
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOvercoming fundamental challenges of the electrochemical nitrogen oxidation reaction
Aditya
Prajapati†
 *ab,
Alexandra
Zagalskaya†
*c,
Natalie
Hwee
ab,
Eric
Krall
d,
Jonathan T.
Davis
bd,
Sneha A.
Akhade
ab and
Jeremy T.
Feaster
*ab
*ab,
Alexandra
Zagalskaya†
*c,
Natalie
Hwee
ab,
Eric
Krall
d,
Jonathan T.
Davis
bd,
Sneha A.
Akhade
ab and
Jeremy T.
Feaster
*ab
aMaterials Science Division, Lawrence Livermore National Laboratory, Livermore, CA 94550, USA. E-mail: prajapati3@llnl.gov; feaster2@llnl.gov
bLaboratory for Energy Applications for the Future (LEAF), Lawrence Livermore National Laboratory, Livermore, CA 94550, USA
cDepartment of Chemical Engineering, University of Massachusetts Amherst, Amherst, MA 01003, USA. E-mail: azagalskaya@umass.edu
dMaterials Engineering Division, Lawrence Livermore National Laboratory, Livermore, CA 94550, USA
First published on 18th November 2025
Abstract
Nitric acid and nitrates play a crucial role in modern agriculture and medicine; however, their commercial production through the Haber–Bosch and Ostwald processes is energy-intensive and generates significant greenhouse gas emissions. Additionally, the rapidly growing global population is expected to significantly increase food demand in the coming decades. Therefore, there is a pressing need to explore sustainable alternatives for synthetic fertilizer production to meet increasing global crop demands while reducing the carbon footprint. The electrochemical nitrogen oxidation reaction (NOR) presents a viable alternative which produces nitrates using renewable electricity. However, NOR is still a nascent reaction that has not been studied extensively, and several key challenges must be addressed as the field develops. In this feature article review, we highlight the set of effective approaches that we have employed for studying this complex reaction and overcoming these challenges, as well as discuss future directions for advancing NOR technology.
 Aditya Prajapati | Dr Aditya Prajapati is a Principal Investigator and Research Scientist in the Materials Science Division at Lawrence Livermore National Laboratory (LLNL). He received his PhD in Chemical Engineering from University of Illinois at Chicago in 2022. With a multidisciplinary approach encompassing electrochemistry, materials engineering, catalysis, advanced manufacturing, Multiphysics modeling, and AI/machine learning, Dr Prajapati's research aims to design energy-efficient systems for electrochemical CO2 reduction, N2 oxidation, and specialty chemical electro-synthesis to improve performance, durability, and scalability. |
 Alexandra Zagalskaya | Dr Alexandra Zagalskaya is an Assistant Professor in the Department of Chemical Engineering at the University of Massachusetts Amherst, where she began her faculty appointment in January 2025. She previously served as a postdoctoral researcher in the Quantum Simulations Group at Lawrence Livermore National Laboratory from 2022 to 2025, working with Dr Tuan Anh Pham. She received her PhD in Materials Engineering from the University of Nebraska-Lincoln in 2022, under the guidance of Dr Vitaly Alexandrov. Her research focuses on the development of advanced models to capture realistic behavior of the electrochemical interfaces. |
 Natalie Hwee | Natalie Hwee is a Staff Scientist in Materials Science Division at Lawrence Livermore National Laboratory (LLNL). Her research interests include electrochemical nitrogen oxidation and CO2 reduction. She also works on the integration of additive manufacturing in reactor design and development of materials. Natalie received her master's in chemistry from University of Oregon in 2022. |
 Eric Krall | Eric Krall is a Staff Scientist in the Materials Science Division at Lawrence Livermore National Laboratory (LLNL), focusing on the intersection of electrochemistry and polymer science. Experienced in physical vapor deposition (PVD), thin film characterization, porous membranes, and electrocatalysis; responsible for material development and characterization for many electrochemistry projects. He received his master's in chemistry (with a focus in polymer science) from the University of Oregon in 2022. |
 Jonathan T. Davis | Dr Jonathan Davis is a Staff Scientist with the Materials Engineering Division at Lawrence Livermore National Laboratory (LLNL). In 2024, he served as an Embassy Science Fellow in Reykjavik, Iceland, advising on strategies for electrochemical hydrogen offtake. He also completed his postdoctoral training at LLNL (2022). Prior to joining the lab, he received his PhD in chemical engineering from Columbia University (2019) studying under Prof. Daniel Esposito. His bachelor's degree is in chemical engineering from The Ohio State University (2014). His research interests include additive manufacturing, reactor design, and transport processes in electrochemical systems. |
 Sneha A. Akhade | Dr Sneha Akhade currently serves as the Hydrogen Lead at the Laboratory for Energy Applications for the Future (LEAF) and is a Staff Scientist in the Materials Sciences Division at Lawrence Livermore National Laboratory. Dr Akhade earned her PhD in Chemical Engineering from Pennsylvania State University in 2016 and MS in Chemical Engineering from Carnegie Mellon University in 2011 and held a prior postdoctoral position at Pacific Northwest National Laboratory. At LLNL, Dr Akhade leads several multiscale modeling efforts in hydrogen carriers for storage and delivery infrastructure, reactive carbon capture, sustainable ammonia production and biomass alcohol upgrading for sustainable plastic production. She works at the intersection of several domains with partner national laboratories, start-ups, and academic institutions and has over 50 peer-reviewed publications, 3 patents and over 40 conference talks. |
 Jeremy T. Feaster | Dr Jeremy T. Feaster is a Principal Investigator and Research Staff Scientist in the Materials Science Division at Lawrence Livermore National Laboratory. At LLNL, his research teams use electrochemical engineering and advanced manufacturing to create new reactors and processes that transform air into fertilizer, remove impurities from wastewater sources, and convert waste carbon into fuels and chemicals. Dr Feaster earned his MS and PhD in Chemical Engineering from Stanford University after completing his BS in Chemical and Biomolecular Engineering at Georgia Tech. He also serves as the executive director for the Jeremy T. Feaster Foundation, Inc., a 501(c)(3) nonprofit organization whose mission is “to amplify a culture of lift as you climb.” Over the past 13 years, the Feaster Foundation has awarded over $20 |
Sustainable nitrate production via nitrogen oxidation
Nitrates (NO3−), the salts of nitric acid, are vital components in many areas of life, including modern agriculture, industry, and medicine.1 The increased demand for nitric acid over the last decade is primarily dictated by the fertilizer industry,2 as most synthetic fertilizers are nitrogen-based compounds. As of 2022, global nitrogen fertilizer consumption reached approximately 110 million metric tons per year.3 In the U.S. alone, fertilizer usage increased by 5.4% between 2022 and 2023, due to the expanding cultivation of cereals and grains. The U.S. nitrogenous fertilizer market was valued at USD 8.82 billion in 2023 and is projected to grow to $15.72 billion USD by 2032, emphasizing the need for more nitrate-based fertilizers in the present and future.4The production of commercial nitric acid today involves a complex, two-step industrial process. First, nitrogen gas (N2) is separated from air, combined with hydrogen gas (H2), then reacted over an iron-based catalyst under high temperatures (400–500 °C) and pressures (100–200 atm) to produce ammonia (NH3) through the Haber–Bosch process. Consequently, NH3 is then oxidized into nitric oxide (NO) over a platinum–rhodium catalyst at 900 °C, then reacted with oxygen and water to form nitric acid. Overall, these two processes are primarily used to produce nitrogen-based fertilizers, which consume ∼3% of the world's energy demand,5 as well as contributes to ∼1.5% of global greenhouse gas emissions, generating about 2.6 tons of carbon dioxide equivalent for every ton of fertilizer produced.6
Motivated to develop alternative, sustainable routes of nitrate production, several approaches have been explored. The current state of the art is indirect in that one must first fully reduce N2 to NH3, then fully oxidize NH3 to form NO3−. Direct oxidation of N2 to nitrates could provide a more energy-efficient path for fertilizer production. Yet, this reaction is incredibly slow at atmospheric pressures due to high stability of N2 in our atmosphere. Various strategies, including biological,7 plasma8 and lightning-based9 approaches, have all been explored. However, most of these attempts are plagued by sluggish kinetics at ambient conditions and often require extreme operating conditions to achieve practical nitrate production rates.
