
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNitrile hydration and α-deuteration of amides catalyzed by a PCNHCP Mn(I) pincer complex
Rohit
Kamte
,
Ranjeesh
Thenarukandiyil
,
Kartick
Dey
,
Natalia
Fridman
and
Graham
de Ruiter
 *
*
Schulich Faculty of Chemistry and the Resnick Sustainability Center for Catalysis, Technion – Israel Institute of Technology, Technion City, Haifa 3200008, Israel. E-mail: graham@technion.ac.il
First published on 29th September 2025
Abstract
Amides are prevalent structural motifs in nature and important functional groups in synthetic organic chemistry. Amide synthesis typically requires harsh reaction conditions necessitating alternative strategies that are atom economic, catalytic, and easy to perform. Here we present the efficient hydration of nitriles, catalyzed by a manganese(I) PCNHCP pincer complex, that furnishes a diverse set of amides with excellent functional group compatibility. The reaction occurs at moderate temperatures (90 °C) and produce the corresponding amides in excellent yields (up to 99%). Deuterated amides can also be accessed via subsequent H/D exchange with D2O using the same catalyst.
In recent years, earth-abundant metals have increasingly been investigated as suitable replacements for precious metal catalysts in organic syntheses.1 Their low toxicity, high availability, and low environmental impact contribute to their sustainable footprint when combined with chemical reactions that exhibit a high atom economy.2 One such reaction is the hydration of nitriles to form amides.
Amides are important constituents in a variety of organic compounds including agrochemicals, pharmaceuticals, and most importantly; biomolecules (i.e., proteins).3 Their synthesis generally relies on the condensation of an amine with a carboxylic acid, ester, or acyl chloride, typically requiring harsh conditions and additives.3,4 As result, developing practical approaches for amide synthesis is particularly attractive. One such approach is the hydrolysis of nitriles that generally requires forcing reaction conditions or is catalyzed by precious metals such as gold,5 ruthenium,6 rhodium,7 platinum,8 and palladium.9
In recent years approaches have been developed that (i) use earth-abundant metal catalysts, (ii) are highly atom economic, and (iii) proceed under mild reaction conditions.10 For example, Milstein and co-workers have shown that secondary amides can be directly synthesized from alcohols and amines through dehydrogenative coupling.11 The same authors used a different approach when demonstrating that primary amides could also easily be accessed through the hydration of nitriles via manganese mediated “template catalysis” (Fig. 1A).12 Piers and co-workers on the other hand, developed the nickel catalyzed hydration of nitriles that is mechanistically distinct from template catalysis (Fig. 1A), and proceeds via a nickel hydroxide intermediate.13 Yet, besides the contributions from Milstein and Piers, there are not many examples of well-defined catalysts that use earth-abundant metals for catalytic nitrile hydration (Fig. 1).14
 | ||
| Fig. 1 (A) Excerpt of the state-of-the-art. (B) Herein reported manganese catalyzed hydration of nitriles. | ||
Instead of relying on metal–ligand cooperativity, our group has focused on developing earth-abundant metal catalysts with strong-field PCNHCP pincer ligands.15 Recently, our group demonstrated that Mn(I) PCNHCP pincer complexes are highly active catalysts for the α-methylation of ketones,16 and the hydrogenation17 and cyano-alkylation of α,β-unsaturated ketones.15c Here we present the efficient hydration of a variety of nitriles catalyzed by our previously reported catalyst [(PCNHCP)Mn(CO)2H] (Fig. 1B; 1).16
The reaction occurs at moderate temperature (90 °C) and exhibits excellent functional group compatibility. The corresponding amides are isolated in excellent yields and could afterwards be deuterated via subsequent H/D exchange. Mechanistic studies indicate a formal Lewis acid catalyzed pathway in which water is reversibly deprotonated, which is further discussed in this report.
