
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A non-innocent, π-extended N-heterobicyclic carbene–thiolate ligand†
Michael
Marquardt
ab,
Laure
Vendier
b,
Alix
Sournia-Saquet
b,
Vincent
Maurel
 c,
Jean-Marie
Mouesca
c,
Stéphanie
Bastin
*b,
Ivan
Castillo
*a and
Vincent
César
*b
c,
Jean-Marie
Mouesca
c,
Stéphanie
Bastin
*b,
Ivan
Castillo
*a and
Vincent
César
*b
aInstituto de Química, UNAM, Circuito Exterior, Ciudad Universitaria México, 04510, Mexico. E-mail: joseivan@unam.mx
bLCC-CNRS, Université de Toulouse, CNRS, UPS, Toulouse, France. E-mail: stephanie.bastin@lcc-toulouse.fr; vincent.cesar@lcc-toulouse.fr
cUniversité Grenoble Alpes, CEA, CNRS, IRIG, SyMMES, 38000 Grenoble, France
First published on 23rd June 2025
Abstract
A bidentate NHC–thiolate ligand based on the imidazo[1,5-a]pyridine scaffold and featuring an extended π-system is described. It is derived from a readily available zwitterionic precursor and the chemistry of the corresponding homoleptic Ni(II) complex is investigated.
Thanks to their beneficial stereoelectronic properties, N-heterocyclic carbenes (NHCs)1,2 constitute a privileged ligand class in organometallic, main-group, materials, nano-, and surface chemistries with outstanding applications in organometallic and organic catalysis.3 They are particularly appealing strongly bounded C-anchors for the design of functional polydentate ligands, leading to a plethora of ligand scaffolds built by the combination of NHC units with heteroelements.4 Interestingly, while the metal–thiolate bond is crucial in nature, playing a key role in important biological processes, such as H2 or N2 activation by hydrogenases or nitrogenases, respectively, only a handful of NHC–thiolate ligands have been reported to date (Fig. 1A–E).5 Merging our interests in bioinspired sulfur-rich ligands6 and in N-heterobicyclic carbenes based on an imidazo[1,5-a]pyridine scaffold (ImPy),7,8 we thus devised to develop the LX-type ligand 1−, in which the thiolate moiety is directly connected to the bicyclic carbenic heterocycle. Featuring an extended π-system, this ligand structure might also be redox-active by stabilizing an electro-generated radical through delocalization. To our knowledge, the redox-active nature of the ImPy scaffold has not been previously reported in the literature.9 We report herein the synthesis, the coordination ability with nickel and the non-innocent character of ligand 1−.
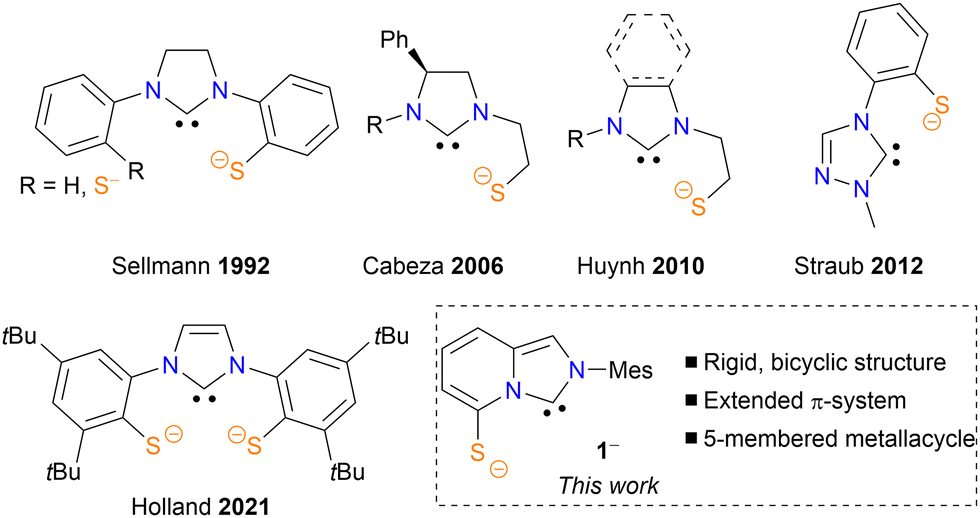 | ||
| Fig. 1 NHC–thiolate ligands. Mes = 2,4,6-trimethylphenyl. | ||
Following a late-stage aromatic nucleophilic substitution strategy previously developed by some of us,7b,10 the gram-scale synthesis of the pro-ligand 1·H was achieved by treating the 5-bromoimidazo[1,5-a]pyridinium bromide [2·H]Br with an excess of Na2S·9H2O in DMF at room temperature (Scheme 1). The air- and water-stable zwitterionic compound 1·H, containing an internal thiophenolate, was isolated in 68% yield through column chromatography. 1·H was fully characterized by spectroscopic and analytical techniques and its molecular formula was firmly established through a single-crystal X-ray diffraction analysis (Fig. 2).11 The zwitterionic character of 1·H was confirmed by elemental analysis, by the absence of a counter-anion in its crystal structure, and by the presence of a nominal thiophenolate group. Nevertheless, the C–S bond length of 1.7061(13) Å evidences a partial C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S double bond character, indicating that the sulfur valence electrons are in resonance with the imidazopyridinium aromatic system. The C–C bonds of the bicyclic heterocycle, on the other hand, form the pattern of alternating single and double C–C bonds typical of ImPy-type compounds. Hence, 1·H should be classified as a “conjugated mesomeric betaine” (CMB) according to the Ollis classification.12
S double bond character, indicating that the sulfur valence electrons are in resonance with the imidazopyridinium aromatic system. The C–C bonds of the bicyclic heterocycle, on the other hand, form the pattern of alternating single and double C–C bonds typical of ImPy-type compounds. Hence, 1·H should be classified as a “conjugated mesomeric betaine” (CMB) according to the Ollis classification.12
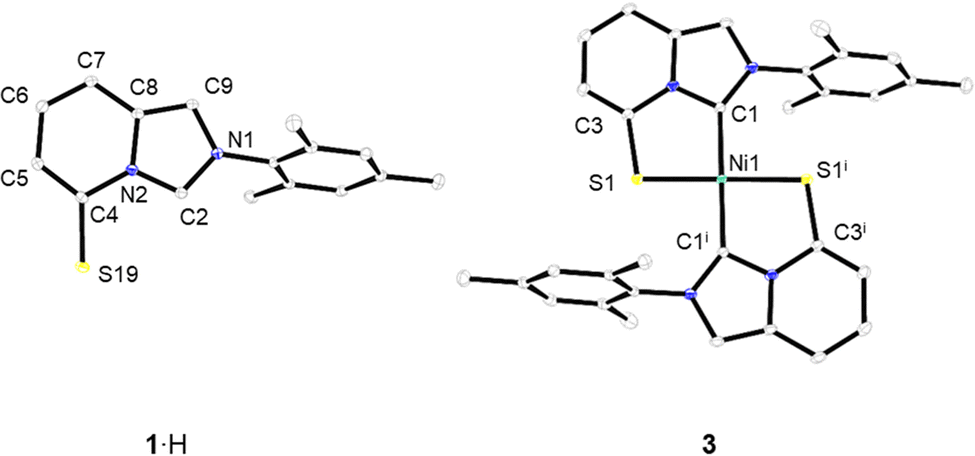 | ||
| Fig. 2 Molecular structures of zwitterionic precursor 1·H (left) and complex 3 (right) (ellipsoids drawn at the 50% probability level; hydrogen atoms have been omitted for clarity). Selected bond lengths (Å) and angles (deg): for 1·H: C4–S19 1.7061(13), C4–C5 1.3743(17), C5–C6 1.4168(18), C6–C7 1.3657(18), C7–C8 1.4139(17), C8–C9 1.3761(17). For 3: Ni1–C1 1.9144(18), Ni1–S1 2.1968(4), C3–S1 1.7273(19), C1–Ni1–S1 86.96(6). | ||
Deprotonation of 1·H with 1 equivalent of potassium bis(trimethylsilyl)amide (KHMDS) in THF at room temperature led to the clean and quantitative generation of the stable anionic NHC [1]K, which was proven by the disappearance of the signal of the N2CH proton in the 1H NMR spectrum and the appearance of a singlet at δC = 205.5 ppm (in THF-d8) in the typical range of free ImPy-type carbenes (206–209 ppm).13
 | ||
| Scheme 1 Synthesis of the zwitterionic precursor 1·H, generation of the free anionic carbene [1]K and synthesis of the homoleptic nickel(II) complex 3. | ||
The free anionic NHC [1]K was then trapped by the addition of 0.5 equiv. of NiCl2(dme) at −35 °C. This resulted in a dark green mixture that was purified by column chromatography to obtain a red solid, identified as [Ni(1)2] (3), in 55% yield. Complex 3 was fully characterized by analytical and spectroscopic techniques, supplemented by an X-ray diffraction analysis on single crystals, grown by slow vapor diffusion of diethyl ether into a concentrated dichloromethane solution of 3. The complex crystallized in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , with a square planar arrangement around Ni(II) defined by the two LX-type chelating ligands 1− (Fig. 2). The perfect planar geometry around the Ni(II) center was evidenced by the torsion angle of 0 degrees (presence of an inversion point in the lattice) between the planes of the bicyclic heterocycles of the two ligand fragments. The coordination of the carbenic carbon atoms N2C of ligands 1 was inferred from the 13C{1H} NMR spectrum, with a chemical shift of δC = 159.3 ppm, which is consistent with related Ni(II)–ImPy complexes.14
, with a square planar arrangement around Ni(II) defined by the two LX-type chelating ligands 1− (Fig. 2). The perfect planar geometry around the Ni(II) center was evidenced by the torsion angle of 0 degrees (presence of an inversion point in the lattice) between the planes of the bicyclic heterocycles of the two ligand fragments. The coordination of the carbenic carbon atoms N2C of ligands 1 was inferred from the 13C{1H} NMR spectrum, with a chemical shift of δC = 159.3 ppm, which is consistent with related Ni(II)–ImPy complexes.14
The electrochemical behaviors of pro-ligand 1·H and complex 3 were investigated by cyclic voltammetry (CV). The zwitterionic precursor 1·H displayed an irreversible oxidation wave at Epa = 0.57 V vs. SCE (Fig. S38, ESI†), which could be assigned to the formation of a sulfur-centered radical. Conversely, the CV of 3 in dichloromethane showed the presence of two reversible waves at E1/2 = 0.39 and 1.19 V vs. SCE (Fig. 3).
 | ||
| Fig. 3 Anodic cyclic voltammogram of complex 3 (Glassy carbon working electrode, 1 mM in CH2Cl2 with 0.1 M Bu4NPF6 as the supporting electrolyte, 100 mV s−1 scan rate; potentials were calibrated against Fc+/Fc used as an internal standard with E1/2 (Fc+/Fc) = 0.46 V vs. SCE). | ||
The first redox process was proved to correspond to a one-electron oxidation resulting in a stable cationic species, [3]+, that was obtained by bulk electrolysis at 0.8 V vs. SCE. As a half-integer species, the electrogenerated complex [3]+ was probed by EPR spectroscopy in frozen CH2Cl2/toluene (1/1) solution and exhibited a rhombic EPR signal with g-tensor eigenvalues of 1.988, 2.028, and 2.147 (gav = 2.054) (Fig. 4). These values were in very good agreement with those computed at the VBP XC level and the DFT calculations provided further insights into the electronic nature of complex [3]+.
 | ||
| Fig. 4 (a) X-band EPR spectrum of the electrochemically generated [3]+ in frozen CH2Cl2/toluene solution (+0.1 M TBAP) at 110 K. Microwave frequency = 9.417556 GHz, mod. ampl. = 0.2 mT, and power = 6.4 mW. (b) g values obtained by simulations15 and DFT calculations. (c) DFT-optimized structure of [3]+ and representation of the SOMO spin α and LUMO spin β (isodensity value of 0.05 a.u.). | ||
Indeed, apart from a small contribution of the nickel center, the SOMO is delocalized over the two sulfur ligands and the π-systems of the imidazopyridine scaffolds of the NHC ligands. This was further corroborated by the Mülliken spin population distribution, where the atomic spin populations are shared by the central Ni metal (∼1/4 of the total) and by the two NHC–thiolate ligands 1− (∼3/4 of the total) (Fig. S40, ESI†). The description of [3]+ is therefore intermediate between a genuine Ni(III) complex and a Ni(II) complex with an oxidized ligand framework. This was also reflected by the experimental isotropic Landé factor of [3]+ (gav = 2.054), which appears to be the weighted average between the free electron value (giso = 2.002) and the typical values of Ni(III) complexes (gav = 2.14–2.18),16 with relative weights (2 to 1) relatively close to the one calculated for the Mülliken spin distribution (3/4 on the ligands and 1/4 on the metal center).
The chemical oxidation of complex 3 was then carried out. Unfortunately, although 3 easily reacted with various organic and inorganic oxidizing agents such as Fc(BF4), (NO)(SbF6), PhICl2 or CuCl2, the nature of the products could not be firmly determined in most of the cases (see the ESI† for more details).
At that point, we were interested in evaluating the propensity of complex 3 to act as a S-donating metalloligand to form heterometallic aggregates. Indeed, metal–thiolate (or sulfide) bonds are widespread in nature and often serve as connecting units between metallic centers in metalloenzymes or metalloproteins.17,18 From a synthetic point of view, several nickel–silver clusters were previously obtained, thanks to the great affinity of Ag+ for coordinated thiolato groups.19 Thus, the reaction between complex 3 and AgSbF6 was tested, which led to the formation of cluster 4, in which two Ag+ centers were sandwiched by two Ni complexes 3 through coordination of the remaining lone pairs of the thiolate ligands (Scheme 2). The molecular structure of the stable complex 4 was firmly established by single crystal X-ray diffraction analysis (Fig. 5). Most notably, the central Ag2Ni2S4 core is perfectly planar and the bond angles and lengths within this plane indicate significant metal–metal bonding interactions between the silver atoms themselves as well as between the nickel and silver centers. Indeed, although the Ag1–Ag1iii bond length [3.1839(15) Å] is a little longer than that in previously reported Ni–Ag clusters [Ag⋯Ag of ∼3.0 Å], it is clearly below the sum of the van der Waals radii of silver atoms [3.42 Å]. Likewise, the Ni–Ag distance [2.9519(11) Å] is intermediate between a covalent Ni–Ag bond [2.69] and a non-bonding distance [3.35 Å].19b
 | ||
| Scheme 2 Reaction of complex 3 with AgSbF6 and HBF4·OEt2. | ||
 | ||
| Fig. 5 Solid-state structure of complex 4 (ellipsoids drawn at the 50% probability level, hydrogen atoms and counter-anions have been omitted for clarity). Selected bond lengths (Å) and angles (degrees): Ni1–C10 1.923(5), Ni1–S1 2.2001(11), C8–S1 1.751(5), Ag1–S1 2.4431(13), Ni1–Ag1 2.9519(11), Ag1–Ag1iii 3.1839(15), S1–Ag1–S1i 151.33(6), C10–Ni1–S1 86.91(14), C8–S1–Ni1 100.05(16), C8–S1–Ag1 98.89(16). | ||
In order to study the reactivity of 3 towards protons and investigate if 3 can serve as a potential proton reducing catalyst, 3 was treated with equimolar amounts of Brønsted acids to isolate and characterize the protonated species. As a result, HBF4·OEt2 proved to have an adequate pKa value to react with 3 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. Thus, when 3 was treated with HBF4·OEt2 in dry dichloromethane at room temperature, an immediate color change to dark green was observed. NMR analysis of the crude product showed the selective formation of one diamagnetic product. Characterization by 1D and 2D NMR methods revealed the presence of a characteristic methylene group at δH = 5.43 ppm and δC = 65.4 ppm (in CDCl3), arising from the uncommon protonation of the C-1 position of the imidazolyl ring of one NHC ligand, leading to the formation of the cationic complex 5 (Scheme 2). Complex 5 is stable for several hours at room temperature in CH2Cl2 but the protonation appeared reversible, since only the starting complex 3 was isolated by crystallization.
:1 ratio. Thus, when 3 was treated with HBF4·OEt2 in dry dichloromethane at room temperature, an immediate color change to dark green was observed. NMR analysis of the crude product showed the selective formation of one diamagnetic product. Characterization by 1D and 2D NMR methods revealed the presence of a characteristic methylene group at δH = 5.43 ppm and δC = 65.4 ppm (in CDCl3), arising from the uncommon protonation of the C-1 position of the imidazolyl ring of one NHC ligand, leading to the formation of the cationic complex 5 (Scheme 2). Complex 5 is stable for several hours at room temperature in CH2Cl2 but the protonation appeared reversible, since only the starting complex 3 was isolated by crystallization.
In summary, we have reported a viable and straightforward route to a new bidentate NHC–thiolate embedded in a π-extended bicyclic aromatic system. Through the synthesis and reactivity of the corresponding square-planar Ni(II) complex, the ligand was shown to be redox-active and chemically non-innocent, allowing the formation of heterometallic clusters and unusual protonated species. This opens up exciting possibilities for the application of this ligand in bio-inspired small molecule activation and catalysis. Studies in these directions are currently underway in our laboratories.
The authors thank UNAM and the CNRS for financial support and the International Research Project MCCM of the CNRS for exchanges between Toulouse and Mexico, and DGAPA-PAPIIT (IN216823). Dr Baptiste Martin (LCC) is gratefully acknowledged for EPR measurements.
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the ESI.†References
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS PubMed.
- H. V. Huynh, Chem. Rev., 2018, 118, 9457–9492 CrossRef CAS PubMed.
- Selected recent reviews: (a) T.-Y. Li, S.-J. Zheng, P. I. Djurovich and M. E. Thompson, Chem. Rev., 2024, 124, 4332–4392 CrossRef CAS PubMed; (b) M. Koy, P. Bellotti, M. Das and F. Glorius, Nat. Catal., 2021, 4, 352–363 CrossRef CAS; (c) Q. Liang and D. Song, Chem. Soc. Rev., 2020, 49, 1209–1232 RSC; (d) Q. Zhao, G. Meng, S. P. Nolan and M. Szostak, Chem. Rev., 2020, 120, 1981–2048 CrossRef CAS PubMed; (e) C. A. Smith, M. R. Narouz, P. A. Lummis, I. Singh, A. Nazemi, C.-H. Li and C. M. Crudden, Chem. Rev., 2019, 119, 4986–5056 CrossRef CAS PubMed; (f) A. Doddi, M. Peters and M. Tamm, Chem. Rev., 2019, 119, 6994–7112 CrossRef CAS PubMed.
- (a) C. Fliedel, A. Labande, E. Manoury and R. Poli, Coord. Chem. Rev., 2019, 394, 65–103 CrossRef CAS; (b) S. Hameury, P. de Fremont and P. Braunstein, Chem. Soc. Rev., 2017, 46, 632–733 RSC; (c) V. Charra, P. de Frémont and P. Braunstein, Coord. Chem. Rev., 2017, 341, 53–176 CrossRef CAS; (d) C. Fliedel and P. Braunstein, J. Organomet. Chem., 2014, 751, 286–300 CrossRef CAS.
- (a) D. Sellmann, W. Prechtel, F. Knoch and M. Moll, Organometallics, 1992, 11, 2346–2348 CrossRef CAS; (b) J. A. Cabeza, I. da Silva, I. del Río and M. G. Sánchez-Vega, Dalton Trans., 2006, 3966–3971 RSC; (c) D. Yuan and H. V. Huynh, Dalton Trans., 2011, 40, 11698–11703 Search PubMed; (d) S. C. Holm, F. Rominger and B. F. Straub, J. Organomet. Chem., 2012, 719, 54–63 CrossRef CAS; (e) A. L. Speelman, K. L. Skubi, B. Q. Mercado and P. L. Holland, Inorg. Chem., 2021, 60, 1965–1974 Search PubMed.
- (a) B. A. Murueta-Cruz, A. Berlanga-Vázquez, D. Martínez-Otero, L. N. Benitez, I. Castillo and A. Mondragón-Díaz, Eur. J. Inorg. Chem., 2021, 2089–2098 Search PubMed; (b) A. Mondragón-Díaz, E. Robles-Marín, B. A. Murueta-Cruz, J. C. Aquite, P. R. Martínez-Alanis, M. Flores-Alamo, G. Aullón, L. N. Benítez and I. Castillo, Chem. – Asian J., 2019, 14, 3301–3312 CrossRef PubMed.
- (a) T. Kittikool, K. Phakdeeyothin, A. Morales, C. Barthes, L. Vendier, S. Yotphan, S. Bontemps, S. Bastin, A. Lledós, O. Baslé and V. César, Chem. Eur., 2024, 2, e202300083 Search PubMed; (b) L. Pallova, L. Abella, M. Jean, N. Vanthuyne, C. Barthes, L. Vendier, J. Autschbach, J. Crassous, S. Bastin and V. César, Chem. Eur. J., 2022, 28, e202200166 CrossRef CAS PubMed.
- For reviews, see: (a) P. Teixeira, S. Bastin and V. César, Isr. J. Chem., 2023, 63, e202200051 Search PubMed; (b) N. U. D. Reshi and J. K. Bera, Coord. Chem. Rev., 2020, 422, 213334 CrossRef CAS.
- A redox non-innocent bidentate ImPy–phenoxide ligand was reported, but ImPy was not involved in the redox process. Please see: R. Kunert, C. Philouze, O. Jarjayes, T. Storr and F. Thomas, Organometallics, 2023, 42, 1550–1560 CrossRef CAS.
- (a) Y. Tang, I. Benaissa, M. Huynh, L. Vendier, N. Lugan, S. Bastin, P. Belmont, V. César and V. Michelet, Angew. Chem., Int. Ed., 2019, 58, 7977–7981 Search PubMed; (b) I. Benaissa, L. Pallova, M.-E. Morantin, T. Lafitte, M. Huynh, C. Barthes, L. Vendier, N. Lugan, S. Bastin and V. César, Chem. – Eur. J., 2019, 25, 13030–13036 CrossRef CAS PubMed; (c) K. Azouzi, C. Duhayon, I. Benaissa, N. Lugan, Y. Canac, S. Bastin and V. César, Organometallics, 2018, 37, 4726–4735 Search PubMed.
- CCDC 2449873–2449875† contain the supplementary crystallographic data for this paper.
- W. D. Ollis, S. P. Stanforth and C. A. Ramsden, Tetrahedron, 1985, 41, 2239–2329 CrossRef CAS.
- M. Alcarazo, S. J. Roseblade, A. R. Cowley, R. Fernández, J. M. Brown and J. M. Lassaletta, J. Am. Chem. Soc., 2005, 127, 3290–3291 Search PubMed.
- R. Kunert, C. Philouze, O. Jarjayes, T. Storr and F. Thomas, Organometallics, 2023, 42, 1550–1560 CrossRef CAS.
- EPR spectra were fitted using Easyspin software: S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed.
- See for examples: (a) J.-P. Cloutier, L. Rechignat, Y. Canac, D. H. Ess and D. Zargarian, Inorg. Chem., 2019, 58, 3861–3874 Search PubMed; (b) I. Bhowmick, A. J. Roehl, J. R. Neilson, A. K. Rappé and M. P. Shores, Chem. Sci., 2018, 9, 6564–6571 Search PubMed; (c) A. Sarkar, S. Das, P. Mondal, B. Maiti and S. Sen Gupta, Inorg. Chem., 2023, 62, 20439–20449 CrossRef CAS PubMed; (d) E. M. Gale, A. C. Simmonett, J. Telser, H. F. SchaeferIII and T. C. Harrop, Inorg. Chem., 2011, 50, 9216–9218 CrossRef CAS PubMed; (e) P. Pirovano, E. R. Farquhar, M. Swart, A. J. Fitzpatrick, G. G. Morgan and A. R. McDonald, Chem. – Eur. J., 2015, 21, 3785–3790 CrossRef CAS PubMed.
- W. Weigand and P. Scholhammer, Bio-inspired catalysis, Metal-sulfur complexes, John Wiley & Sons, 2014 Search PubMed.
- M. Gennari and C. Duboc, Acc. Chem. Res., 2020, 53, 2753–2761 CrossRef CAS PubMed.
- (a) A. Obanda, K. Valerius, J. T. Mague, S. Sproules and J. P. Donahue, Organometallics, 2020, 39, 2854–2870 Search PubMed; (b) S. R. Biltek, A. C. Reber, S. N. Khanna and A. Sen, J. Phys. Chem. A, 2017, 121, 5324–5331 CrossRef CAS PubMed; (c) S. R. Biltek, S. Mandal, A. Sen, A. C. Reber, A. F. Pedicini and S. N. Khanna, J. Am. Chem. Soc., 2013, 135, 26–29 CrossRef CAS PubMed; (d) T. Konno, M. Usami, M. Hirotsu, T. Yoshimura and T. Kawamoto, Chem. Commun., 2004, 2296–2297 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2449873–2449875. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5cc02841h |
| This journal is © The Royal Society of Chemistry 2025 |