The electrochemical nitrogen oxidation reaction (NOR) for producing nitric acid or nitrates demonstrates significant potential as an environmentally friendly method for fertilizer production under ambient conditions. This approach utilizes nitrogen directly from air, eliminating the need for pre-synthesized ammonia as a chemical feedstock. The modular nature of NOR enables localized, small-scale nitrate production, reducing reliance on centralized industrial infrastructure and long-distance transport. Moreover, this technology aims to bypass multiple unit operations needed for ammonia synthesis and subsequent oxidation to nitric acid.
While the NOR holds promise for sustainable fertilizer production, limited research has been conducted on studying this reaction. Several groups of catalysts have been investigated, including metal oxides,10–14 thin films,15,16 mixed metal materials,17–20 and functionalized metals.19,21,22 However, most reports demonstrate extremely low concentrations of nitrate (<0.1 mM) and molar production rates (<1 µmol NO3−/h).18,23 As a result, there is a lack of clarity on the viability of this reaction, let alone how to perform NOR experiments or simulations in an accurate and reproducible manner. This stems from several major challenges that make the NOR uniquely difficult to investigate and that must be overcome if one is to successfully synthesize nitrate from N2.
We have identified four fundamental challenges that have severely limited the ability to perform NOR reliably (Fig. 1). The first challenge of NOR lies in the activation of N2. The N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N triple bond is one of the strongest, and innovative approaches are needed to develop catalysts and reaction conditions that can effectively weaken and break this robust bond at mild conditions. The second fundamental challenge for NOR is the complex pathway of reactions that lead to a decrease in selectivity to nitrate, as well as lower overall selectivity of the NOR compared to water splitting. The equilibrium potential for the NOR is within 0.1 V of the equilibrium potential for the oxygen evolution reaction (OER), which presents a challenge to selectively drive the NOR without simultaneously promoting the OER and resulting in reduced efficiency and increased energy consumption. The third challenge for NOR is the hindered mass transport of the reactant gas to the catalyst surface. This is primarily due to the low solubility of N2 in water at standard pressure and temperature that restricts the amount of N2 available to the catalyst surface in aqueous media. The fourth challenge for NOR is NOR product detection, quantification and validation. Electrochemical nitrogen chemistries are susceptible to false positives due to contaminants.23 Since NOR can result in both gas and liquid products, it is of chief importance to establish rigorous experimental protocols that account for these impurities and also capture the full spectrum of reaction products.
N triple bond is one of the strongest, and innovative approaches are needed to develop catalysts and reaction conditions that can effectively weaken and break this robust bond at mild conditions. The second fundamental challenge for NOR is the complex pathway of reactions that lead to a decrease in selectivity to nitrate, as well as lower overall selectivity of the NOR compared to water splitting. The equilibrium potential for the NOR is within 0.1 V of the equilibrium potential for the oxygen evolution reaction (OER), which presents a challenge to selectively drive the NOR without simultaneously promoting the OER and resulting in reduced efficiency and increased energy consumption. The third challenge for NOR is the hindered mass transport of the reactant gas to the catalyst surface. This is primarily due to the low solubility of N2 in water at standard pressure and temperature that restricts the amount of N2 available to the catalyst surface in aqueous media. The fourth challenge for NOR is NOR product detection, quantification and validation. Electrochemical nitrogen chemistries are susceptible to false positives due to contaminants.23 Since NOR can result in both gas and liquid products, it is of chief importance to establish rigorous experimental protocols that account for these impurities and also capture the full spectrum of reaction products.
 | ||
| Fig. 1 Schematic showing the four fundamental challenges that must be overcome to unlock NOR: NN bond stability, selectivity of NOR over OER, mass transport of N2 to the catalyst surface, and detection, quantification and validation of NOR products. | ||
This feature article describes our approach to each of the aforementioned technical challenges in detail. We begin by examining the intrinsic limitations in nitrogen activation, then delve into the foundational reaction pathways and determine the appropriate key figures of merit when evaluating NOR experiments. We then discuss strategies to enhance conversion and selectivity efficiency through reactor engineering and mechanistic determination of catalyst behavior. Advances in mass transport engineering are explored as a potential solution to nitrogen solubility and diffusion constraints. Finally, protocols on product quantification and validation are highlighted, emphasizing the importance of high-sensitivity analytical tools and contamination control. Altogether, this article outlines a holistic framework for studying NOR effectively while addressing these four fundamental challenges, integrating experimental and theoretical approaches and identifying strategies and opportunities for overcoming technical gaps and challenges in the field of electrochemical NOR.
Challenge 1: activation of the N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e002.gif) N triple bond
N triple bond
It is no surprise that performing NOR has been difficult given the incredible stability of the reactant N2 molecule. The NN triple bond of the nitrogen molecule (N2) is one of the strongest covalent bonds in nature, with a bond dissociation energy of 941 kJ mol−1. The exceptional strength of the NN bond presents a significant kinetic barrier to activation, rendering nitrogen chemically unreactive under standard conditions. Atomistic scale modeling using density functional theory (DFT) calculations can give us insight into how to approach activating N2, as well as elucidate the reaction mechanism for the NOR.
Accurately modeling the initial adsorption and activation of N2 is crucial yet challenging. It requires highly precise electronic structure methods. Anand et al.24 explored the uncatalyzed conversion of N2 to HNO3 using the VASP and Gaussian09 computational packages. Their study included benchmarking commonly used functionals such as B3LYP, M06, M06-2X, and WB97XD in Gaussian, as well as PBE and RPBE in VASP. The latter is known to provide the best treatment of adsorption properties on solid surfaces. They also examined the effects of Grimme's D3 dispersion correction and implicit solvation on the computational results. However, the oxidation of nitrogen is a complex electrochemical process that takes place at the electrode–electrolyte interface, where the interplay between dynamic molecular interactions, local electric fields, and solvation effects critically governs the reaction pathways and kinetics. To accurately capture these phenomena, computational modeling of the electrochemical interface needs a highly detailed and nuanced approach. Conventional models often fall short in representing the inherent complexity of the interface, particularly the influence of the electric double layer and the role of explicit solvation.
