef CAS.
- P. Ríos, M. S. See, O. Gonzalez, R. C. Handford, A. Nicolay, G. Rao, R. D. Britt, D. K. Bediako and T. D. Tilley, Chem. Commun., 2024, 60, 8912 RSC.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349 RSC.
- T. R. Holman, M. P. Hendrich and L. Que, Jr., Inorg. Chem., 1992, 31, 937 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., 1976, A32, 751 CrossRef CAS.
- (a) S. S. Massoud, C. C. Ledet, T. Junk, S. Bosch, P. Comba, R. Herchel, J. Hošek, Z. Trávníček, R. C. Fischer and F. A. Mautner, Dalton Trans., 2016, 45, 12933 RSC; (b) H. Adams, D. Bradshaw and D. E. Fenton, Inorg. Chim. Acta, 2002, 332, 195 CrossRef CAS; (c) C. Pathak, D. Kumar, M. K. Gangwar, D. Mhatre, T. Roisnel and P. Ghosh, J. Inorg. Biochem., 2018, 185, 30 CrossRef CAS PubMed; (d) T. Koga, H. Furutachi, T. Nakamura, N. Fukita, M. Ohba, K. Takahashi and H. Ōkawa, Inorg. Chem., 1998, 37, 989 CrossRef CAS; (e) S. Uozumi, H. Furutachi, M. Ohba, H. Ōkawa, D. E. Fenton, K. Shindo, S. Murata and D. J. Kitko, Inorg. Chem., 1998, 37, 6281 CrossRef CAS; (f) D. Volkmer, A. Hörstmann, K. Griesar, W. Haase and B. Krebs, Inorg. Chem., 1996, 35, 1132 CrossRef CAS PubMed; (g) R. M. Buchanan, M. S. Mashuta, K. J. Oberhausen, J. F. Richardson, Q. Li and D. N. Hendrickson, J. Am. Chem. Soc., 1989, 111, 4497 CrossRef CAS; (h) P. Chaudhuri, H.-J. Küppers, K. Wieghardt, S. Gehring, W. Haase, B. Nuber and J. Weiss, J. Chem. Soc., Dalton Trans., 1988, 1367 RSC.
- E. Garribba and G. Micera, J. Chem. Educ., 2006, 83, 1229 CrossRef CAS.
- S. S. Massoud, M. Spell, C. C. Ledet, T. Junk, R. Herchel, R. C. Fischer, Z. Trávníček and F. A. Mautner, Dalton Trans., 2015, 44, 2110 RSC.
- P. Comba, S. Hausberg and B. Martin, J. Phys. Chem. A, 2009, 113, 6751 CrossRef CAS PubMed.
- (a) L. Gutierrez, G. Alzuet, J. A. Real, J. Cano, J. Borrás and A. Castiñeiras, Inorg. Chem., 2000, 39, 3608 CrossRef CAS PubMed; (b) L. K. Thompson, S. S. Tandon, F. Lloret, J. Cano and M. Julve, Inorg. Chem., 1997, 36, 3301 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2455269–2455272. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5cc03435c |
| This journal is © The Royal Society of Chemistry 2025 |