
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntroducing the substituted azobispyrrole framework: synthesis and properties†
Adil Alkaş a,
Roberto M. Diaz-Rodrigueza,
Steve O. Sequeiraa,
James W. Hilborna,
Mmasinachi Atansia,
Em C. Sullivana,
Emily B. Browna,
Rosinah Liandrah Gaparea,
Bulent Mutusac,
Katherine N. Robertsonb and
Alison Thompson*a
a,
Roberto M. Diaz-Rodrigueza,
Steve O. Sequeiraa,
James W. Hilborna,
Mmasinachi Atansia,
Em C. Sullivana,
Emily B. Browna,
Rosinah Liandrah Gaparea,
Bulent Mutusac,
Katherine N. Robertsonb and
Alison Thompson*a
aDepartment of Chemistry, Dalhousie University, PO Box 15000, Halifax, Nova Scotia B3H 4J3, Canada. E-mail: Alison.Thompson@dal.ca
bDepartment of Chemistry, Saint Mary's University, Halifax, NS B3H 3C3, Canada
cDepartment of Chemistry & Biochemistry, University of Windsor, 401 Sunset Ave., Windsor N9B 3P4, Ontario, Canada
First published on 3rd July 2025
Abstract
An unprecedented azobispyrrole framework is introduced, wherein two aryl-substituted pyrroles are joined by an azo (–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–) linkage. Functionalisation was demonstrated via pyrrolic N-methylation and by –BF2 complexation using the coordinating abilities of the pyrrolic and azo nitrogen atoms. Control of co-planarity enables tunability with maximal absorption and emission spanning almost 300 nm.
N–) linkage. Functionalisation was demonstrated via pyrrolic N-methylation and by –BF2 complexation using the coordinating abilities of the pyrrolic and azo nitrogen atoms. Control of co-planarity enables tunability with maximal absorption and emission spanning almost 300 nm.
With emergent applications reliant on synthetic versatility and the quest for enhanced tunability, the exploration of novel azo frameworks is vibrant.1 The potentiality of heteroaryl azo dyes, including the promise of activation using red light, has led to a multitude of next-generation frameworks demonstrating advantages over the better understood and more widely utilised azobenzenes.2 In terms of azo compounds featuring nitrogen-containing heterocycles, recent reports describe the azo functionality flanked by phenylazothiazoles3 and pyrazoles,4 as well as indoles and pyrroles.5–7 Although the pyrrole heterocycle features in recently-reported azo compounds bearing one8 and two9 4,4-difluoro-4-borata-3a-azonia-4a-aza-s-indacene (BODIPY) units, the incorporation of multiple pyrrolic units into azo compounds is significantly underdeveloped.5,10–13 As such, preparing and characterising pyrrole-containing azo compounds offers significant allure.
Isolation of the substituted azobispyrrole framework reported herein began with the search for F-aza-BODIPYS featuring significant steric bulk across the pyrrolic backbone. Aza-dipyrrins featuring tetra-aryl substitution patterns are typically prepared by reacting the corresponding nitrobutanone with an ammonia source.14 Accordingly, we attempted to prepare aza-dipyrrin 4 (Scheme 1) by heating 4-nitro-1,3-diarylbutanone 2, bearing a bulky mesityl group, with NH4OAc in glacial acetic acid.15 Two solid materials were thus isolated; one red and one purple, the latter matching our expectations for the anticipated aza-dipyrrin 4. As regards the red material, 1H and 13C NMR spectral data indicated the presence of phenyl and mesityl units, and a pyrrolic N–H feature. Furthermore, the 15N NMR data indicated two distinct nitrogen environments; as represented by resonances at 125 ppm and 526 ppm. Mass spectrometric analysis of the red material revealed a sodiated molecular cation consistent with a species composed of two pyrroles, two phenyl groups, two mesityl groups and a total of four N atoms (two of which were presumed to be part of pyrrolic heterocycles), together suggesting the formation of the dipyrrolic azo compound 3 (Scheme 1): this composition was confirmed via single-crystal X-ray diffraction analysis (Fig. 1). Given the steric demand of the mesityl groups, it appears reasonable that azobispyrrole formation is competitive with aza-dipyrrin production16,17 since the symmetrical azobispyrrole 3, with two nitrogen atoms separating the two pyrrole units, positions the mesityl groups more distant to each other than in the aza-dipyrrin 4. Despite extensive variation of reaction conditions (solvent, temperature, mode of heating, time, stoichiometry, nitrogen source), the isolated yield of 3 could not be enhanced beyond 30%.