The whole reaction pathway is thermodynamically uphill and requires a remarkably high limiting potential of 3.23 V vs. RHE to become plausible, highlighting the critical need for the development of highly efficient catalysts to drive and facilitate this reaction.24 The initial step involves the adsorption of N2 onto the catalyst surface, a process strongly influenced by the intrinsic properties of the catalyst, including its chemical composition, crystallographic orientation (Fig. 2A), surface coverage (Fig. 2B), and the applied potential (Fig. 2C). Following adsorption, N2 undergoes oxidation on the catalyst surface to form N2O. For the reaction to proceed efficiently, the binding strength of both intermediates to the catalyst surface must be carefully balanced. Specifically, the intermediates must be bound strongly enough to prevent desorption as N2(g) or N2O(g) into the solution but not so strongly as to inhibit subsequent oxidation steps leading to the formation of NxOy species. Fig. 2C highlights the importance of identifying the structures that are representative of experimental conditions, serving as a basis for detailed and comprehensive analysis. This approach is essential for minimizing the frequently observed discrepancies between simulations and experimental results.
 | ||
| Fig. 2 (A) N2/N2O binding on stoichiometric PtO2 low index facets. (B) N2/N2O scaling relationship computed at different surface coverages. The insets represent the surface structures of PtO2(100). (C) Computational surface phase diagram of PtO2(100) in aqueous media. A cyclic voltammetry curve on PtO2 at a scan rate of 50 mV s−1 is also shown. Reproduced with permission from ref. 27. | ||
It is important to note that while thermodynamic free energy profiles provide valuable insights into the energetics of the initial and final states, greater emphasis should be placed on the high-energy transition states and the reaction kinetics. The accurate modeling of these processes is further complicated by the necessity of incorporating potential dependence into simulations. Among current quantum chemical approaches, grand-canonical nudged elastic band (NEB) simulations and ab initio molecular dynamics (AIMD) show significant promise for predictive accuracy. However, these methods are computationally demanding. Addressing these aspects is crucial for understanding and overcoming the kinetic bottlenecks that govern the overall reaction pathway.
To achieve a more realistic representation, it is essential to incorporate explicit solvent molecules and ionic species into simulations. These components not only shape the local electric field distribution but also dynamically modulate the stability and reactivity of intermediate species through solvation effects and ion-pairing interactions. However, the inclusion of explicit solvation significantly increases the computational cost and complexity of the models, as it requires accounting for large-scale atomistic configurations, long-range electrostatic interactions, and time-dependent changes under applied potential. Advanced methodologies, such as constant-potential AIMD in conjunction with enhanced sampling techniques, are therefore required to probe the intricate dynamics of the interface and accurately predict the energetics and mechanisms of nitrogen oxidation reactions.
Challenge 2: complex mechanistic pathways and competing oxygen evolution reaction (OER)
The next challenge is a direct consequence of nitrogen's poor solubility. In aqueous electrochemical systems, water molecules dominate the environment. Consequently, the oxygen evolution reaction (OER) naturally competes with NOR for the active sites on the catalyst. The preferential occurrence of OER over NOR is driven by the higher availability of water molecules compared to dissolved nitrogen. This parasitic OER significantly hinders the observation and efficiency of nitrogen conversion at the catalyst surface, making it difficult to achieve selective NOR.Fig. 3 illustrates the intricate reaction network associated with NOR, encompassing both chemical and electrochemical steps, as well as the potential products arising from nitrogen oxidation. This reaction network for NOR in aqueous solution shows the number of transferred electrons for various nitrogen species as they are progressively increased from the lowest oxidation state 0 in molecular nitrogen, N2, to the highest oxidation state +5 in nitric acid, HNO3. As dashed arrows indicate, most reaction intermediates can be formed via proton-coupled electron transfer (PCET) steps and then undergo chemical transformations to targeted HNO3. Furthermore, Table 1 provides a comprehensive list of possible anodic electrochemical reactions, detailing the corresponding free energy changes and the number of electrons transferred per reaction. Understanding the key factors that govern the formation of desired products is therefore crucial for optimizing NOR selectivity and advancing nitrogen electrochemistry.
 | ||
| Fig. 3 Overview of electrochemical (solid arrows) and chemical (dashed arrows) nitrogen oxidation reactions based on oxidation state. Reproduced with permission from ref. 27. | ||
| Anode electrochemical reaction | E 0 (vs. RHE) (V) | # e/product |
|---|---|---|
| N2 + H2O → N2O + 2e− + 2H+ | +1.77 | 2 |
| N2 + 2H2O → 2NO + 4e− + 4H+ | +1.68 | 2 |
| N2 + 4H2O → 2HNO2 + 6e− + 6H+ | +1.48 | 3 |
| N2 + 3H2O → N2O3 + 6e− + 6H+ | +1.48 | 6 |
| N2 + 4H2O → 2NO2 + 8e− + 8H+ | +1.36 | 4 |
| N2 + 4H2O → N2O4 + 8e− + 8H+ | +1.36 | 8 |
| N2 + 6H2O → 2NO3− + 10e− + 12H+ | +1.31 | 5 |
| N2 + 5H2O → N2O5 + 10e− + 10H+ | +1.35 | 10 |
| 2H2O → O2 + 4e− + 4H+ | +1.23 | 4 |
| 2H2O → H2O2 + 2e− + 2H+ | +1.76 | 2 |
In the recent years, researchers have focused on noble metals and their oxides, as well as transition metal materials, as potential NOR catalysts. Recent theoretical studies have sought to elucidate the mechanisms and intermediates involved in NOR, while also examining their competition with the OER. These investigations are primarily focused on identifying catalysts that can selectively enhance NOR. For instance, TiO2, PdO2 and SnO2, known for weaker *O adsorption, was suggested as potential candidates for NOR.24,25 The NOR mechanisms, activity, and selectivity of various phases, including rutile (110) and anatase (100) and (101), were investigated on metal oxides.26 The findings highlighted the anatase PtO2(100) facet, covered by a monolayer of *OH, as a promising catalyst with an estimated nitrate faradaic efficiency of 62.8%. In our computational analysis, the PtO2(100) facet was identified as exhibiting the strongest adsorption for both *N2 and *N2O species. We also highlight that the reaction intermediates may involve lattice oxygen as a result of N–N bond cleavage, and the role of defects on the reaction energetics needs further investigation.27
Defect engineering was investigated as a promising way to activate nitrogen.15,28 The enhanced NOR performance was demonstrated on oxygen vacancy-enriched niobium oxide nanobelts (Au–Nb2O5−x),29 promoting N2 adsorption on oxygen vacancy sites. Faradaic efficiency (FE) of 6.70% at 1.36 V and a maximum yield of 767.92 µg h−1 mgcat−1 at 1.44 V were obtained on cation vacancy-rich RuO2 in sulfuric acid electrolyte,13 and DFT calculations suggested the cooperative behavior of OER intermediates in the rate-determining step of *NN(OH) formation. Doping 2.79 wt% Ru into TiO2 structure showed the accelerated *N2 conversion to *NO due to the upshift of d band center in Ru sites.15 Further, over-doping of Ru ions resulted in formation of RuO2 on the TiO2 surface, forming active sites for dominant OER, thus suppressing NOR. Similarly, the positive impact on nitrogen electrooxidation was found in Ru-doped Pd nanoparticles (Pd0.9Ru0.1),10 compared to pristine Pd and Ru metal catalysts. Tensile strain was suggested as a strategy to facilitate NOR activity in PdO2.11 The synergetic effect was demonstrated for Ru-nanoclusters-coupled Mn3O412 led to approximately 2 eV reduction in the kinetic barrier for the rate-determining step of *NN(OH) formation, compared to Mn3O4. This resulted in FE of 28.87% and NO3− yield of 35.34 µg h−1 mgcat−1 at 1.6 and 2.0 V, respectively. A series of ZnFexCo2−xO4 spinel oxides17 were studied for NOR performance, and the optimal ZnFe0.4Co1.6O4 composition demonstrated the production of approximately 130 µg h−1 g−1 of nitrates at 1.6 V vs. RHE, with average current densities around 0.04 mA cm−2 in an open beaker three-electrode setup. DFT calculations performed on the identified catalyst, along with ZnFe2O4 and ZnCo2O4, reveal lower free energy barriers, indicating a synergistic interaction between Fe and Co atoms. Specifically, Fe plays a crucial role in facilitating the formation of the first N–O bond on the *N site, while Co contributes to stabilizing the adsorbed OH− species, thereby enabling the subsequent formation of the second and third N–O bonds. Similarly, atomically dispersed Fe atoms on N-doped carbon nanosheet achieved 35.63% nitrate production at a rate of 2.45 µmol cm−2 h−1 at 2.1 V vs. RHE, with an average current density of 0.2 mA cm−2 in an H-cell.30 In addition to these surface-mediated electrochemical mechanisms, alternative pathways have recently been explored in which reactive oxygen species act as intermediates for N2 activation. Dong et al.31 demonstrated an indirect NOR route where *OH radicals generated in situ oxidize N2. This decouples N2 activation from the conventional OER competition. Such radical-mediated mechanisms introduce a different approach to NOR where the local environment variable such as radical lifetime becomes just as important as the catalyst surface itself. Along similar lines, electro-Fenton-like processes as a means of generating *OH radicals have also been reported to drive NOR.32–34 While these studies remain at a formative stage as well, they certainly broaden the mechanistic landscape and further highlight the diversity of pathways through which NOR might be achieved. In the framework of addressing the complexity of this reaction network, our work used DFT and AIMD in combination with enhanced sampling techniques to show that under anodic bias the OH-terminated PtO2(100) surface binds N2 and N2O most strongly, with calculated free-energy barriers favoring chemical *N2O formation over direct electrochemical water attack. Guided by these predictions and our isotope labeling experiments using 15N2, we confirmed a clear signal for NO visa mass spectrometry and N2O via FTIR, unambiguously confirming N–N bond cleavage via a *N2O intermediate. This approach probes into deconvoluting the competing electrochemical and chemical pathways- and yet, it represents only an initial foray into the complex mechanistic landscape of NOR.