We became interested in the hydration of nitriles because of the facile H/D exchange between our previously reported complex 1 and D2O (Fig. 2). Addition of 20 equiv. of D2O to a solution of [(PCNHCP)Mn(CO)2H] (1) in THF resulted in the clean formation of [(PCNHCP)Mn(CO)2D] (2) as judged by 1H, 2H, and 31P NMR spectroscopy (Fig. S1–S3). The H/D exchange suggest that complex 1 can, at least transiently generate hydroxide anions that are beneficial for nitrile hydration as shown by Piers and co-workers.13 Indeed, our optimization protocol (Table S1) shows that heating a solution of benzyl cyanide in THF or tert-butanol in the presence of H2O (5 equiv.) and catalyst 1 (1 mol%), resulted in the quantitative formation of 2-phenylacetamide. Using other manganese Lewis acids such as Mn(OTf)2(CH3CN)2, or other manganese complexes such as [(PCNHCP)Mn(CO)2Br] (3) or [(PCNHCP)Mn(CO)3][PF6] (4), did not result in the formation of any detectable product (Table S1)
 | ||
| Fig. 2 H/D exchange on manganese complex 1 by D2O. | ||
With the optimized conditions established, we proceeded to establish the scope of the herein reported hydration reaction with a variety of electronically and sterically differentiated nitriles (Table 1). As shown in Table 1, various electron deficient benzyl cyanides could be converted to their corresponding 2-phenylacetamides 5b–5d in near quantitative yields (98–99%). Likewise, 2-phenoxyacetonitrile was quantitatively converted to 2-phenoxyacetamide 5e (Table 1). Besides benzyl cyanides, (substituted) benzonitriles could also be selectively converted to their corresponding benzamides (5f–5o). With these substrates, both electron withdrawing groups (e.g., –F, –Cl, –Br, –CF3) and electron donating groups (e.g., –OMe) are well tolerated and their corresponding benzamides 5i–5n were all isolated in excellent yields (Table 1). Functional groups such as ketones (5o), alcohols (5p) or heteroaromatics (5p–5s) were also compatible with our established protocol yielding the corresponding amides in excellent yields. Even simple aliphatic nitriles could be converted to their corresponding amides(5t–5y), although sterically encumbered nitriles (5v and 5z) or nitriles bearing long alkyl chains (5x) showed somewhat diminished yields (Table 1).
| a Reactions were performed in a 38 mL Ace® pressure tube with nitrile (1.0 mmol), catalyst (1 mol%), and 5.0 equiv. of H2O in tBuOH (1 mL) at 90 °C for 24 hours. Yields were determined by 1H NMR spectroscopy using 1,3,5-trimethoxybenzene as internal standard. |
|---|

|
Compared to Milstein's Mn–PNP pincer catalyst, which hydrates nitriles through a metal–ligand cooperativity,12 involving ketimido and enamido intermediates, our Mn(I)–PCNHCP pincer complex promotes nitrile hydration via a Lewis-acid pathway without the need for such cooperativity. As such our system still enables a broad substrate scope and high yields under mild and additive-free conditions. Notably, our approach uniquely enables isotope labeling at the amide stage, where the activated α-methylene of the amide undergoes selective H/D exchange in the presence of D2O and catalyst 1. For example, 2-phenylacetamide (5a) was quantitatively deuterated in THF under mild conditions (Fig. S73 and S74). Whereas Milstein's Mn–PNP system achieves α-deuteration at the nitrile stage under specific solvent regimes, our Mn–PCNHCP catalyst provides a complementary late-stage route, granting practical access to deuterated amides directly from the amide products, providing a green methodology for facile H/D exchange.18
With the substrate scope established, we next sought to elucidate the underlying mechanism of the herein reported nitrile hydration reaction. While prior studies on the cyanoalkylation of α,β-unsaturated ketones suggested that complex 1 can activate nitriles at elevated temperatures, such pathways are unlikely here due to the absence of an activatable α-CH group in many substrates.15c Instead, we propose that, based on our H/D exchange experiments (Fig. 2), hydroxide ions are formed upon deprotonation of water by complex 1. This hypothesis is further supported by the fact that H2 inhibits the reaction (Scheme S11), suggesting that water activation is crucial in forming the reactive intermediate I (Scheme 1).19 After the initial activation step, the bound nitrile undergoes nucleophilic attack at the nitrile-carbon by the hydroxide ion, forming an imide intermediate (III), which tautomerizes to the manganese amide IV (Scheme 1). Protonation of intermediate IV, releases the amide and regenerates the active catalyst (I).
