
Unsymmetrical bis(anilido)xanthene ligands: development and use in the preparation of magnesium diamide complexes†
Matthew J.
Evans
 and
Cameron
Jones
*
and
Cameron
Jones
*
School of Chemistry, Monash University, P.O. Box 23, Melbourne, Victoria 3800, Australia. E-mail: cameron.jones@monash.edu
First published on 2nd July 2025
Abstract
Unsymmetrical bis(anilido)xanthene ligands, decorated by a 2,4,6-tricyclohexylanilido substituent and a less bulky anilido group, are readily prepared in moderate yields from a 4,5-dibromo-substituted xanthene. These ligands are used for the preparation of magnesium(II) complexes, the reductions of which in methylcyclohexane and benzene are described.
Xanthene bridged scaffolds feature diverse and tuneable structural and electronic properties that have led to their application in materials,1 medicinal2 and chemical sciences.3 Within the context of organometallic chemistry, substituted derivatives of xanthene have been widely utilised as neutral, monoanionic and dianionic ligands, which are suitable for the stabilisation of metals in various oxidation states. Their rigidity and amenability to modification enables control over the coordination environment at the metal centre(s). An archetypal example here is the neutral xanthphos ligand (I, Fig. 1), transition metal complexes of which have been extensively employed in homogeneous catalysis (e.g. Suzuki–Miyaura and Buchwald–Hartwig reactions).4 With regard to related anionic ligand systems, and of most relevance to this study, the first bis(anilido)xanthene ligand (II, R = iPr, R′ = H) was prepared by Emslie and co-workers in 2007.5 This, and similar bis(anilido)xanthenes, were subsequently used to form complexes with metals from across the periodic table (e.g. Mg,6 Al,7 K,8 Ca,6,9 Sr,9 Ba,9b Mn,10 Fe,11 Co,11 Y,7 Zr,12 La,13 Sm,14 Dy,14 Yb,14 Lu,13 Th,5,15 U16).
 | ||
| Fig. 1 Selected examples of previously reported xanthene bridged ligand systems. | ||
Recently, bis(anilido)xanthenes have further shown their value with the stabilisation of main group elements in low oxidation states. For example, Aldridge and Goicoechea enlisted Emslie's ligand II (R = iPr, R′ = H) to prepare the first isolable aluminyl anion (an AlI species) and a gallyl anion (a GaI species) in 2018,17–19 with the InI analogue soon to follow.20 Several years later, the same ligand system featured in neutral GeII and SnII diamide complexes, the reduction of which afforded anionic GeI and SnI radicals.21 In 2022, our group developed a much bulkier analogue of Emslie's ligand, viz.II (R/R′ = Cy, NONTCHP), and a derived calcium complex [(NONTCHP)Ca(η6-toluene)], which when reduced with potassium metal, under dinitrogen, gave a rare calcium-dinitrogen compound K2[{(NONTCHP)Ca}2(μ-η2,η2-N2)].22 The next year, the first example of a magnesium–dinitrogen complex K2[{(NONTCHP)Mg}2(μ-κ1,κ1-N2)] III was similarly synthesized.23 Both reduction reactions were calculated to proceed via metal(I) radicals, K[(NONTCHP)M·], which are sterically frustrated from forming metal–metal bonds due to the bulk of the diamide ligand, and therefore preferentially reduce dinitrogen to [N2]2-. Strong evidence for this came in 2024, when we reduced a less bulky magnesium diamide, [(NONTrip)Mg] (NONTrip = II R/R′ = iPr), which did not give an [N2]2− complex, but instead a magnesium(I) system, K2[{(NONTrip)Mg}2] IV, with a very long Mg–Mg covalent bond (3.137(2) Å).24 Both III and IV have proved powerful reagents for the reductive activation of gaseous small molecules (e.g. H2, CO, CO2, C2H4), P4 and typically inert arenes.9b,23–25
Given that the contrasting nature of the reduction products III and IV results from steric differences between their N-aryl substituents, it seemed a logical extension to investigate the outcome of the reduction of a magnesium complex incorporating a bis(anilido)xanthene ligand that is unsymmetrically substituted with one TCHP (2,4,6-tricyclohexylphenyl) and one Trip (2,4,6-triisopropylphenyl), or smaller, aryl group. However, although there is one report on an unsymmetrical amido/anilido-xanthene ligand (V),26 unsymmetrical bis(anilido)xanthene ligands are unknown. In this contribution, we detail the straightforward syntheses of such ligands, and the formation and reduction of their magnesium(II) complexes.