Challenge 3: mass transport of nitrogen to the catalyst surface in aqueous solutions
The third major challenge is the slow mass transport of nitrogen gas due to its poor solubility in water. N2 is a stable, nonpolar molecule with a high vapor pressure, which leads to its low solubility in water (20 mg L−1) at standard temperature and pressure (STP). Additionally, the traditional H-cell or flow cell configurations used in aqueous electrochemistry – where N2 gas is dissolved into the electrolyte and is expected to diffuse to the catalyst layer – results in large (∼100 µm) diffusion boundary layers,35,36 which only exacerbates this challenge. We can calculate the limiting current density, which represents the maximum current that can be passed due to mass transport limitations, using the following equation: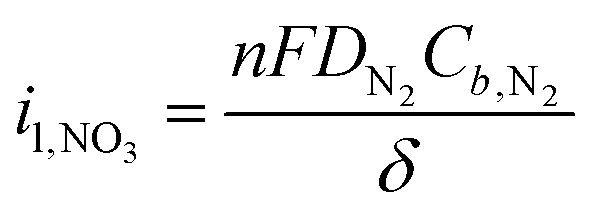 | (1) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1), DN2 is the diffusivity of N2 in water (2 × 10−5 cm2 s−1), Cb,N2 is the bulk concentration of N2 in water (<1 mM), and δ is the hydrodynamic diffusion boundary layer defined by the electrochemical reactor geometry. If we assume that each molecule of NO3− requires 5 electrons (Table 1) and the boundary layer in a traditional H-cell is 100 µm, we arrive at a limiting current density of 0.689 mA cm−2. This limitation poses a significant barrier to achieving the desired reaction rates and efficiencies in aqueous environments. Furthermore, as the vast majority of the NOR field to date has been conducted in these types of electrochemical cells, it is unsurprising that only very small amounts of nitrate have been detected and the NOR has been difficult to validate.
485 C mol−1), DN2 is the diffusivity of N2 in water (2 × 10−5 cm2 s−1), Cb,N2 is the bulk concentration of N2 in water (<1 mM), and δ is the hydrodynamic diffusion boundary layer defined by the electrochemical reactor geometry. If we assume that each molecule of NO3− requires 5 electrons (Table 1) and the boundary layer in a traditional H-cell is 100 µm, we arrive at a limiting current density of 0.689 mA cm−2. This limitation poses a significant barrier to achieving the desired reaction rates and efficiencies in aqueous environments. Furthermore, as the vast majority of the NOR field to date has been conducted in these types of electrochemical cells, it is unsurprising that only very small amounts of nitrate have been detected and the NOR has been difficult to validate.
Gas diffusion electrodes (GDEs) play a pivotal role in addressing the transport limitations of conventional electrochemical systems, particularly in reactions like NOR, where low gas solubility and diffusivity significantly constrain reaction rates.37 GDEs enable direct gas-phase delivery of reactants such as N2 to the catalyst interface, bypassing the slow diffusion and large boundary layers of H-cell and flow cell designs. This approach ensures a higher local concentration of nitrogen at the active sites, facilitating improved reaction kinetics and enhanced selectivity.38
In addition to improving mass transport, GDEs provide an optimal environment for tuning the three-phase boundary, where gas, liquid, and solid interfaces intersect. GDEs can help maximize the utilization of active sites and minimizing undesired side reactions, such as OER. The integration of GDEs into vapor-fed reactor architectures has demonstrated remarkable advancements in achieving high current densities for NOR. We utilized the first such use of a GDE-based reactor in our previous work27 in order to overcome the abovementioned limitations (Fig. 4). This reactor design exhibits higher catalytic activity compared to the conventional planar or non-hydrophobic porous catalysts that rely on the availability of dissolved N2 in the electrolyte. This design not only mitigates the issues of low gas solubility but also enables better control of operating conditions, such as reactant flow rates and pressure. As NOR research advances, the continued development and optimization of GDEs will be instrumental in bridging the gap between laboratory-scale discoveries and industrial-scale applications, paving the way for a sustainable nitrogen economy.
 | ||
| Fig. 4 Our experimental design of a NOR system includes several features to improve NO3− production, including (1) gas flow compartment for N2 or air to enter the reactor, (2) GDE design for the anode to improve diffusion of N2 to the catalyst surface, (3) small volume of electrolyte to increase surface area to volume ratio, leading to increased NO3− detection, (4) membrane-less design to avoid nitrate impurities, and (5) hydrogen-producing GDE cathode with a separate gas flow channel to quantify cathode products and ensure no nitrate is being consumed. Reproduced with permission from ref. 27. | ||
The advent of advanced manufacturing (AM) has introduced a paradigm shift in the landscape of reactor design across the domain of electrocatalysis.39 Specifically, incorporating AM in the design and fabrication of reactors can potentially help bridge the gaps highlighted in this article. AM offers significant advantages in optimizing reactor design to enhance mass transport of reactant species, either by fine-tuning the reactor geometry or by introducing efficient flow pathways in stagnant-electrolyte cells. Moreover, the capability for rapid prototyping through AM can address challenges related to reactor variability across different research groups by accelerating the design and standardization of NOR electrochemical reactors (Fig. 5). A notable example of AM's potential is demonstrated by Corral et al.,40 who used custom printing to create an electrochemical reactor capable of efficiently converting CO2 to ethylene at high current densities. AM devices have accelerated reaction engineering across multiple domains: in water oxidation, AM-enabled microfluidic reactors tailor local flow fields and improve oxygen bubble removal, significantly boosting reaction efficiency.41,42 These systems allow precise control over local hydrodynamics, which is critical for mitigating concentration gradients and enhancing reaction rates. In biomass electrooxidation, AM-fabricated reactors have been used to optimize the electrode–electrolyte interface integrating compact design with lower electrolyte volume to rapidly assess the intrinsic kinetics of lignocellulosic intermediates;43 for methane-to-methanol synthesis, biocatalytic polymers within a silicon lattice were created using AM exhibited enhanced gas transport pathways that significantly improve product yields.44
 | ||
| Fig. 5 Rapid and informed iteration of electrochemical reactors via AM offer significant advantages in reactor design, including sub-millimeter control over reactor features, manufacturing times on the scale of hours, not weeks, straightforward ability to introduce complex parts and features into the reactor design, and two orders of magnitude cheaper manufacture compared to conventional approaches. Reproduced with permission from ref. 35. | ||
Such efforts underscore the importance of AM in enhancing and understanding reactor kinetics, thus contributing to the development of new electrochemical technologies. These advancements are essential for building a fundamental understanding of electrocatalytic processes and for driving innovation in reactor design, ultimately leading to more efficient and scalable electrochemical systems.