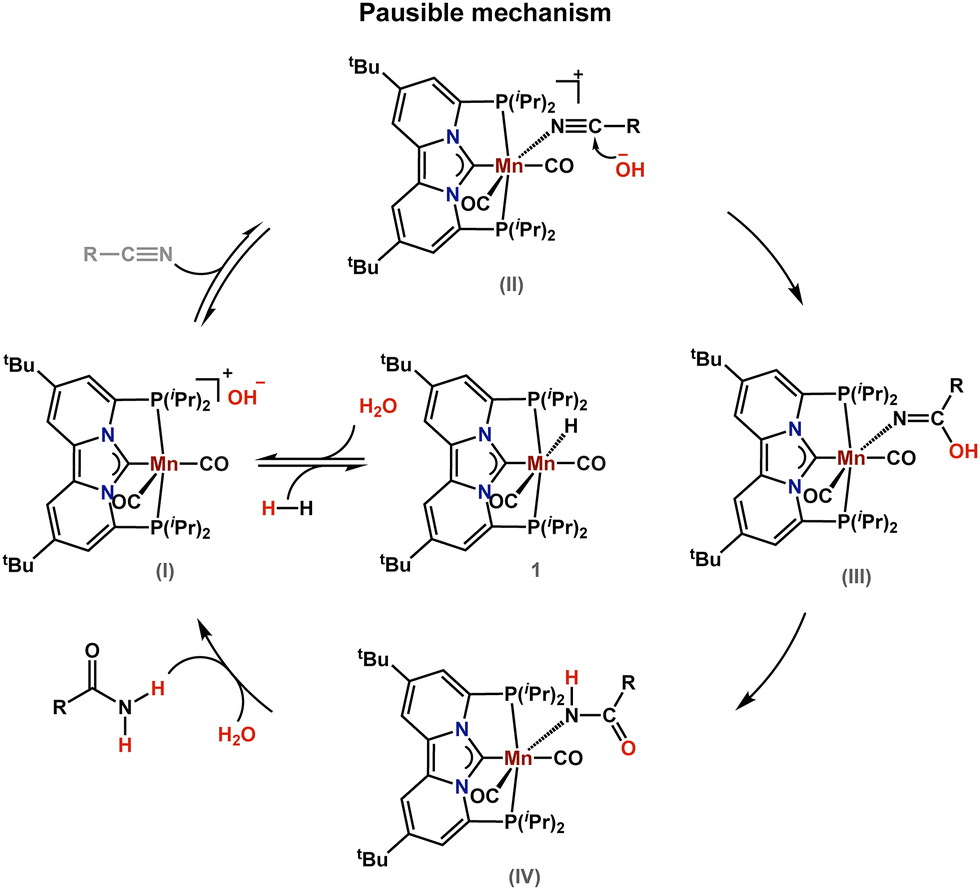 | ||
| Scheme 1 Plausible mechanism for the manganese catalyzed hydration of nitriles. | ||
The involvement of IV in the herein proposed mechanistic scenario is based on additional control experiments that demonstrated that IV could also be isolated after heating a solution of 1 (1.0 equiv.), benzyl cyanide (8.0 equiv.), and excess H2O (1.0 mL) in tert-butanol, at 90 °C for 48 h. The identity of IV was unequivocally established by X-ray crystallography (Fig. 3) and via independent synthesis (Fig. S6 and S7). Furthermore, IV catalyzes the hydration of benzyl cyanide to 2-phenyl-acetamide in 95% yield, supporting its role in the catalytic cycle (Scheme S8).
 | ||
| Fig. 3 Solid state structure of [(PCNHCP)Mn(Co)2(BA)] (IV). Ellipsoids are shown at the 30% probability level. Co-crystallized solvent molecules, amides, and hydrogen atoms are not shown for clarity. | ||
To gain mechanistic insight, we performed detailed kinetic studies on the model reaction of phenylacetonitrile in THF-d8. Monitoring substrate consumption at different catalyst loadings revealed exponential decay profiles, and linear fits upon plotting the natural logarithm of the substrate concentration versus time, confirming a first-order dependence. Subsequent kinetic studies also established first-order dependence with respect to catalyst, substrate, and H2O, affording an overall third-order rate law that does not indicate catalyst saturation behavior (Fig. S9–S11). Furthermore, performing Temperature dependent experiments between 70 and 100 °C yielded activation parameters of ΔG‡ = 26.9 kcal mol−1 and ΔS‡ = –36.1 cal mol−1 K−1. The relatively low barrier and large negative entropy suggest a highly ordered transition state in which substrate, catalyst, and hydroxide are preorganized, consistent with hydroxide attack as the rate-determining step.6g,20 This hypothesis was further supported by the observed secondary kinetic isotope effect of KH/KD = 1.538 measured with H2O and D2O. The observed secondary kinetic isotope effect also rules out protonolysis of the manganese-amide (IV) as the rate determining step, as the KH/KD = 1.538 is relatively small.20 Overall, these kinetic data support the plausible mechanism shown in Scheme 1, involving nucleophilic attack of transiently generated hydroxide ions on the bound nitrile.
In conclusion, we have demonstrated a practical method for accessing amides via a manganese catalyzed hydration reaction of the corresponding nitriles. Our methodology is compatible with a wide variety of functional groups and can be applied to a range of aromatic and aliphatic nitriles. By using D2O the corresponding α-deuterated amides could be accessed, either via hydration of the nitrile, or via H/D exchange at the amide. Mechanistic studies involving reaction kinetics, Deuterium-labeling, and intermediate characterization suggest that the manganese-catalyzed hydration reaction proceeds through water deprotonation, followed by nucleophilic attack of the resulting hydroxide ion on the metal-coordinated nitrile.
Conflicts of interest
There are no conflicts to declare.Data availability
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Associated Professor Graham de Ruiter (graham@technion.ac.il). The published article includes all datasets generated or analyzed during this study.Supplementary information: Synthetic procedures and characterization data. See DOI: https://doi.org/10.1039/d5cc02804c.
CCDC 2451223 [IV] contains the supplementary crystallographic data for this paper.21
Notes and references
- (a) R. M. Bullock, J. G. Chen, L. Gagliardi, P. J. Chirik, O. K. Farha, C. H. Hendon, C. W. Jones, J. A. Keith, J. Klosin, S. D. Minteer, R. H. Morris, A. T. Radosevich, T. B. Rauchfuss, N. A. Strotman, A. Vojvodic, T. R. Ward, J. Y. Yang and Y. Surendranath, Science, 2020, 369, eabc3183 CrossRef; (b) M. Fritz and S. Schneider, in The Periodic Table II: Catalytic, Materials, Biological and Medical Applications, ed. D. M. P. Mingos, Springer International Publishing, Cham, 2019, pp. 1–36 DOI:10.1007/430_2019_48.
- (a) J. R. Ludwig and C. S. Schindler, Chem, 2017, 2, 313–316 CrossRef; (b) P. L. Holland, Chem, 2017, 2, 443–444 CrossRef.
- C. L. Allen and J. M. J. Williams, Chem. Soc. Rev., 2011, 40, 3405–3415 RSC.
- V. R. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471–479 Search PubMed.
- R. S. Ramón, N. Marion and S. P. Nolan, Chem. – Eur. J., 2009, 15, 8695–8697 Search PubMed.
- (a) S. Murahashi, S. Sasao, E. Saito and T. Naota, J. Org. Chem., 1992, 57, 2521–2523 CrossRef; (b) K. Yamaguchi, M. Matsushita and N. Mizuno, Angew. Chem., Int. Ed., 2004, 43, 1576–1580 CrossRef PubMed; (c) R. García-Álvarez, J. Díez, P. Crochet and V. Cadierno, Organometallics, 2010, 29, 3955–3965 CrossRef; (d) W.-C. Lee and B. J. Frost, Green Chem., 2012, 14, 62–66 RSC; (e) E. Tomás-Mendivil, F. J. Suárez, J. Díez and V. Cadierno, Chem. Commun., 2014, 50, 9661–9664 RSC; (f) B. Guo, J. G. de Vries and E. Otten, Chem. Sci., 2019, 10, 10647–10652 RSC; (g) S. Yadav and R. Gupta, Inorg. Chem., 2022, 61, 15463–15474 CrossRef CAS PubMed.
- (a) A. Goto, K. Endo and S. Saito, Angew. Chem., Int. Ed., 2008, 47, 3607–3609 CrossRef CAS; (b) E. Tomás-Mendivil, R. García-Álvarez, C. Vidal, P. Crochet and V. Cadierno, ACS Catal., 2014, 4, 1901–1910 CrossRef; (c) P. Daw, A. Sinha, S. M. W. Rahaman, S. Dinda and J. K. Bera, Organometallics, 2012, 31, 3790–3797 CrossRef CAS.
- (a) T. Ghaffar and A. W. Parkins, Tetrahedron Lett., 1995, 36, 8657–8660 CrossRef CAS; (b) X.-b Jiang, A. J. Minnaard, B. L. Feringa and J. G. de Vries, J. Org. Chem., 2004, 69, 2327–2331 CrossRef CAS; (c) T. J. Ahmed, B. R. Fox, S. M. M. Knapp, R. B. Yelle, J. J. Juliette and D. R. Tyler, Inorg. Chem., 2009, 48, 7828–7837 CrossRef CAS; (d) X. Xing, C. Xu, B. Chen, C. Li, S. C. Virgil and R. H. Grubbs, J. Am. Chem. Soc., 2018, 140, 17782–17789 CrossRef CAS PubMed.
- S. Zhang, H. Xu, C. Lou, A. M. Senan, Z. Chen and G. Yin, Eur. J. Org. Chem., 2017, 1870–1875 CAS.
- (a) J. M. Pérez, R. Postolache, M. Castiñeira Reis, E. G. Sinnema, D. Vargová, F. de Vries, E. Otten, L. Ge and S. R. Harutyunyan, J. Am. Chem. Soc., 2021, 143, 20071–20076 Search PubMed; (b) R. A. Farrar-Tobar, S. Weber, Z. Csendes, A. Ammaturo, S. Fleissner, H. Hoffmann, L. F. Veiros and K. Kirchner, ACS Catal., 2022, 12, 2253–2260 CrossRef CAS PubMed; (c) P. J. Chirik, Acc. Chem. Res., 2015, 48, 1687–1695 CAS.
- N. A. Espinosa-Jalapa, A. Kumar, G. Leitus, Y. Diskin-Posner and D. Milstein, J. Am. Chem. Soc., 2017, 139, 11722–11725 CAS.
- (a) Q.-Q. Zhou, Y.-Q. Zou, S. Kar, Y. Diskin-Posner, Y. Ben-David and D. Milstein, ACS Catal., 2021, 11, 10239–10245 CAS; (b) M. Vogt, A. Nerush, M. A. Iron, G. Leitus, Y. Diskin-Posner, L. J. W. Shimon, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2013, 135, 17004–17018 CAS; (c) A. Nerush, M. Vogt, U. Gellrich, G. Leitus, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2016, 138, 6985–6997 CAS; (d) S. Tang and D. Milstein, Chem. Sci., 2019, 10, 8990–8994 CAS.
- J. Borau-Garcia, D. V. Gutsulyak, R. J. Burford and W. E. Piers, Dalton Trans., 2015, 44, 12082–12085 CAS.
- (a) J. A. Garduño, A. Arévalo, M. Flores-Alamo and J. J. García, Catal. Sci. Technol., 2018, 8, 2606–2616 RSC; (b) X. Wen, J. He, H. Xi, Q. Zheng and W. Liu, Asian J. Org. Chem., 2022, 11, e202100781 CrossRef CAS; (c) M. Prejanò, M. E. Alberto, N. Russo and T. Marino, Organometallics, 2020, 39, 3352–3361 CrossRef; (d) N. H. Anderson, J. M. Boncella and A. M. Tondreau, Organometallics, 2018, 37, 4675–4684 CrossRef CAS; (e) J. H. Kim, J. Britten and J. Chin, J. Am. Chem. Soc., 1993, 115, 3618–3622 CAS; (f) Z. Li, L. Wang and X. Zhou, Adv. Synth. Catal., 2012, 354, 584–588 CrossRef CAS.
- (a) S. Garhwal, A. Kaushansky, N. Fridman, L. J. W. Shimon and G. D. Ruiter, J. Am. Chem. Soc., 2020, 142, 17131–17139 CrossRef CAS PubMed; (b) T. S. Mohammad, Y. Jin, S. Raje, K. Młodzikowska-Pieńko, Z.-X. Yu and G. de Ruiter, J. Am. Chem. Soc., 2025, 147, 15195–15204 CrossRef CAS PubMed; (c) K. Dey, A. Gorai, K. Młodzikowska-Pieńko, N. Fridman, I. Avigdori, R. Gershoni-Poranne and G. de Ruiter, Angew. Chem., Int. Ed., 2025, 64, e202423275 CrossRef CAS PubMed.
- R. Thenarukandiyil, R. Kamte, S. Garhwal, P. Effnert, N. Fridman and G. de Ruiter, Organometallics, 2023, 42, 62–71 CrossRef CAS.
- K. Dey and G. de Ruiter, Org. Lett., 2024, 26, 4173–4177 CrossRef CAS PubMed.
- S. Ning, C. Wu, L. Zheng, M. Liu, Y. Zhang, X. Che and J. Xiang, Green Chem., 2023, 25, 9993–9997 CAS.
- P. Daw, A. Sinha, S. M. W. Rahaman, S. Dinda and J. K. Bera, Organometallics, 2012, 31, 3790–3797 CAS.
- T. Hirano, K. Uehara, K. Kamata and N. Mizuno, J. Am. Chem. Soc., 2012, 134, 6425–6433 CAS.
- CCDC 2451223: Experimental Crystal Structure Determination, 2025 DOI:10.5517/ccdc.csd.cc2n8pqc.
| This journal is © The Royal Society of Chemistry 2025 |