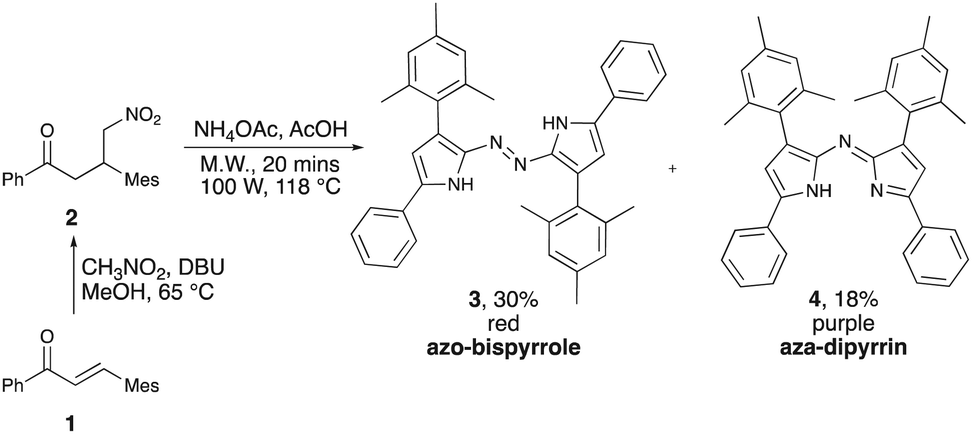 | ||
| Scheme 1 Synthesis of tetra-aryl azobispyrrole 3. | ||
 | ||
| Fig. 1 Structure of azobispyrrole 3. Thermal ellipsoids drawn at the 50% probability level. Only one half of the molecule is uniquely defined. A second molecule, the solvent molecule and the hydrogen atoms have been removed for clarity. | ||
Azobispyrrole 3 crystallised in the centrosymmetric triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with one half of each of two unique molecules in the asymmetric unit, along with one molecule of water. In the solid state, the azo functionality is Z- (trans) in configuration and is essentially co-planar with the flanking pyrrole rings. However, the two phenyl rings make 22° and 23° angles, in molecules 1 and 2 respectively, with the pyrrole-NN-pyrrole core plane and this arrangement is stabilised via hydrogen bonding to water (see ESI† for full details). As expected, the bulky mesityl groups lie almost perpendicular to the core plane. No short stacking interactions were observed in the structure of 3, or in any of the solids studied.
with one half of each of two unique molecules in the asymmetric unit, along with one molecule of water. In the solid state, the azo functionality is Z- (trans) in configuration and is essentially co-planar with the flanking pyrrole rings. However, the two phenyl rings make 22° and 23° angles, in molecules 1 and 2 respectively, with the pyrrole-NN-pyrrole core plane and this arrangement is stabilised via hydrogen bonding to water (see ESI† for full details). As expected, the bulky mesityl groups lie almost perpendicular to the core plane. No short stacking interactions were observed in the structure of 3, or in any of the solids studied.
Given the voraciousness of the search for electronically tunable azo compounds, synthesis of the substituted azobispyrrole framework represents a considerable advance. As such, the reactivity of azobispyrrole 3 was explored. Treatment of 3 with methyl iodide, under basic conditions (Scheme 2), gave rise to the methylated analogues 5a and 5b. A crystal structure of the dimethylated azobispyrrole 5a was refined as a pseudo-merohedral twin in the triclinic space group P (see ESI† for full details), and a planar pyrrole-NN-pyrrole motif observed. The phenyl groups are twisted out of the core plane courtesy of steric interactions with the N-methyl groups, and the mesityl groups are of course again out of plane with the central core.
 | ||
| Scheme 2 Synthesis of methylated azobispyrroles. | ||
With boron-containing heterocyclic complexes, including those featuring azo functionality,18,19 exhibiting tunable emission properties,20,21 attention then turned to the synthesis of a boron difluoride adduct of 3. A solution of the azobispyrrole 3 in CH2Cl2 was treated with BF3·OEt2 in the presence of NEt3, rendering loss of the red colour and isolation of a blue solid (Scheme 3). Despite surviving the aqueous work-up, the –BF2 adduct 6 proved to be slightly sensitive to moisture in the solid state, with decomposition to the ligand 3 ensuing.
 | ||
| Scheme 3 Synthesis of azobispyrrole –BF2 adduct 6. | ||
The –BF2 adduct 6 crystallised in the non-centrosymmetric orthorhombic space group Pna21, with one molecule of the product and one molecule of acetonitrile solvent within the asymmetric unit (Fig. 2). The non-centrosymmetric space group highlights the stereochemical consequences of the diastereotopic fluoro substituents at boron, courtesy of the atropisomerism stemming from restricted rotation of the sterically bulky mesityl groups. The boron is complexed such as to form a five-membered ring that is reliant on the Z-configuration across the NN double bond. Formation of a five-membered ring involving boron and nitrogen atoms, as well as the pyrrolic construct, is somewhat reminiscent of the BOIMPY motif.22 Furthermore, the two nitrogen atoms not bound to boron within 6 are involved in hydrogen bonding along with the nitrogen atom of the acetonitrile solvate, resulting in a five-membered ring featuring the N–H⋯N motif: as such, five-membered rings are evident on both sides of the central azo unit. Insertion of an additional –BF2 unit, utilising the uncomplexed nitrogen atoms in 6, proved challenging such that isolation of the desired product was elusive.
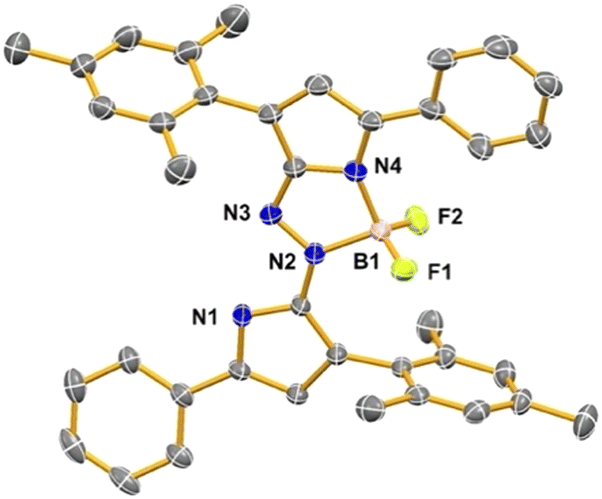 | ||
| Fig. 2 Structure of –BF2 adduct 6. Thermal ellipsoids drawn at the 50% probability level. The solvent molecule and the hydrogen atoms have been removed for clarity. | ||
The entire phenyl-pyrrole-NN-pyrrole-phenyl motif in 6 is essentially planar, complementing the inherent three-dimensionality of the tetrahedrally-coordinated boron atom and the mesityl ring planes positioned ∼80° relative to this main plane (see ESI† for full details). The mean plane defined by all the central core (non-hydrogen) atoms plus all carbon atoms of the two phenyl rings has a mean deviation of 0.22 Å, a relatively small value considering the number of included atoms. The inclusion of the phenyl rings in the same plane as the central core in 6 facilitates conjugation and manifests in a red-shifted absorption profile cf. the parent compound 3. The co-planarity of the phenyl rings with the central core also allows the formation of a bifurcated hydrogen bond between the ortho-H of the phenyl ring and both fluoro atoms of the BF2 group on one side of the molecule, and on the other side of the molecule H⋯N intermolecular interactions also stabilise crystal packing.
The solvatochromic absorption properties of solutions of azobispyrroles 3, 5 and 6 in various solvents are shown in Table 1 (see ESI† for full details, Fig. S1–S30, ESI†), and the relative absorption profiles of solutions in MeCN are plotted in Fig. 3. The aryl substitution of 3 engenders a significant bathochromic shift in the absorption maximum cf. the parent compound with λmax![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) abs = 429 nm,10 an effect which is maintained for the methylated analogues 5a and 5b. Solutions of the –BF2 adduct 6 exhibit a λmax absorption value of 666 nm, representing a ∼160 nm bathochromic shift cf. the free ligand 3. This is attributable to the stabilising effect of the –BF2 group upon co-planarisation and π-electron delocalisation within the core framework, as well as to the additional conjugation brought about by the phenyl groups being co-planar with the central pyrrole-NNpyrrole core in 6 cf. 3 and 5a, as evident in the solid state (Fig. 2).
abs = 429 nm,10 an effect which is maintained for the methylated analogues 5a and 5b. Solutions of the –BF2 adduct 6 exhibit a λmax absorption value of 666 nm, representing a ∼160 nm bathochromic shift cf. the free ligand 3. This is attributable to the stabilising effect of the –BF2 group upon co-planarisation and π-electron delocalisation within the core framework, as well as to the additional conjugation brought about by the phenyl groups being co-planar with the central pyrrole-NNpyrrole core in 6 cf. 3 and 5a, as evident in the solid state (Fig. 2).
| Solvent | UV-Vis absorption maxima (λmax abs nm) | Molar absorptivity ε (mol L−1 cm−1) × 104 | Fluorescence emission (λmax em nm)ab | Stokes shift (nm) | |
|---|---|---|---|---|---|
| a Excited at 504 nm.b In all cases, emission was sufficiently weak to prevent calculation of quantum yield. | |||||
| 3 | MeCN | 504 | 6.3 | 602 | 98 |
| 522 | 6.3 | ||||
| DMF | 504 | 5.1 | 583 | 79 | |
| 532 | 5.2 | 606 | |||
| THF | 504 | 5.1 | 565 | 61 | |
| 529 | 4.9 | 603 | |||
| CH2Cl2 | 513 | 6.1 | 576 | 63 | |
| 534 | 6.1 | ||||
| 4 | MeCN | 570 | 4.0 | 635 | 65 |
| DMF | 576 | 3.8 | 634 | 58 | |
| 5a | MeCN | 510 | 2.8 | ||
| DMF | 514 | 2.8 | 599 | 85 | |
| 5b | MeCN | 512 | 3.7 | 594 | 82 |
| DMF | 518 | 3.7 | 596 | 78 | |
| 6 | MeCN | 666 | 4.7 | — | — |
| DMF | 672 | 4.8 | — | — | |
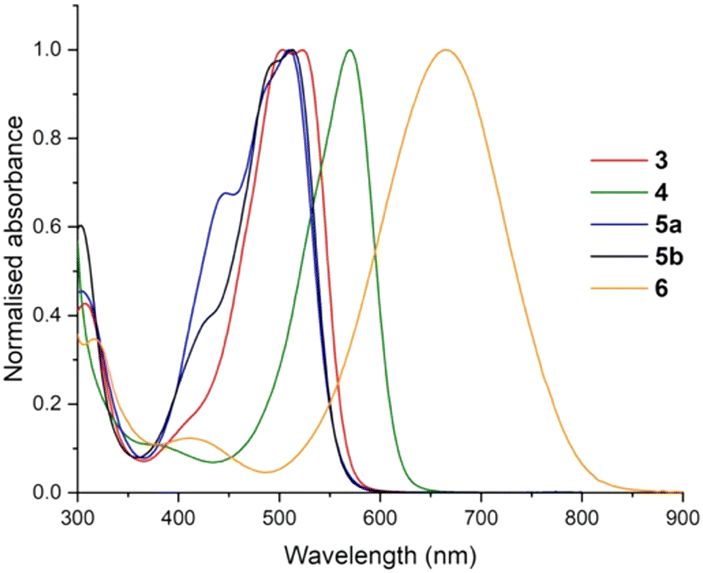 | ||
| Fig. 3 Normalised absorption spectra for 3–6 in MeCN. | ||
The stability of 3 in various solvents was then determined (Fig. S31–S39, ESI†). Solutions of 3 in MeCN and DMF were found to be more stable than those in CH2Cl2. Furthermore, solutions of 3 maintained in the dark and/or under anhydrous conditions were more stable than those exposed to sunlight. Exposure to light led only to decomposition, with no evidence for photoswitching of the azo group between Z and E configurations: this is unsurprising, given the significant steric crowding that would occur in an E configuration. The absorption properties of 3 were then explored in a range of solvents of varying polarity and viscosity, with significant solvatochromism observed (Fig. S40 and S41, ESI†). Intriguingly, azobispyrrole 3 exhibited only very weak fluorescence when soluble, yet visibly stronger fluorescence when present as a suspension or as a solid (Table S1, ESI†). Two different colours of fluorescence emission were sometimes, dependent upon mode of preparation, apparent to the naked eye, one more reddish and one more orange (Fig. S42, ESI†), yet the observed colour seemed not to necessarily correlate to the viscosity or polarity of the medium. The most stable suspensions were formed in dipropylene glycol and PEG-400, which gave relatively homogeneous dispersions persisting over many days (Table S1, ESI†). A suspension of 3 in sulfolane persisted a few days while less viscous media resulted in suspensions that quickly settled.
The origins of the variable and nonlinear responses with concentration of solutions and suspensions of azobispyrrole 3 were further investigated given the possibilities of aggregation-induced emission23 and the solid-state emission of organic chromophores.24 The emission spectra of solutions of 3 at various concentrations in MeCN were recorded (Fig. 4, left). As concentration was increased, emission at 625 nm (excitation at 325 nm) increased in a positive non-linear fashion cf. that at 415 nm (Fig. 4, right). The bathochromically-shifted emission at higher concentrations suggests an aggregation effect; and this was confirmed by comparison of emission intensity at 625 nm of 3 in MeCN versus 3 in PEG (Fig. S43, ESI†). Similarly, the large Stokes shift values suggest structurally distinctive ground and excited states, and inspire thorough analysis.25–27
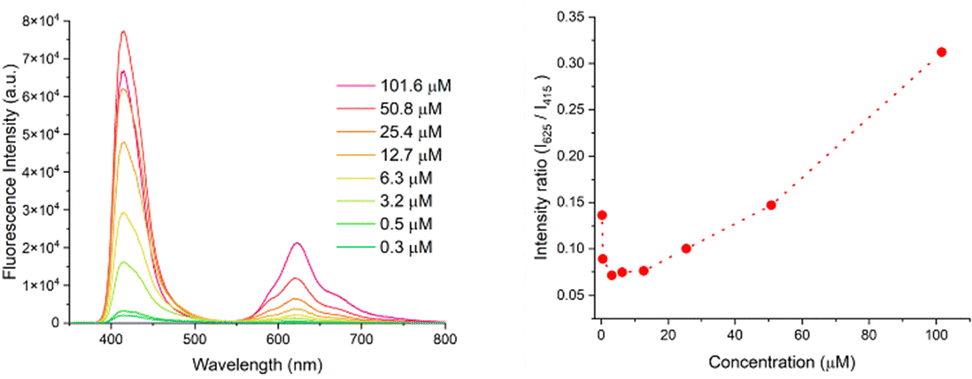 | ||
| Fig. 4 Left = emission spectra of azobispyrrole 3 in MeCN at various concentrations, with excitation at 365 nm; right = ratio of emission at 625 nm versus 415 nm at various concentrations, with excitation at 365 nm. | ||
Visual analysis of mixtures containing higher concentrations of 3 suggested emission associated with the presence of suspended material at higher concentrations (Table S1 and Fig. S44, ESI†). Presumably aggregation of 3 occurs as a result of π-stacking of the delocalised framework, although enhancement by hydrogen bonding, as seen in the solid-state structure for 3, is presumably also feasible in solution. Significant non-linear responses with concentration were observed for absorption and for emission upon the addition of MeOH or water to a solution of 3 in MeCN, thereby reducing the solubility and enhancing aggregation (Fig. S45–S48, ESI†). In contrast, when a solution of 3 in MeCN was treated with increasing amounts of toluene, loss of emission intensity at 625 nm was observed. This suggests reduced aggregation due to disruption of π-stacking by the toluene. Finally, although solid samples of 3 were seen to fluoresce slightly as a powder and on silica TLC plates, spectroscopic analysis of solid prepared by air-drying a drop of a solution of 3 in MeCN revealed only weak emission cf. the original solution from which the solid sample was prepared (Fig. S49, ESI†). However, the solid samples gave rise to nearly equal distributions of emission peaks corresponding to monomer (λmax 415 nm) and aggregates (λmax 625 nm).
In summary, the unprecedented tetra-aryl azobispyrrolic framework is introduced herein. Characterisation of 3 reveals that absorption and emission parameters are solvent dependent. Furthermore, solutions of azobispyrrole 3 exhibit concentration-dependent behaviour that is modified via the disruption of π-stacking by toluene, and through the addition of polar solvents. Reactivity of the azobispyrrole construct is briefly explored, demonstrating the potential for tunability of photochemical properties through post-synthetic modification as well as via variation of the key nitrobutanone starting material. Characterisation of each of the frameworks via single crystal X-ray diffraction analysis reveals the potential to extend π-delocalisation by manipulating the orientation of the flanking terminal α-substituents of the pyrroles, which presents another opportunity by which to achieve significant tunability of the photochemical potential of the novel azobispyrrole framework. Future work involves development of enhanced synthetic methods to azobispyrroles, such as to enable a thorough exploration of the tunability of photochemical properties through variation of substituents that influence conjugation across the framework.
Conceptualisation and investigation: AA, RD-R, SOS, JWH, MA, ECS, EBB, RLG, BM, KNR and AT. Funding acquisition, administration and supervision: AT. Writing and editing: AA, RD-R, JWH, EBB, RLG, BM, KNR and AT. We thank Dr M. Lumsden and Mr X. Feng (Dalhousie University) for sharing expertise in NMR spectroscopy and mass spectrometry.
This research was supported, in part, by: the Canada Research Chairs Program (950-232829); Canada Foundation for Innovation (JELF 39824); Research Nova Scotia (Research Opportunities Fund 2020-1208); Dalhousie University; NSERC of Canada via Discovery Grants, Undergraduate Student Research Awards and CREATE Training Program in BioActives (510963); Walter C. Sumner Memorial Fellowship to EBB; and Dalhousie's Southern African Student Education Project (SASEP) scholarship to RLG.
Conflicts of interest
There are no conflicts to declare.Data availability
Data supporting this article have been included as part of the ESI.† Crystallographic data for 3, 5a and 6 have been deposited at the CCDC under 2410353–2410355.Notes and references
- F. A. Jerca, V. V. Jerca and R. Hoogenboom, Nat. Rev. Chem., 2022, 6, 51–69 CrossRef PubMed.
- T. Dang, Z.-Y. Zhang and T. Li, J. Am. Chem. Soc., 2024, 146, 19609–19620 Search PubMed.
- R. Lin, P. K. Hashim, S. Sahu, A. S. Amrutha, N. M. Cheruthu, S. Thazhathethil, K. Takahashi, T. Nakamura, T. Kikukawa and N. Tamaoki, J. Am. Chem. Soc., 2023, 145, 9072–9080 CrossRef CAS PubMed.
- J. Calbo, C. E. Weston, A. J. P. White, H. S. Rzepa, J. Contreras-García and M. J. Fuchter, J. Am. Chem. Soc., 2017, 139, 1261–1274 CrossRef CAS PubMed.
- S. Crespi, N. A. Simeth and B. König, Nat. Rev. Chem., 2019, 3, 133–146 CrossRef CAS.
- T.-B. Zhang, F. Wang, J.-Y. Ouyang, Z.-W. Luo, J.-H. Qin, J.-H. Li and X.-H. Ouyang, Org. Lett., 2024, 26, 461–466 CrossRef CAS PubMed.
- Y. Li, B. O. Patrick and D. Dolphin, J. Org. Chem., 2009, 74, 5237–5243 CrossRef CAS PubMed.
- T. Zhang, B. Yang, T. Jiang, X. Kong, X. Huo, Y. Ma, K. Yang, M. Liu, Y. Liu, Z. Yao, H. Yu, H. Liu, K. Zhang and Y. Liu, J. Med. Chem., 2025, 68, 3020–3030 CrossRef CAS PubMed.
- H. Yokoi, S. Hiroto and H. Shinokubo, Org. Lett., 2014, 16, 3004–3007 CrossRef CAS PubMed.
- Z. Yoshida, H. Hashimoto and S. Yoneda, J. Chem. Soc. D, 1971, 1344–1345 RSC.
- J. Del Nero and B. Laks, Synth. Met., 1999, 101, 440–441 CrossRef.
- G. Zotti, S. Zecchin, G. Schiavon, A. Berlin, G. Pagani, A. Canavesi and G. Casalbore-Miceli, Synth. Met., 1996, 78, 51–57 CrossRef CAS.
- M. I. Bruce, A. Burgun, M. Jevric, J. C. Morris, B. K. Nicholson, C. R. Parker, N. Scoleri, B. W. Skelton and N. N. Zaitseva, J. Organomet. Chem., 2014, 756, 68–78 CrossRef CAS.
- Y. Ge and D. F. O’Shea, Chem. Soc. Rev., 2016, 45, 3846–3864 RSC.
- L. Jiao, Y. Wu, Y. Ding, S. Wang, P. Zhang, C. Yu, Y. Wei, X. Mu and E. Hao, Chem. – Asian J., 2014, 9, 805–810 CrossRef CAS PubMed.
- M. A. T. Rogers, J. Chem. Soc., 1943, 590–596 RSC.
- M. Grossi, A. Palma, S. O. McDonnell, M. J. Hall, D. K. Rai, J. Muldoon and D. F. O’Shea, J. Org. Chem., 2012, 77, 9304–9312 CrossRef CAS PubMed.
- Q. Qi, S. Huang, X. Liu and I. Aprahamian, J. Am. Chem. Soc., 2024, 146, 6471–6475 CrossRef CAS PubMed.
- Q. Qiu, Q. Qi, J. Usuba, K. Lee, I. Aprahamian and G. G. D. Han, Chem. Sci., 2023, 14, 11359–11364 RSC.
- J. He, X. Chang, C. Zou, Y. Yu, S. Han, C. Wu, S. Ou, W. Lu and K. Li, J. Org. Chem., 2023, 88, 14836–14841 CrossRef CAS PubMed.
- E. B. Kömüşdoğan, S. Batool, E. Şahin, E. Yildirim, M. Işık and C. Tanyeli, Chem. Commun., 2025, 61, 576–579 RSC.
- B. Lee, B. G. Park, W. Cho, H. Y. Lee, A. Olasz, C.-H. Chen, S. B. Park and D. Lee, Chem. – Eur. J., 2016, 22, 17321–17328 CrossRef CAS PubMed.
- J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429–5479 CrossRef CAS PubMed.
- M. K. Bera, P. Pal and S. Malik, J. Mater. Chem. C, 2020, 8, 788–802 RSC.
- K. Tanaka and Y. Chujo, NPG Asia Mater., 2015, 7, e223 CrossRef CAS.
- Z. Liu, Z. Jiang, M. Yan and X. Wang, Front. Chem., 2019, 7, 712 CrossRef CAS PubMed.
- Y. Yang, X. Su, C. N. Carroll and I. Aprahamian, Chem. Sci., 2012, 3, 610–613 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterisation data. CCDC 2410353–2410355. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5cc02884a |
| This journal is © The Royal Society of Chemistry 2025 |