Advancements in catalyst design, reaction engineering, and system optimization are essential for translating laboratory-scale findings into scalable and sustainable NOR processes, ultimately contributing to a greener chemical industry. While notable progress has been made in developing NOR catalysts, overcoming the limitations of current experimental setups is crucial for accurate evaluation of performance. Addressing these challenges requires innovative approaches to activate the N–N triple bond under mild conditions, enhance nitrogen solubility and transport in aqueous systems, and suppress competing reactions like OER. Future research should focus on improving nitrogen solubility and transport in electrochemical systems and developing more controlled reactor environments to ensure accurate measurement of catalytic activities.
Just as efficient transport of N2 to the catalyst interface is essential for enabling NOR, the downstream management of the products also presents a challenge. In most aqueous systems, the NOR systems might generate a mixture of nitrates, nitrites, and possible short-lived NOx species, rather than just pure nitric acid. Direct collection of HNO3 has not yet been realized under ambient electrochemical conditions and achieving this will likely require coupling downstream separation processes such as ion-exchange, capacitive deionization, or membrane-based nitrate concentration. Each of these processes introduce additional energy and cost penalties that call for an integrated reactor-separator design. While the future research initially would focus on improving the transport of N2 in these electrochemical systems, consequently, addressing separation and purification is also essential for NOR to become a practical alternative to the Ostwald process.
Challenge 4: NOR product detection, quantification and validation
NOR is a complex reaction that follows multiple pathways, therefore understanding the conditions that lead to specific products can significantly improve the efficiency and selectivity of the reaction. Fig. 3 illustrates the possible products of both the chemical and electrochemical anodic reactions. Notably, the thermodynamic equilibrium potentials for these reactions range between two critical 4e− and 2e− water oxidation reactions: oxygen (O2) at +1.23 V vs. RHE and hydrogen peroxide (H2O2) at +1.77 V vs. RHE at 25 °C and 1 atm, respectively. Thus, accurately detecting and quantifying the products is crucial for a comprehensive understanding of N2 oxidation.Establishing robust experimental protocols
Current literature on experimental NOR predominantly employs open beaker or H-cell electrochemical reactors. These conventional reactors have been instrumental in studying fundamental electrocatalysis but can be vulnerable to atmospheric and reactor component nitrate contamination, potentially leading to overestimation of catalytic activities. This issue is reminiscent of challenges faced in the electrochemical nitrogen reduction (NRR) field and therefore calls for a need to develop robust experimental protocols to avoid similar pitfalls as we learned from the NRR research community.In a recent study by Dong et al.,31 they showcase robust experimental methodologies, including systematic catalyst synthesis, detailed structural characterization, and advanced operando techniques such as in situ electrochemical Raman spectroscopy and differential electrochemical mass spectrometry (DEMS). These methods ensure precise monitoring of reaction intermediates and products, minimizing ambiguities associated with side reactions or environmental contaminants. For instance, the careful calibration of nitrogen species via15N-labeled experiments and comprehensive ion chromatography validates the reported catalytic performance with high confidence.
Such rigorous practices are critical in electrochemical research to avoid over-interpretation or misinterpretation of results. By eliminating confounding variables and accurately quantifying reaction outcomes, researchers can reliably correlate observed performance metrics with underlying catalytic phenomena. This not only bolsters scientific credibility but also lays a robust foundation for scaling laboratory findings to practical applications. Future advancements in electrochemical nitrogen oxidation will greatly benefit from the adoption of similarly stringent experimental protocols.23,45,46 Given the wide range of potential NOR products, it is crucial to establish robust experimental protocols to confidently support mechanistic hypotheses. While we acknowledge that having access to a variety of characterization and quantification equipment can be expensive and time-consuming, the lack of certain facilities should not hinder researchers from sharing their ideas. However, in such cases, it is recommended that researchers carefully bound their conclusions based on the available resources to best accelerate scientific discoveries in this nascent field of NOR.
One of the fundamental requirements for aqueous NOR is the quantification of the parasitic oxygen evolution reaction (OER) using a gas chromatography (GC) instrument. This is considered the bare minimum for product analysis, as it provides quantitative insight into the OER faradaic efficiency (FE). If researchers find that the FE does not total 100% and if neither the catalyst nor the electrolyte are degrading, they can attribute the remaining FE to NOR. This approach (as seen in Fig. 6) offers a quick and preliminary way to screen electrochemical systems for NOR activity, facilitating the identification of promising candidates for further investigation.
 | ||
| Fig. 6 Detailed schematic of the experimental setup for electrochemical NOR. Reproduced with permission from ref. 27. | ||
To truly probe into the mechanistic insights of NOR, further analytical techniques should be employed. Ion chromatography can determine the concentration of nitrates produced, while NMR with N-15 isotope labeling, and mass spectrometry combined with FTIR can detect short-lived intermediates. These advanced techniques will deepen our understanding of the NOR process, driving innovation and efficiency in electrochemical nitrogen oxidation.
Importance of N-15 isotope labeling
N-15 isotope labeling is crucial for understanding the role of potential contaminants in electrochemical nitrogen oxidation (NOR). This technique involves using N-15 enriched N2 gas instead of the common N-14. The electrochemical reaction products can then be analyzed with standard techniques, and NMR can be employed to detect N-15 based stable NOR products.Nitrogenous impurities are easily misinterpreted in experiments, leading to erroneous conclusions about the source of nitrogen in the reaction products. Isotopically labeled nitrogen is an excellent method to eliminate such misinterpretations. Since the only source of N-15 in the system is the supplied N-15 N2 gas, any N-15 based NOR products detected can be confidently attributed to the supplied gas. If the reaction products originate from other nitrogen-based impurities present in the reactor, N-15 based NOR products should not be detected. This method provides a robust check to confirm that the NOR activity is predominantly derived from the gaseous N2 supplied and not from contaminants. By using N-15 isotope labeling, researchers can ensure the accuracy of their results, leading to a more reliable understanding of the NOR process and its potential for practical applications.