The synthesis of unsymmetrical bis(aniline)xanthene pro-ligands, (NONTCHP/Ar)H2 (4-(TCHP-NH)-5-(Ar-NH)-2,7-diethyl-9,9-dimethylxanthene; Ar = Mes or Trip; Mes = 2,4,6-trimethylphenyl), is depicted in Scheme 1. Dibromoxanthene A22 is first converted to 4,5-diazido-2,7-diethyl-9,9-dimethyl-xanthene (B) and then subsequently reduced with lithium aluminium hydride to yield 4,5-diamino-2,7-diethyl-9,9-dimethyl-xanthene (C) in moderate to high yields over 2 steps. This route to C is achievable on a 10 gram scale, and is based on a previous report by Metz and co-workers who detailed a similar smaller scale (< 1 g) route to 4,5-diaminoxanthene directly from xanthene.27 Moreover, previous syntheses of 2,7-disubstituted-4,5-diaminoxanthenes are generally low yielding and/or utilise precious metal catalysts,11,28 while our method is convenient, high yielding, cheap and does not require catalysts.
 | ||
| Scheme 1 Synthesis of unsymmetrical bis(aniline)xanthene pro-ligands. (i) nBuLi (2 equiv.), THF, −78 °C. (ii) Tosylazide (2 equiv.), THF, −78 °C – RT, 18 h. (iii) LiAlH4 (3 equiv.), Et2O, reflux, 2 h. (iv) (TCHP)Br (1 equiv.), Pd(OAc)2 (4 mol%), DPEPhos (6 mol%), NaOtBu (1.4 equiv.). (v) (Mes)Br (1 equiv.) or (Trip)Br (1 equiv.), Pd(OAc)2 (4 mol%), DPEPhos (6 mol%), NaOtBu (1.4 equiv.). Inset: Molecular structure of (NONTCHP/Trip)H2. Thermal ellipsoids shown at 15% probability; hydrogen atoms (except amine protons) omitted; cyclohexyl and isopropyl groups shown in wireframe for clarity. | ||
Syntheses of symmetrical bis(aniline)xanthene pro-ligands such as (NONTCHP)H2 normally proceed via cross-coupling reactions between dibromoxanthenes, e.g.A, and an aniline. However, we have found that preparing unsymmetrical systems, (NONTCHP/Trip)H2, by sequential cross-coupling reactions of A with (TCHP)NH2 and another aniline is problematic. Hence, an alternative route was developed whereby diaminoxanthene C was cross-coupled with one equivalent of (TCHP)Br, affording a moderate yield of the mono-substituted product D. Subsequent cross-coupling reactions with the aryl bromides, (Mes)Br or (Trip)Br, gave the unsymmetrical bis(aniline)xanthene pro-ligands, (NONTCHP/Ar)H2 (Ar = Mes or Trip), as colourless solids in moderate yields after work-up (see ESI† for further details).
So as to draw comparisons with the aforementioned reduced species, III and IV, we sought to prepare magnesium(II) precursor complexes containing bis(anilido)xanthene ligands of intermediate steric bulk, which could be subsequently reduced with potassium metal. To this end, the pro-ligands (NONTCHP/Ar)H2 were deprotonated with one equivalent of Mg(CH2SiMe3)2 to give 1 (Ar = Mes, 65% yield) and 2 (Ar = Trip, 98% yield) as colourless and pale yellow solids respectively (Scheme 2). The 1H NMR spectrum (C6D6) of each complex exhibits signals for four chemically inequivalent xanthene arene protons, which is consistent with the proposed dimeric solid-state structure of 1, but would also be expected if the compounds are monomeric in solution.
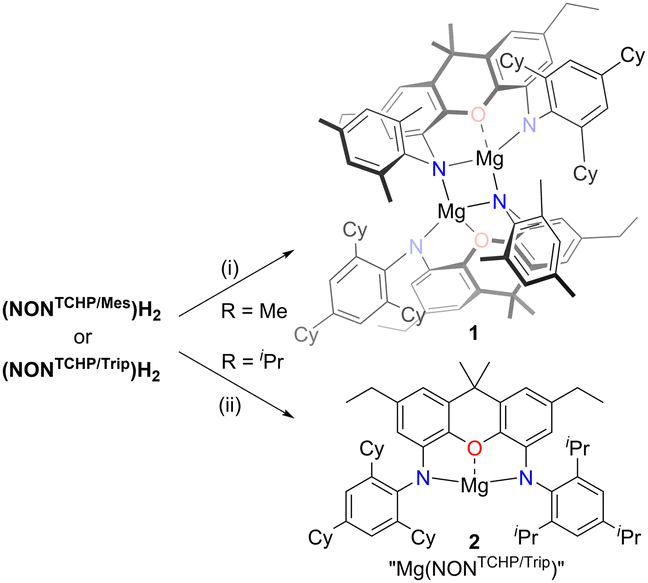 | ||
| Scheme 2 Synthesis of magnesium(II) complexes 1 and 2. (i) Mg(CH2SiMe3)2, C6H6, 80 °C. (ii) Mg(CH2SiMe3)2, methylcyclohexane, 80 °C. | ||
Suitable crystals of 2 could not be obtained from hexane or benzene solutions due to its high solubility in these solvents, but 1 could be crystallised from benzene. The molecular structure of 1 (see Fig. S47, ESI†) reveals it to be an N-mesityl bridged dimer, which is similar to that previously reported for propane bridged dianilido complexes [Mg(NCNAr)]2 (NCNAr = [{(Ar)NCH2}2CH2]2−, Ar = Trip or TCHP).29 This is in contrast to the related complex [Mg(NONDip)]2 (NONDip = II, R = iPr, R′ = H) which dimerises through bridging Mg⋯π(Dip) arene contacts,6 and to the arene capped magnesium(II) complex Mg(NONTCHP)(η2-C6H6),9b both of which are crystallised from benzene. The inability of both 1 and 2 to form isolable benzene-capped complexes may suggest that they exist as N- or arene-bridged dimers in both aliphatic and arene solutions. If this is the case for 2, any intermolecular association can be disrupted by addition of the Lewis bases 1,3,4,5-tetramethylimidazol-2-ylidene (TMC) or THF to the compound. This results in the formation of the corresponding monomeric Lewis adducts Mg(NONTCHP/Trip)(TMC)2 (3) and Mg(NONTCHP/Trip)(THF)2 (4), respectively (Scheme S1, ESI†). Both complexes were characterised in the solid-state by X-ray diffraction measurements, which showed them to have similar heavily distorted square-based pyramidal magnesium geometries (see Fig. S48 and S49, ESI†).
With 1 and 2 in hand, reductions of the compounds were investigated, in order to compare the nature of the products with III and IV. Accordingly, complexes 1 and 2 were treated with K/KI30 (5% w/w, 4 equivalents) in methylcyclohexane over 18 hours. Compound 1 was found to be unreactive towards K/KI under these reaction conditions, presumably as the strength of its dimeric structure towards dissociation protects its Mg centres from reaction with potassium. In contrast, 2 does react with K/KI, but this generated a complex mixture of products, none of which could be identified by NMR spectroscopy or crystallised from the reaction mixture. Given that 1 and 2 do not form complexes with benzene, we turned to that solvent to further assess their reduction. That is, solutions of compounds 1 and 2 in benzene were reacted with K/KI (4 equivalents), resulting in intensely dark red coloured reaction solutions after 18 hours. Analysing the reaction mixtures by 1H NMR spectroscopy revealed mixtures of ill-defined products, including dihydrogen, in both cases. For sake of comparison, the reduction of Mg(NONTCHP) in benzene was also shown to give a complex mixture of products, with dissolved dihydrogen present. Although the reduction of 1 failed to give any crystalline products after work-up, single crystals of 5 could be obtained from the reaction involving 2 in a low (5%) but reproducible yield (Scheme 3).
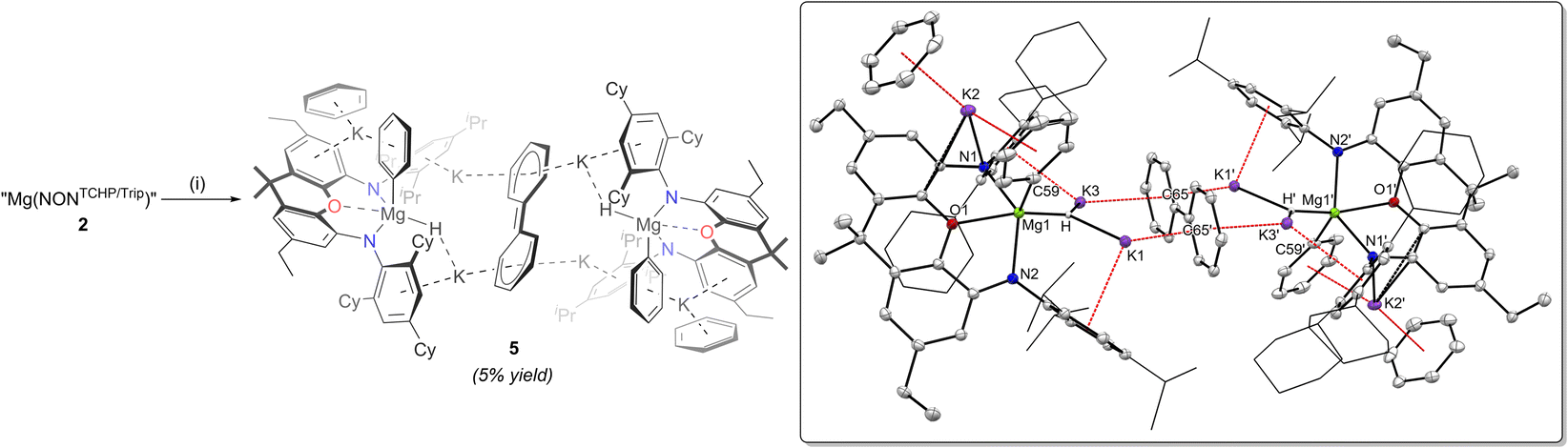 | ||
| Scheme 3 Reduction of complex 2 with K/KI to give 5 and other unidentified reduction products. (i) K/KI (3 equiv.), C6H6, 16 h, RT. Inset: Molecular structure of 5. Thermal ellipsoids shown at 20% probability; hydrogen atoms omitted; cyclohexyl and isopropyl groups shown as wireframe for clarity. Selected bond lengths (Å) and angles (°): Mg1–N1 2.169(3), Mg1–N2 2.157(3), Mg1–C59 2.204(5), Mg1–O1 2.142(3), Mg1–H 1.855(15), C65–C65’ 1.399(7), C65–C66 1.470(4), C65–C70 1.454(5), C66–C67 1.365(5), C67–C68 1.421(6), C68–C69 1.418(6), N1–Mg–N2 138.12(13), O1–Mg1–H 155.9(7). | ||
The solid-state structure of 5 shows it to comprise two formally dianionic [(NONTCHP/Trip)Mg(H)(Ph)]2− units which exhibit distorted square pyramidal geometries (τ5 = 0.30) at each magnesium centre. A partially delocalised biphenyl dianion, [C12H10]2−, bridges the two units, through interactions with four potassium cations, while each of the two remaining K+ centres exhibit arene coordination to a [(NONTCHP/Trip)Mg(H)(Ph)]2− unit and a molecule of benzene. The structure of 5 is related to reported complexes of rare earth and alkaline earth metals (Y,31 Sm,32 Yb,32 Ca,33 Sr33), which incorporate the biphenyl dianion ligand. Furthermore, and similar to the formation of 5, an electride, “K+[Li{N(SiMe3)2}e−]”, prepared by ball-milling, has recently been shown by Lu and co-workers to react with benzene to give a black solution, from which the biphenyl dianion bridged complex [(Li)(K)2{N(SiMe3)2}2]2[μ-η12-(C12H10)] was isolated.34
It is not clear what the mechanism of formation of 5 entails, but the complexity of the product, which incorporates three anionic fragments seemingly derived from benzene, makes a meaningful computational analysis of the reaction pathway improbable. With that said, it likely involves an initial reduction of 2 to give a magnesium(I) radical K[(NONTCHP/Trip)Mg·], similar to the calculated intermediate in the formation of III.23 This could then doubly reduce benzene to give a “Birch-like” intermediate K2[{(NONTCHP/Trip)Mg}2(μ-C6H6)], analogous to the reaction of III with benzene, which yields more sterically protected K2[{(NONTCHP)Mg}2(μ-C6H6)].25a The reduced intermediate K2[{(NONTCHP/Trip)Mg}2(μ-C6H6)] could then undergo a benzene C–H activation reaction, yielding magnesium hydride and magnesium phenyl products, e.g. K[(NONTCHP/Trip)MgH] and K[(NONTCHP/Trip)MgPh], ultimately leading to the observed dianionic units [(NONTCHP/Trip)Mg(H)(Ph)]2− in 5 (see Scheme S2, ESI†). Indeed, it has been previously shown that in situ generated magnesium(I) radicals can similarly reduce benzene to isolable [C6H6]2− containing products, the reduced benzene fragment of which can then be C–H activated, yielding magnesium hydride and magnesium phenyl compounds.35 With regard to the formation of the biphenyl dianion in 5, the proposed intermediate K2[{(NONTCHP/Trip)Mg}2(μ-C6H6)] could alternatively react with a further molecule of benzene to give the biphenyl dianion via C–C bond formation and dihydrogen elimination processes. This enhanced reactivity, relative to alkali metal free magnesium “Birch-like” products,35 could be explained by Mg/K mixed-metal cooperativity, which has been previously shown to boost reactivity relative to monometallic systems.36 Further precedent for this mechanistic proposal comes from a study by Harder and co-workers, who showed that calcium and strontium complexes of the benzene dianion [C6H6]2− can similarly react with benzene to give the biphenyl dianion and H2.33 Whatever the mechanism is for the formation of 5, it most likely involves a number of potentially competing pathways and intermediate redistribution steps, which is consistent with the low yield of the compound.
In summary, we have presented a convenient approach to synthesise the first examples of unsymmetrical bis(anilido)xanthene ligands, NONTCHP/Ar. It is likely that this method should be readily adaptable to access a variety of other unsymmetrical bis(anilido)xanthenes, which will allow an expansion of the already broad coordination chemistry of related symmetrical bis(anilido)xanthenes. Preliminary investigations into the coordination chemistry of NONTCHP/Ar have yielded four magnesium(II) complexes of the ligands. The reduction of one of these (NONTCHP/Trip)Mg 2 with K/KI in benzene affords a low yield of the unusual complex 5, which contains two dianionic magnesium(II) fragments [(NONTCHP/Ar)Mg(H)(Ph)]2− bearing phenyl and hydride ligands. These are bridged by a biphenyl dianion, with the charges on all anionic units being balanced by six potassium cations. Mechanisms for the formations of the anions in 5 have been proposed. We are currently investigating the application of unsymmetrical bis(anilido)xanthene ligands for the stabilisation of low oxidation state main-group compounds.
C. J. thanks the Australian Research Council and the Air Force Office of Scientific Research (FA2386-21-1-4048) for funding. We acknowledge the Monash Analytical Platform and the Australian Synchrotron for access to the MX2 beamlines.
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the ESI.†Notes and references
- (a) S. Kamino and M. Uchiyama, Org. Biomol. Chem., 2023, 21, 2458–2471 RSC; (b) P. Wight and Xanthene Dyes, Kirk-Othmer Encyclopedia of Chemical Technology, 2000 Search PubMed.
- (a) A. G. Ghahsare, Z. S. Nazifi and S. M. R. Nazifi, Curr. Org. Synth., 2019, 16, 1071–1077 CrossRef PubMed; (b) M. Maia, D. I. S. P. Resende, F. Durães, M. M. M. Pinto and E. Sousa, Eur. J. Med. Chem., 2021, 210, 113085 CrossRef.
- (a) P. C. J. Kamer, P. W. N. M. van Leeuwen and J. N. H. Reek, Acc. Chem. Res., 2001, 34, 895–904 CrossRef PubMed; (b) G. M. Adams and A. S. Weller, Coord. Chem. Rev., 2018, 355, 150–172 CrossRef CAS.
- P. W. N. M. van Leeuwen and P. C. J. Kamer, Catal. Sci. Technol., 2018, 8, 26–113 RSC.
- C. A. Cruz, D. J. H. Emslie, L. E. Harrington, J. F. Britten and C. M. Robertson, Organometallics, 2007, 26, 692–701 CrossRef.
- J. S. McMullen, A. J. Edwards and J. Hicks, Dalton Trans., 2021, 50, 8685–8689 RSC.
- K. S. A. Motolko, D. J. H. Emslie and H. A. Jenkins, Organometallics, 2017, 36, 1601–1608 CrossRef.
- N. R. Andreychuk and D. J. H. Emslie, Angew. Chem., Int. Ed., 2013, 52, 1696–1699 CrossRef.
- (a) R. Huo, A. J. Armstrong, G. R. Nelmes, D. J. Lawes, A. J. Edwards, C. L. McMullin and J. Hicks, Chem. – Eur. J., 2024, 30, e202400662 CrossRef PubMed; (b) M. J. Evans, D. T. Nguyen, J. M. Parr, J. Mullins, R. Mondal, T. Rajeshkumar, L. Maron and C. Jones, Chem, 2025 DOI:10.1016/j.chempr.2025.102650.
- S. Wolff, M. J. Evans, T. Rajeshkumar, D. T. Nguyen, K. B. Krause, C. Herwig, L. Maron, C. Jones and C. Limberg, ChemRxiv, 2025, preprint, chemrxiv-2025-3wss3 DOI:10.26434/chemrxiv-2025-3wss3.
- A. Nicolay, M. S. Ziegler, L. Rochlitz and T. D. Tilley, Polyhedron, 2020, 180, 114420 CrossRef.
- K. S. A. Motolko, J. S. Price, D. J. H. Emslie, H. A. Jenkins and J. F. Britten, Organometallics, 2017, 36, 3084–3093 CrossRef.
- K. S. A. Motolko, D. J. H. Emslie and J. F. Britten, RSC Adv., 2017, 7, 27938–27945 RSC.
- A. Hauser, L. Münzfeld, S. Schlittenhardt, R. Köppe, C. Uhlmann, U.-C. Rauska, M. Ruben and P. W. Roesky, Chem. Sci., 2023, 14, 2149–2158 RSC.
- (a) C. A. Cruz, D. J. H. Emslie, C. M. Robertson, L. E. Harrington, H. A. Jenkins and J. F. Britten, Organometallics, 2009, 28, 1891–1899 CrossRef; (b) C. A. Cruz, D. J. H. Emslie, L. E. Harrington and J. F. Britten, Organometallics, 2008, 27, 15–17 CrossRef.
- N. R. Andreychuk, S. Ilango, B. Vidjayacoumar, D. J. H. Emslie and H. A. Jenkins, Organometallics, 2013, 32, 1466–1474 CrossRef.
- J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, Nature, 2018, 557, 92–95 CrossRef.
- M. Asay, C. Jones and M. Driess, Chem. Rev., 2011, 111, 354–396 CrossRef PubMed.
- (a) J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, Angew. Chem., Int. Ed., 2021, 60, 1702–1713 CrossRef; (b) M. P. Coles and M. J. Evans, Chem. Commun., 2023, 59, 503–519 RSC.
- L. P. Griffin, M. A. Ellwanger, A. E. Crumpton, M. M. D. Roy, A. Heilmann and S. Aldridge, Angew. Chem., Int. Ed., 2024, 63, e202404527 CrossRef.
- (a) F. Krämer, M. S. Luff, U. Radius, F. Weigend and F. Breher, Eur. J. Inorg. Chem., 2021, 3591–3600 CrossRef; (b) L. F. Lim, M. Judd, P. Vasko, M. G. Gardiner, D. A. Pantazis, N. Cox and J. Hicks, Angew. Chem., Int. Ed., 2022, 61, e202201248 CrossRef.
- R. Mondal, K. Yuvaraj, T. Rajeshkumar, L. Maron and C. Jones, Chem. Commun., 2022, 58, 12665–12668 RSC.
- R. Mondal, M. J. Evans, T. Rajeshkumar, L. Maron and C. Jones, Angew. Chem., Int. Ed., 2023, 62, e202308347 CrossRef PubMed.
- R. Mondal, M. J. Evans, D. T. Nguyen, T. Rajeshkumar, L. Maron and C. Jones, Chem. Commun., 2024, 60, 1016–1019 RSC.
- (a) M. J. Evans, J. Mullins, R. Mondal and C. Jones, Chem. – Eur. J., 2024, 30, e202401005 CrossRef CAS PubMed; (b) D. T. Nguyen, R. Mondal, M. J. Evans, J. M. Parr and C. Jones, Angew. Chem., Int. Ed., 2025, 64, e202500264 CrossRef CAS; (c) M. J. Evans and C. Jones, Chem. Soc. Rev., 2024, 53, 5054–5082 RSC.
- F. Kallmeier, A. J. R. Matthews, G. R. Nelmes, N. R. Lawson and J. Hicks, Dalton Trans., 2024, 53, 12450–12454 RSC.
- A. E. Metz, K. Ramalingam and M. C. Kozlowski, Tetrahedron Lett., 2015, 56, 5180–5184 CrossRef CAS PubMed.
- (a) F. Kallmeier, G. R. Nelmes, C. L. McMullin, A. J. Edwards and J. Hicks, Chem. Sci., 2025, 16, 10750–10758 RSC; (b) F. B. Schwarz, T. Heinrich, J. O. Kaufmann, A. Lippitz, R. Puttreddy, K. Rissanen, W. E. S. Unger and C. A. Schalley, Chem. – Eur. J., 2016, 22, 14383–14389 Search PubMed; (c) J. Yin, A. N. Khalilov, P. Muthupandi, R. Ladd and V. B. Birman, J. Am. Chem. Soc., 2020, 142, 60–63 CrossRef CAS; (d) K.-J. Chang, Y.-J. An, H. Uh and K.-S. Jeong, J. Org. Chem., 2004, 69, 6556–6563 CrossRef CAS.
- D. T. Nguyen, C. Helling and C. Jones, Chem. – Asian J., 2024, 19, e202400498 CrossRef CAS PubMed.
- J. Hicks, M. Juckel, A. Paparo, D. Dange and C. Jones, Organometallics, 2018, 37, 4810–4813 Search PubMed.
- M. D. Fryzuk, J. B. Love and S. J. Rettig, J. Am. Chem. Soc., 1997, 119, 9071–9072 CrossRef CAS.
- Y. Xiao, X.-K. Zhao, T. Wu, J. T. Miller, H.-S. Hu, J. Li, W. Huang and P. L. Diaconescu, Chem. Sci., 2021, 12, 227–238 RSC.
- J. Mai, M. Morasch, D. Jędrzkiewicz, J. Langer, B. Rösch and S. Harder, Angew. Chem., Int. Ed., 2023, 62, e202212463 CrossRef CAS PubMed.
- N. Davison, J. A. Quirk, F. Tuna, D. Collison, C. L. McMullin, H. Michaels, G. H. Morritt, P. G. Waddell, J. A. Gould, M. Freitag, J. A. Dawson and E. Lu, Chemistry, 2023, 9, 576–591 CrossRef CAS.
- (a) D. D. L. Jones, I. Douair, L. Maron and C. Jones, Angew. Chem., Int. Ed., 2021, 60, 7087–7092 CrossRef CAS; (b) J. C. Mullins, K. Yuvaraj, Y. Jiang, G. P. Van Trieste Iii, A. Maity, D. C. Powers and C. Jones, Chem. – Eur. J., 2022, 28, e202202103 CrossRef CAS; (c) T. X. Gentner, B. Rösch, G. Ballmann, J. Langer, H. Elsen and S. Harder, Angew. Chem., Int. Ed., 2019, 58, 607–611 CrossRef CAS.
- T. X. Gentner, A. R. Kennedy, E. Hevia and R. E. Mulvey, ChemCatChem, 2021, 13, 2371–2378 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2456733–2456740. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5cc03202d |
| This journal is © The Royal Society of Chemistry 2025 |