Mass spectrometry
Mass spectrometry is a vital tool in the arsenal of advanced analytical techniques used for electrochemical NOR. Its quick response time provides valuable information about the intermediates formed during NOR, whether they are stable or short-lived, up to a certain temporal resolution. This capability is crucial for gaining deeper insights into reaction pathways by identifying the intermediates. By elucidating the formation and consumption of intermediates, mass spectrometry helps researchers understand the mechanistic details of NOR, ultimately contributing to the optimization and efficiency of the process.Importance of FTIR
In our study, Fourier-transform infrared (FTIR) spectroscopy played a crucial role in identifying and quantifying nitrogen-containing products formed during the electrochemical nitrogen oxidation reaction (NOR). By capturing high-resolution spectra of gas-phase nitrogen oxides, FTIR enabled precise differentiation among species such as NO, NO2, N2O, and nitrate intermediates. This level of detail provided direct insights into reaction pathways, aiding in the validation of catalytic activity and selectivity.FTIR is an indispensable tool in electrochemical NOR due to its ability to offer non-invasive, real-time monitoring of gaseous and aqueous products. Unlike traditional techniques, FTIR allows for the simultaneous analysis of multiple reaction products, making it highly effective for studying complex reaction networks. Furthermore, when paired with isotopic labeling (e.g., 15N), FTIR enhances mechanistic understanding by enabling tracking of nitrogen atoms through the reaction sequence.
By integrating FTIR into the analytical workflow, researchers can ensure accurate product identification and minimize ambiguities in product quantification. This robust methodology is vital for advancing the development of NOR catalysts and optimizing reaction conditions, ultimately contributing to the design of sustainable nitrogen conversion processes.
As depicted in Fig. 7, for our published work,27 we assembled a NOR screening platform integrating DFT and AIMD insights with a vapor-fed reactor, online FTIR, isotope-labeling, and GDE-based mass-transport control—to elucidate and validate the coupled chemical/electrochemical pathways of nitrogen oxidation. By utilizing computation and experiment in a single workflow, we established what we think is a rigorous methodology for NOR benchmarking and mechanistic deconvolution. Future efforts can build on this foundation to probe solvent effects, catalyst–electrolyte interactions, and alternative reaction pathways that remain unexplored.
 | ||
| Fig. 7 A comprehensive diagram depicting the essential equipment for screening, evaluating, and benchmarking electrochemical systems for NOR ensuring the implementation of best practices in this emerging field. Reproduced with permission from ref. 27. | ||
Advancements in nitrate production for electrochemical nitrogen oxidation
The field of electrochemical NOR is still in its infancy and faces several experimental challenges that need to be addressed for commercial scalability. In this section, we review recent advancements in NOR, focusing on nitrate production rates, and discuss the implications for future research and development.Comparative analysis of nitrate production rates
We compare the nitrate production rate of our electrochemical system with previous NOR reports. As highlighted in Fig. 8, our system27 produces nitrates at an order of magnitude higher than recent literature, demonstrating a considerable advancement in the field. Despite these promising outcomes, electrochemical nitrogen oxidation remains in its early stages and has yet to be scaled for commercial implementation. For this technology to be commercially viable in fertilizer production, the nitrate concentration must meet specific requirements. Based on the nitrogen content in typical fertilizers, we estimate that an acceptable range for nitrates in liquid-distributed fertilizers is between 14–35 mM. Our previous work showcases the potential of engineering the electrode–electrolyte-reactor system to achieve these higher concentrations of nitrate, moving closer to practical application. Fig. 8 and Table 2 provide a comparative analysis of our nitrate production rate with those reported in the literature. Our system achieves the highest rate of nitrate production to date, underscoring its significance. While reporting high FE is crucial in this emerging field, it is not the sole metric for success. Low production rates can hinder the commercial realization of an electrochemical process. Therefore, achieving both high FE and high production rates is essential for practical applications. While FE remains a key performance indicator, NOR's intertwined chemical and electrochemical pathways may make single-product FE reports potentially misleading. Because of this, our work reported the cumulative FE across all nitrogen-oxide products. Electrochemical NOR FE can be approximated as 100% – OER FE; however, the field is not yet mature enough to quantitatively decouple electrochemical and purely chemical contributions. We therefore encourage the community to develop and adopt pathway-resolved decoupling methods and to report electrochemical and chemical efficiencies separately if possible, fostering more robust and reliable benchmarks that will accelerate NOR's practical advancement. | ||
| Fig. 8 Comparison of the performance of our work with previously reported NOR experiments as a function of applied potential. Reproduced with permission from ref. 27. | ||
| Catalyst | Applied potential (V vs. RHE) | Rate of nitrate production (×10−8 mol h−1) |
|---|---|---|
| ZnFexCO(2−x)O4 | 1.60 | 6.41 |
| Ru0.1Pd0.9O2 | 1.70 | 7.46 |
| PdO2 | 1.75 | 17.67 |
| Ru/TiO2 | 1.80 | 3.14 |
| Pd/MXene | 2.03 | 1.96 |
| Fe/SnO2 | 1.97 | 27.33 |
| Ru–Mn3O4 | 2.00 | 11.23 |
| B13C2 | 2.20 | 38.32 |
| Pt plate | 2.37 | 12.00 |
| RuO2 | 1.44 | 63.94 |
| Our work | 2.47 | 300 |
The findings from our work lay the groundwork for further exploration and optimization in the field of electrochemical nitrogen oxidation. Translating NOR performance from laboratory demonstrations to commercially scalable production will require overcoming challenges associated with selectivity, product separation, reactor durability, and standardized benchmarking. By addressing the current challenges and employing reactor design, electrode–electrolyte-reactor engineering, and advanced analytical techniques, future research can achieve higher efficiencies and production rates, moving closer to commercial viability. This progress is essential for developing scalable and sustainable NOR processes, ultimately contributing to a greener chemical industry.
Perspective and outlook
This feature article highlights recent advancements in NOR research while identifying gaps that must be bridged to achieve commercial viability. By focusing on a multidisciplinary approach that combines experimental innovation, computational insights, and reactor engineering, the NOR field is poised to deliver groundbreaking solutions for sustainable fertilizer production. Ultimately, advancing NOR technology is not just a scientific challenge but a critical step toward achieving global sustainability goals and fostering a greener future. We suggest the following future directions to bridge the existing knowledge gaps and accelerate the development of scalable NOR systems.To establish a robust feedback loop for catalyst screening in the NOR process, an iterative framework integrating computational predictions, experimental validation, and data-driven optimization is essential. High-throughput screening methods enable the exploration of expansive chemical and compositional spaces, facilitating the rapid identification of promising NOR catalysts while deriving fundamental design principles for further development. Key catalyst properties, such as adsorption energies, reaction barriers, and thermodynamic stability, can be systematically predicted using automated DFT calculations. Additionally, electronic structure descriptors correlating with catalytic performance must be identified and refined. The integration of machine learning (ML) approaches provides a pathway to generate extensive datasets characterizing catalyst performance with reduced computational demand. Notably, while research efforts have predominantly focused on catalytic activity, it is imperative to address the often-overlooked challenge of catalyst stability under high anodic potentials. Many catalysts exhibit rapid dissolution in aqueous environments under such conditions, highlighting the need for a balanced consideration of both activity and durability in catalyst design. Integrating experimental data with multiscale modeling techniques can capture the behavior of realistic catalyst phases under dynamic conditions, but also iteratively guide experimental design, accelerate material screening, and uncover rate limiting steps. High-throughput synthesis techniques, such as automated combinatorial catalyst preparation and robotic ink formulation, combined with high-throughput electrochemical testing platforms, provide an efficient pathway for rapidly evaluating large libraries of NOR catalysts. These approaches allow simultaneous screening of activity, selectivity, and stability across diverse materials under controlled conditions. By generating expansive datasets, they facilitate the identification of performance trends and accelerate the refinement of catalyst design principles.
Characterizing complex dynamic interfaces requires the integration of computational and experimental approaches to capture both structural and mechanistic insights under realistic conditions. AIMD simulations play a pivotal role in this effort by offering atomistic-level information on the structure and dynamics of the electric double layer, which is critical for understanding interfacial phenomena such as charge distribution, ion solvation, and adsorption behavior.
To address the challenges of rare event sampling and accurately characterize reaction pathways, the application of enhanced sampling techniques, such as metadynamics, umbrella sampling, or slow growth methods, is essential. These methods enable the exploration of free energy surfaces, identification of reaction intermediates, and estimation of kinetic barriers for processes occurring in aqueous environments.
Another promising avenue is the development of ML potentials which combine the accuracy of quantum mechanical calculations with the computational efficiency needed for larger-scale or longer-time simulations. These potentials can be trained on high-fidelity AIMD datasets and used to extend the simulation timescales or system sizes, providing more comprehensive insights into the dynamic behavior of interfaces. The inclusion of ML-based models allows for scalable simulations of complex systems, thereby bridging the gap between atomic-scale understanding and macroscopic observables.
In addition to probing structural and mechanistic properties, computational tools can be employed to simulate experimental observables. For instance, simulated FTIR spectra derived from AIMD trajectories can provide information about molecular vibrations and interfacial interactions. These simulated spectra can be directly compared with experimental FTIR data, serving as a critical validation step and enabling the refinement of computational models to ensure they accurately capture the interfacial behavior.
AM has revolutionized reactor design by enabling precise customization of geometry and flow dynamics to improve mass transport and reaction efficiency. Multiphysics modeling can complement AM by simulating reactor transport dynamics, including fluid flow, ion migration, and gas diffusion, to optimize reactor performance. By integrating AM and Multiphysics modeling, researchers can design reactors that address key challenges, such as nitrogen solubility limitations and competing side reactions like OER. These advancements are vital for ensuring consistent and scalable NOR processes across different setups.
Ensuring the accuracy of experimental results is critical in NOR research, particularly given the challenges posed by contamination and side reactions. N-15 isotope labeling provides a robust method for verifying that reaction products originate from supplied nitrogen gas rather than contaminants. By analyzing products using NMR and mass spectrometry, researchers can confidently attribute observed nitrogenous products to electrochemical reactions. Mass spectrometry is instrumental for real-time detection of reaction intermediates, offering insights into transient species that govern NOR pathways. Combined with FTIR spectroscopy, this technique can provide a comprehensive understanding of molecular vibrations and interfacial interactions. Simulated FTIR spectra derived from AIMD trajectories can further validate experimental data, bridging the gap between theoretical predictions and practical observations.
The ultimate goal of NOR research is to achieve high nitrate production rates and faradaic efficiencies while maintaining low energy consumption. Strategies such as defect engineering, doping, and tuning catalyst surface properties must be explored further. Optimizing the balance between catalytic activity and durability remains a critical challenge. Furthermore, establishing robust experimental protocols and standardizing methodologies across research groups will ensure reproducibility and comparability of results. Collaborative efforts between academia and industry can drive innovation, enabling the translation of laboratory-scale discoveries into commercial-scale solutions. To provide perspective, we show an energy intensity calculation with a simple expression:
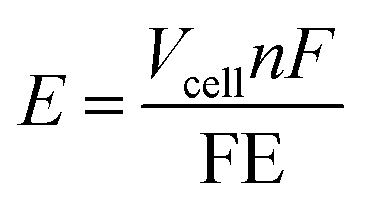 | (2) |
 | ||
| Fig. 9 Contour plot of energy intensity for the NOR as a function of cell voltage and FE. The white line marks the total energy intensity of the conventional nitric acid production from the combination of Haber–Bosch and Ostwald processes. | ||
Advancing NOR technology requires a synergistic approach encompassing catalyst development, reactor engineering, and advanced characterization techniques. By addressing the outlined challenges and leveraging emerging technologies, the field can pave the way for scalable and sustainable nitrate production, contributing significantly to global efforts in climate action and food security.
Conflicts of interest
There are no conflicts to declare.Data availability
No primary research results, software or code have been included, and no new data were generated or analyzed as part of this review.Acknowledgements
This work was performed under the auspices of the U.S. Department of Energy by Lawrence Livermore National Laboratory under Contract DE-AC52-07NA27344 and was supported by Laboratory Directed Research and Development funding under project 22-DR-016. Computational resources were provided by Laboratory Directed Research and Development funding under Project 22-DR-016 with IM release number LLNL-JRNL-2006014.References
- B. H. R. Suryanto, K. Matuszek, J. Choi, R. Y. Hodgetts, H.-L. Du, J. M. Bakker, C. S. M. Kang, P. V. Cherepanov, A. N. Simonov and D. R. MacFarlane, Science, 2021, 372, 1187–1191 CrossRef CAS PubMed.
- J. Lim, C. A. Fernández, S. W. Lee and M. C. Hatzell, ACS Energy Lett., 2021, 6(10), 3676–3685 CrossRef CAS.
- IFASTAT, https://www.ifastat.org/databases/graph/1_1, (accessed June 6, 2025).
- Nitrogenous Fertilizer Market Size & Share Report, 2032, https://www.snsinsider.com/reports/nitrogenous-fertilizer-market-6098, (accessed June 6, 2025).
- L. Lu, J. S. Guest, C. A. Peters, X. Zhu, G. H. Rau and Z. J. Ren, Nat. Sustainability, 2018, 1, 750–758 CrossRef.
- X. Yang, H. Yu, L. Zhang, X. Liu and X. Ding, Sci. China Mater., 2025, 68, 744–754 CrossRef.
- N. Cherkasov, A. O. Ibhadon and P. Fitzpatrick, Chem. Eng. Process., 2015, 90, 24–33 CrossRef CAS.
- K. H. R. Rouwenhorst, F. Jardali, A. Bogaerts and L. Lefferts, Energy Environ. Sci., 2021, 14, 2520–2534 RSC.
- Y. Wang, A. W. DeSilva, G. C. Goldenbaum and R. R. Dickerson, J. Geophys. Res.: Atmos., 1998, 103, 19149–19159 CrossRef CAS.
- T. Li, S. Han, C. Wang, Y. Huang, Y. Wang, Y. Yu and B. Zhang, ACS Catal., 2021, 11, 14032–14037 CrossRef CAS.
- S. Han, C. Wang, Y. Wang, Y. Yu and B. Zhang, Angew. Chem., Int. Ed., 2021, 60, 4474–4478 CrossRef CAS PubMed.
- Z. Nie, L. Zhang, X. Ding, M. Cong, F. Xu, L. Ma, M. Guo, M. Li and L. Zhang, Adv. Mater., 2022, 34, 2108180 CrossRef CAS PubMed.
- F. Xu, X. Liu, L. Zhang, M. Guo, M. Li, X. Ding and L. Zhang, Adv. Energy Mater., 2023, 13(22), 2300615 CrossRef CAS.
- Y. Zhang, T. Xu, Y. Shang, G. Zhang and Z.-F. Yan, J. Colloid Interface Sci., 2023, 650, 669–675 CrossRef CAS PubMed.
- M. Kuang, Y. Wang, W. Fang, H. Tan, M. Chen, J. Yao, C. Liu, J. Xu, K. Zhou and Q. Yan, Adv. Mater., 2020, 32(26), 2002189 CrossRef CAS PubMed.
- L. Zhang, M. Cong, X. Ding, Y. Jin, F. Xu, Y. Wang, L. Chen and L. Zhang, Angew. Chem., Int. Ed., 2020, 59, 10888–10893 CrossRef CAS PubMed.
- C. Dai, Y. Sun, G. Chen, A. C. Fisher and Z. J. Xu, Angew. Chem., Int. Ed., 2020, 59(24), 9418–9422 CrossRef CAS PubMed.
- H. Iriawan, G. Leverick, B. Alkan, D. Delgado, J. Eom, H. Xu, S. Yu, L. Giordano, A. Trunschke, I. E. L. Stephens and Y. Shao-Horn, ACS Catal., 2025, 15, 6315–6333 CrossRef CAS.
- W. Fang, C. Du, M. Kuang, M. Chen, W. Huang, H. Ren, J. Xu, A. Feldhoff and Q. Yan, Chem. Commun., 2020, 56, 5779–5782 RSC.
- Y. Chen, H. Liu, N. Ha, S. Licht, S. Gu and W. Li, Nat. Catal., 2020, 3, 1055–1061 CrossRef CAS.
- J. Lan, M. Luo, J. Han, M. Peng, H. Duan and Y. Tan, Small, 2021, 17, 2102814 CrossRef CAS PubMed.
- A. Adalder, S. Paul, B. Ghorai, S. Kapse, R. Thapa, A. Nagendra and U. K. Ghorai, ACS Appl. Mater. Interfaces, 2023, 15, 34642–34650 CrossRef CAS.
- M. Belotti, F. Matamala-Troncoso, A. I. McKay, T. M. Vu, D. R. MacFarlane and A. N. Simonov, Joule, 2025, 9(5), 101924 CrossRef CAS.
- M. Anand, C. S. Abraham and J. K. Nørskov, Chem. Sci., 2021, 12, 6442–6448 RSC.
- E. Tayyebi, Á. B. Höskuldsson, A. Wark, N. Atrak, B. M. Comer, A. J. Medford and E. Skúlason, J. Phys. Chem. Lett., 2022, 13(26), 6123–6129 CrossRef CAS PubMed.
- J. Long, D. Luan, X. Fu, H. Li, H. Jing and J. Xiao, ACS Catal., 2024, 4423–4431 CrossRef CAS.
- A. Prajapati, A. Zagalskaya, N. Hwee, J. T. Davis, H.-Y. Jeong, J. Moreno, J. Ynzunza, S. A. Akhade and J. T. Feaster, Chem. Catal., 2024, 101220 Search PubMed.
- H. Zheng, Z. Ma, Y. Liu, Y. Zhang, J. Ye, E. Debroye, L. Zhang, T. Liu and Y. Xie, Angew. Chem., Int. Ed., 2024, 63, e202316097 CrossRef CAS PubMed.
- Y. Zhang, F. Du, R. Wang, X. Ling, X. Wang, Q. Shen, Y. Xiong, T. Li, Y. Zhou and Z. Zou, J. Mater. Chem. A, 2021, 9, 17442–17450 RSC.
- Y. Guo, S. Zhang, R. Zhang, D. Wang, D. Zhu, X. Wang, D. Xiao, N. Li, Y. Zhao, Z. Huang, W. Xu, S. Chen, L. Song, J. Fan, Q. Chen and C. Zhi, ACS Nano, 2021, 16(1), 655–663 CrossRef PubMed.
- K. Dong, Y. Yao, H. Li, H. Li, S. Sun, X. He, Y. Wang, Y. Luo, D. Zheng, Q. Liu, Q. Li, D. Ma, X. Sun and B. Tang, Nat. Synth., 2024, 3, 763–773 CrossRef CAS.
- J. Ebenezer, P. Velayudham and A. Schechter, Adv. Energy Mater., 2025, 15, 2501583 CrossRef CAS.
- M. L. Krebs and F. Schüth, J. Am. Chem. Soc., 2024, 146, 30753–30757 CrossRef CAS PubMed.
- Z. Wang, J. Liu, H. Zhao, W. Xu, J. Liu, Z. Liu, J. Lai and L. Wang, Chem. Sci., 2023, 14, 1878–1884 RSC.
- E. L. Clark, J. Resasco, A. Landers, J. Lin, L.-T. Chung, A. Walton, C. Hahn, T. F. Jaramillo and A. T. Bell, ACS Catal., 2018, 8, 6560–6570 CrossRef CAS.
- I. Bagemihl, C. Bhatraju, J. R. van Ommen and V. van Steijn, ACS Sustainable Chem. Eng., 2022, 10, 12580–12587 CrossRef CAS PubMed.
- D. Wakerley, S. Lamaison, J. Wicks, A. Clemens, J. Feaster, D. Corral, S. A. Jaffer, A. Sarkar, M. Fontecave, E. B. Duoss, S. Baker, E. H. Sargent, T. F. Jaramillo and C. Hahn, Nat. Energy, 2022, 7, 130–143 CrossRef CAS.
- K. Liu, W. A. Smith and T. Burdyny, ACS Energy Lett., 2019, 4, 639–643 CrossRef CAS PubMed.
- A. Prajapati, C. Hahn, I. M. Weidinger, Y. Shi, Y. Lee, A. N. Alexandrova, D. Thompson, S. R. Bare, S. Chen, S. Yan and N. Kornienko, Nat. Commun., 2025, 16, 2593 CrossRef CAS PubMed.
- D. Corral, J. T. Feaster, S. Sobhani, J. R. DeOtte, D. U. Lee, A. A. Wong, J. Hamilton, V. A. Beck, A. Sarkar, C. Hahn, T. F. Jaramillo, S. E. Baker and E. B. Duoss, Energy Environ. Sci., 2021, 14, 3064–3074 RSC.
- O. A. Ibrahim, M. Navarro-Segarra, P. Sadeghi, N. Sabaté, J. P. Esquivel and E. Kjeang, Chem. Rev., 2022, 122, 7236–7266 CrossRef CAS PubMed.
- Y. J. Kim, A. Lim, J. M. Kim, D. Lim, K. H. Chae, E. N. Cho, H. J. Han, K. U. Jeon, M. Kim, G. H. Lee, G. R. Lee, H. S. Ahn, H. S. Park, H. Kim, J. Y. Kim and Y. S. Jung, Nat. Commun., 2020, 11, 4921 CrossRef CAS.
- A. Prajapati, N. Govindarajan, W. Sun, J. Huang, H. Bemana, J. T. Feaster, S. A. Akhade, N. Kornienko and C. Hahn, ACS Catal., 2024, 14, 10122–10131 CrossRef CAS.
- C. D. Blanchette, J. M. Knipe, J. K. Stolaroff, J. R. DeOtte, J. S. Oakdale, A. Maiti, J. M. Lenhardt, S. Sirajuddin, A. C. Rosenzweig and S. E. Baker, Nat. Commun., 2016, 7, 11900 CrossRef CAS PubMed.
- Z. D. Rosli, S. Tesana, N. Malone, M. I. Ahmed, V. Jovic, S. Giddey, J. V. Kennedy and P. Gupta, ACS Catal., 2025, 15, 3134–3142 CrossRef CAS.
- S. Z. Andersen, V. Čolić, S. Yang, J. A. Schwalbe, A. C. Nielander, J. M. McEnaney, K. Enemark-Rasmussen, J. G. Baker, A. R. Singh, B. A. Rohr, M. J. Statt, S. J. Blair, S. Mezzavilla, J. Kibsgaard, P. C. K. Vesborg, M. Cargnello, S. F. Bent, T. F. Jaramillo, I. E. L. Stephens, J. K. Nørskov and I. Chorkendorff, Nature, 2019, 570, 504–508 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |