DOI:
10.1039/D5CC02761F
(Feature Article)
Chem. Commun., 2025,
61, 14324-14339
Linear open-shell 3d-metal silylamides – a versatile tool in coordination chemistry
Received
15th May 2025
, Accepted 14th July 2025
First published on 15th July 2025
Abstract
Linear 3d-metal complexes with open-shell metal ions have fascinated coordination chemists for decades. Originally the focus was on metal(II) species, yet the synthetic possibilities to acquire their reduced analogues, together with the introduction of NHCs as stabilizing co-ligands, have expanded the structural knowledge and feasible oxidation states to +I and even 0. Despite a certain wealth of structural data, insights into the reactivity or even catalytic applications of such compounds remained comparably thin for a long time given the intrinsic lability of these coordinatively and electronically unsaturated compounds. In this feature article we review the emerging chemistry and reactivity of silylamide based species, as mostly for these a comprehensive, broader picture of their reactivity patterns has been developed in recent years. Together with related, selected reactivity examples of other linear metal complexes, this review will serve as an entry point for interested molecular organic/inorganic chemists to join the scientific endeavours within this field.
Introduction
Low-coordinate open-shell 3d-metal complexes, more precisely linear complexes, have fascinated inorganic chemists for decades, as reflected in a number of reviews over time.1,2 The interest is founded foremost in acquiring a fundamental understanding of an electronically and coordinatively highly unsaturated metal ion. Furthermore, these molecules may provide general insights into elusive low-coordinate intermediates in catalysis. Seminal research on low-coordinate 3d-metal complexes dates back to the 1960s when Bürger and Wannagat used the hexamethyldisilazanide ligand (–N(SiMe3)2) for the synthesis of the di- or trivalent 3d-transition metal complexes (Mn(N(SiMe3)2)n) (n = 2, 3).3,4 They were unaware at the time that the divalent (M(N(SiMe3)2)2) actually forms dimers (Mn–Co)5,6 or solvent adducts (Cr–Ni)6–12 but can occur as monomeric in solution.10,12–14 Evidence for actual linear open-shell 3d-metal complexes was obtained in the gas phase via electron diffraction studies in 1985 for Mn(CH2tBu)2.15 In the same year Mn(C(SiMe3)3)2 (Fig. 1, left) was structurally characterized by single-crystal X-Ray diffraction analysis.16 In the following decades a variety of two-coordinate 3d-metal(II) complexes bearing different sterically encumbering anionic ligands X− such as terphenyls,17 (thio)phenolates,18 primary arylamides19–21 or secondary (aryl)silylamides (Fig. 1) were reported.21,22 These complexes are often stabilized by intramolecular metal–arene interactions,2 with interligand dispersion forces being a further contributing factor.23
 |
| | Fig. 1 Seminal examples of linear open-shell 3d-metall(II) compounds. | |
Despite the well-developed synthetic access, comprehensive reactivity studies on linear complexes is still lacking. For a long time it allowed only for a fragmented insight into the reactivity of a two-coordinate metal(II) ion. A fundamental problem is associated with maintaining the integrity of the central MX2 fragment. The reaction with substrates is often associated with insertion into the M–X bond, subsequent rearrangements and/or redox state dis/comproportionation, often in an unpredictable manner. A representative example is the reaction of Fe(Aryl)2 with CO (Scheme 1): it leads amongst others to purely organic products and aryl free metal carbonyls.24
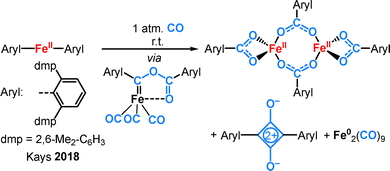 |
| | Scheme 1 Deconstruction of a linear iron(II) aryl complex by reaction with CO under M–L bond insertion, redox disproportionation and complete ligand rearrangement. | |
Furthermore, the coordinatively and electronically deficient nature of a two-coordinate 3d-metal(II) ion lacks to a certain degree the electrons/reducibility for many bond activation and cleavage processes. This was overcome by reduction of the metal ion to the monovalent and even zerovalent state, aided by N-heterocyclic carbenes (NHC) as well as cyclic amino alkyl carbenes (cAAC, Fig. 2).25 Here the π-accepting capabilities of carbenes stabilize the low-valent metal center via back donation into the empty p-orbital of the carbene.25,26 The electron-accepting nature of especially cAACs is exemplified for Mn(cAAC)2:27 Here, the metal is not zero-valent but adopts the oxidation state +I with a 4s1d5 configuration. The metal(I) ion couples antiferromagnetically with an electron that resides equally on both cAAC ligands. For Fe and Co, delocalisation of spin density onto the carbene ligands is less pronounced (approx. 0.25e−)28,29 and is mostly absent for Ni due to its stable d10 configuration.30 For cationic complexes [ML2]+ (L = NHC or cAAC; M = Cr, Fe–Ni)28,31 the spin density is predicted to be metal centered due to charge-dependent d-orbital contraction.
 |
| | Fig. 2 Seminal examples of linear open-shell 3d-metal(I) complexes. | |
Scope
In this perspective we will give an overview of the synthesis of linear metal(II/I) complexes, their electronic properties and emerging reactivity patterns. We will mostly regard compounds containing silylamide ligands (=X), namely those of the type [MX2]0,− and MX(NHC). The focus on silylamide complexes is due to the fact that reports on respective metal complexes as stable platforms for substrate activation are the most prevalent and recent, thus allowing the extraction of predictable knowledge for future endeavours. Suitable studies on related and other complexes will be addressed where appropriate or at the end. By that we aim to inspire interested chemists to further exploit the chemistry of these seemingly simple and intriguing yet synthetically challenging class of molecules.
Synthesis
Homoleptic metal(II/I) amides.
The formation of neutral linear metal(II) complexes bearing two anionic ligands in general, and those using amide ligands in particular, is straightforward (Scheme 2). Reaction of a metal(II) halide in Et2O (or other weakly coordinating solvents) with two equivalents of the respective alkali metal (AM) salt of the ligand yields the envisioned complexes.32–40 To prevent dimerization via bridging ligands sterically bulky disilylamide –N(SiR3)2 (R = Me, iPr and/or Ph) or aryl/silylamide L = –N(Dipp)SiR3 (R = Me, iPr, Ph and/or 2-propenyl)37,41 ligands are used. This works generally well for Mn to Ni, whereas for the early transition metals V and Cr intramolecular C–H bond activation is observed.35,39 For the heavier CuII, the reaction leads to reduction of the metal by the amide ligands to yield the diamagnetic, monovalent [CuL2]−. Only by the unintuitive use of CuICl the isolation of the highly unstable, paramagnetic [CuL2] was achieved under redox disproportionation of the metal.42 Reduction of divalent [ML2] to the anionic metal(I) derivatives with commonly KC8 is performed in the presence of suitable chelating agents, such as crown-ethers, crypt.222 or even donor solvents (THF or DME), to mask the potassium cation and prevent decomposition. Only for aryl/silylamides it was found that K+ can be stabilized via the arene rings in an intra- or intermolecular fashion.43,44 Lighter alkali metals can be used as reductants too. However, rapid degradation in the absence of suitable chelators was observed which is likely due to insufficient stabilisation via AM+–arene interactions.43
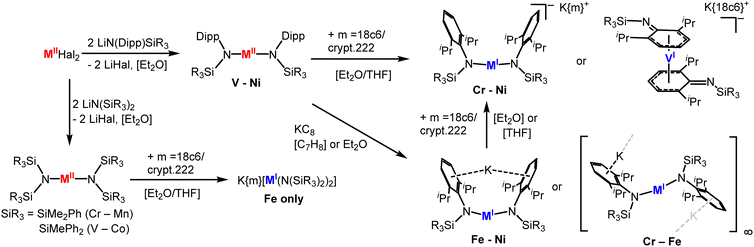 |
| | Scheme 2 Overview of the synthesis of two-coordinate metal(II) and metal(I) silylamides. | |
M(NR2)2 type complexes (R = SiMe3) with comparably less bulky disilazanide ligands form dimers in the solid state (Mn–Co) or are isolated as mononuclear solvent adducts (Cr–Cu6–10,45), yet exist in part as monomers in solution (Scheme 3). This behaviour allows for their reduction with KC8 to [M(NR2)2]− (= [MI]− for R = SiMe3) for Cr–Ni in the presence of crypt.222 or 18c6.13,32,46 The masking agents are mandatory as otherwise oxidation state redistribution to metal(II) and metal(0) species is observed:46 it is likely induced by the competition between the 3d-metal(I) and AM cations for the amide ligands. For [Ni(NR2)2]− a different synthetic approach proved synthetically more viable, given the notoriously labile Ni(NR2)2 precursor.4,10 In monovalent [Ni(PPh3)2NR2], which is obtained from reduction and ligand exchange of [Ni(PPh3)2Cl2] with LiNR2, both phosphines can be easily replaced by KNR2 in the presence of 18-crown-6 or crypt.222 (Scheme 3).13 The synthesis of these unencumbered metal(I) complexes can be performed in THF without solvent coordination, which hints to the electron rich nature of the metal(I) ion in these anionic complexes. Furthermore, the anionic charge (partially) compensates the reduced steric encumbrance of the ligands and gives an open metal site for substrate activation. The reduced steric protection leads for [Mn(NR2)2]− to dimerization in the solid state with formation of an unsupported Mn–Mn bond.32 This kind of behaviour is in stark contrast to the amide bridged neutral parent compounds. It can be reasoned by the electron configuration of 3d54s1 for MnI, and is in line with the observations made for the few other Mn(I)-Mn(I) dimers.47 It hints to the decomposition pathway of these less encumbered metal(I) silylamides: dimerization may initiate amide ligand loss to give [M2(NR2)2(μ2-NR2)]− (Scheme 3), which was isolated in case of nickel.13 Subsequent redox disproportionation and ligand distribution then yield in [M(NR2)3]− and amorphous metal M0.32
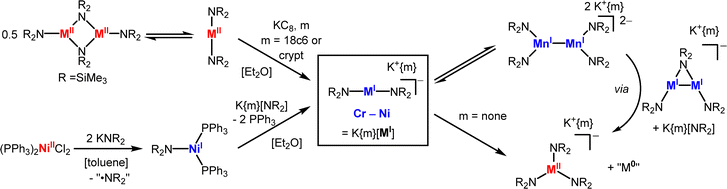 |
| | Scheme 3 Overview over the synthesis of two-coordinate 3d-metal(I) hexamethyldisilazanides (R = SiMe3). | |
NHC based metal(I) amides.
There are four synthetic paths to the subtitle compounds (Scheme 4): reduction of the mixed NHC/L ligated metal(II) halide (A), which can also be generated in situ (B) via partial ammonolysis using bulky IDipp or IMes.48–50 The steric effects of the employed NHC was found to be consequential for iron as the attempted reduction of [(IAd)FeCl(N{Dipp}SiMe3)], that bears the smaller carbene IAd (1,3-diadamantyl-imidazolin-2-ylidene), yielded in decomposition.51 The degradation was prevented by the presence of N2, and gave a dinuclear, N2-bridged iron(I) complex (Scheme 4). The halide/amide ligand exchange can also be performed in the monovalent state, mostly notably using an NHC ligated metal(I) halide (C).52–54 Otherwise, the NHC can be introduced at last by using complexes of the type KML2 (D) either via in situ ammonolysis and NHC addition (Ni, NHC = IDipp) or direct reaction with the respective imidazolium salts (Cr, NHC = IMes, IDipp).55,56 For the iron complex Fe(IDipp)(N{Dipp}SiMe3), 3, (IDipp = 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene) (Scheme 5) its further reduction was attempted.49 In the obtained complex 4 the zerovalent metal does not bind to the carbon donor of the NHC but prefers the interaction with a flanking arene substituent.
 |
| | Scheme 4 Top: General synthetic pathways to neutral, linear 3d-metal(I) (silyl)amides depicted using IDipp (IDipp = 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene). Bottom: Effect of N2 atmosphere on the reduction of 1. | |
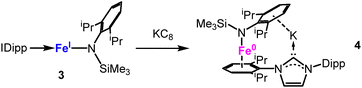 |
| | Scheme 5 Reduction of the linear iron(I) silylamide complex Fe(IDipp)(N{Dipp}SiMe3), 3. | |
Cationic linear metal amide complexes.
One-electron oxidation of the above-shown neutral amide based complexes into their cationic form was possible for chromium.
Oxidation of [Cr(N(Dipp)SiiPr3)2], Si5,33 leads to cationic [Si5]+ that displays a shorter metal-amide bond with a persistent strong secondary metal–arene interaction due to its electronic deficiency (Scheme 6). The seemingly very similar [Cr(N(Dipp)tBu)2], C5,57 does not exhibit such secondary interactions – even upon oxidation ([C5]+). Here, it is attributed to a stronger π-donating effect of the alkyl amide ligands. Oxidation of the heteroleptic complexes Ni(IDipp)(NHDipp*), 6 (Dipp* = 2,6-Dipp-C6H3),53 invokes stronger secondary metal–ligand interactions ([6]+, Scheme 7, top), overall illustrating nicely the impact of the complexes’ charge and oxidation state of the metal on its electron deficiency in these linear complexes. Reducing the steric bulk for the nickel complexes Ni(IDipp)(NHAryl) results in a C–H bond activated complex ([9]+, Aryl = Mes* =2,6-Mes-C6H3)53 or for Aryl = Dipp in disruption of the aromaticity of the arylimido ligand and an allyl-like coordination of the ligand ([10]+).52 For the silylamide complex [Ni(IDipp)N(SiMe3)2], 11, oxidation effectuates a methyl-shift from the ligand to the metal ([12]+, Scheme 7, bottom).52
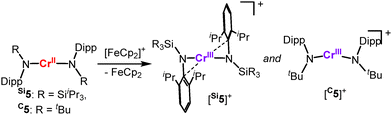 |
| | Scheme 6 Oxidation of the linear chromium(II) complexes Si5 and C5. | |
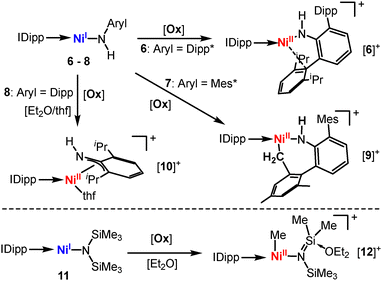 |
| | Scheme 7 Oxidation of neutral linear nickel(I) amides and resulting cationic metal(II) complexes. | |
Structural features and electronic as well as magnetic properties of two-coordinate metal complexes
Structure and ligand field considerations.
For linear 3d-metal complexes a (near) linear L–M–L would be initially expected, due to the absence of Jahn–Teller distortion for linear molecules. Nonetheless, deviations from linearity were shown to require little energy.34 Perfect linearity is rarely observed in the solid state and is mostly a result of a crystallographic inversion centre on the metal atom. As such the L–M–L angles range from 165 to 179°, and were commonly imposed by secondary metal–ligand or inter-ligand dispersion interactions. The low-coordinate nature of the metal ion results in a small ligand field splitting. It is computed between 4000 to 8000 cm−1 and results in exclusively high-spin metal ions.34,44,58–60 The general d-orbital order of linear metal(II) complexes of the type MX2 (X = anionic σ-donor ligand) is shown in Fig. 3A. The non-bonding, energetically degenerate dxy and dxz orbitals are the lowest, followed by the likewise degenerate dx2−y2 and dyz.2 The dz2 orbital is expected to be the highest in energy as it is aligned with the principal bond axis. However, the dz2 is overall close to dxy/dyz due to varying amounts of mixing with the 4s orbital (Fig. 3B).34,59–61 Depending on the electron count it can create orbitally degenerate states and by that spin–orbital coupling is maintained.20,62 This behaviour is in contrast to higher-coordinate metal ions where orbital degeneracy is mostly quenched due to Jahn–Teller distortions. Even more remarkable, the weak ligand field can effectuate a non-Aufbau configuration as the energy separation between the orbitals is less than the electron–electron repulsion. It was first shown for the linear cobalt(II) methanide complex Co(C{SiMe2ONaph}3)2 (Fig. 3C) which exhibits an orbital angular momentum of L = 3 – the theoretical maximum for 3d metals.59,63 With reduction to the monovalent state, the 3d/4s mixing becomes more pronounced, and leads to a partially inverse orbital situation with the 3dz2 + 4s now being the lowest. This kind of situation was computed for [Fe(C{SiMe3}3)2]− (Fig. 3D), and experimentally supported by electron density analysis.60 Changing to silylamide ligands as in [Fe(NR2)2]−, the degeneracy of the dxz and dyz orbitals is partially lifted due to the interaction with the co-planar amide ligands (Fig. 3E).32 Artificial bending of the principal ligand axis in [Fe(NR2)2]− type complexes was shown to have little impact down to 140°.63 For smaller angles, the AILFT orbitals no longer coincide with the N–M–N pseudo axis, and the orbital order associated with the linear ligand field is lost.63 Comparison of the computed ligand field splitting for the different metals in the amide complexes further shows the effect of a stronger spin–orbit coupling: when going to the heaver elements (Cr → Co) the ligand influence onto the magnitude of the orbital splitting is lowered.32 Ligand field analysis of linear metal(0) ions is missing so far, yet even stronger 4s/3d-mixing is likely.
 |
| | Fig. 3 (A) Exemplary d-orbital splitting scheme of a linear 3d-metal(II) complex with σ-donor ligands. (B) calculated d-orbital splitting of the iron(II) silylmethanide Fe(C{SiMe3}3)2 under consideration of 4s orbital involvement.60 (C) Non-Aufbau electron configuration of a d7-ion calculated for [CoII(C{SiMe2Onaph}3)2].59 (D) Calculated d-orbital splitting of the iron(I) silylmethanide [Fe(C{SiMe3}3)2]−.58 (E) d-Orbital splitting of the metal(I) silylamides [MI(N{SiMe3}3)2]− (M = Cr–Co).58 (F) Magnetic relaxation pathways (depicted for a MJ = ±7/2 system). | |
Magnetism.
The magnetic free ion behaviour, resembling the situation of lanthanide ions,20,32,46,63,64 was exploited for creating 3d-metal based single-ion magnets (SIMs). In short, SIMs are paramagnetic compounds that display slow relaxation of magnetisation that originates from the intrinsic properties of a molecular, mononuclear system.65 The core feature of SIM's is the presence of large magnetic anisotropy that arises from partially filled, energetically degenerate orbitals or low-lying excited states. As such the overall spin of the molecule can be oriented in the presence of an external magnetic field. The magnetisation persists after removal of the external field if a considerable barrier (Ueff) is present. For 3d-metal based SIM's the effective barrier for spin reversal is generally defined as Ueff = D × S2 where D denotes the axial zero-field splitting parameter.65 If D < 0 it creates a double well situation (Fig. 3E) with lowest-lying +MS and −MS states being separated by the barrier Ueff. Substantial efforts have been spent on 3d-metal based SIMs to maximize D, while maintaining a high S. Furthermore, non-Kramers half-integer systems are deemed superior due to suppression of through-barrier tunnelling. For linear metal complexes it was however shown that the relaxation barrier corresponds directly to the energy spacing between the ground and first excited spin–orbit coupled MJ states (Fig. 3F). The seminal example of a linear SIM constituted [Fe(C{SiMe3}3)2]− with an effective relaxation barrier of Ueff = 226 cm−1.58,66 It aligns with the energy gap between the two lowest magnetic states (MJ = ±7/2, ±5/2) of approximately 240 cm−1. The gap correlates directly with the spin orbit coupling constant ζ via 2D = ζ/MS (ζ = 361 cm−1 and MS = 3/2) for a free iron(I) ion. [Fe(C{SiMe3}3)2]− showed magnetic blocking (at 4.5 K) – unprecedented by a mononuclear 3d-metal complex at the time. In a similar vein the ground-breaking S = 3/2 complex Co(C(SiMe2ONaph)3)2 exhibits an even higher Ueff of 450 cm−1,59 which also corresponds to the ground/first excited state energy spacing (MJ = ±9/2, ±7/2). The presence of a S = 3/2 ion in a linear environment is however not a sufficient criterion for a high Ueff: for the related [Fe(N{SiMe3}2)2]− a reduced Ueff (only up to 64 cm−1) and no magnetic blocking was determined although the computed energy spacing between the ground and first excited MJ states was close to that of [Fe(C{SiMe3}3)3]−.32 Subsequently, steric encumbrance was revealed as a further crucial factor (as in [Fe(N{SiMePh2}2)2]− or [KFe(N{Dipp}SiMe3)] with its ligands being interlocked by secondary K+-coordination) which mitigates vibrational distortions perpendicular to the X–Fe–X axis.32,46 For the cationic cyclic alkyl amino carbene iron(I) complex [Fe(cAAC)2]+ the negative impact of π-interactions was discussed for the observed absence of a sizable relaxation barrier (Ueff < 20 cm−1).67 The related cobalt complex [Co(IMes)2]+ (IMes = (1,3-bis(2,4,6-trimethylphenyl)imidazol-2-ylidene)) exhibits also a rather low barrier (Ueff = 22.4 cm−1).68
Reactivity.
General coordination behaviour.
As intuitively expected, the general coordination behaviour of linear complexes should be dictated by their coordinative and electronic unsaturation. Indeed, in the neutral and moreover cationic state the complexes behave mostly as Lewis acids. This property is however lost in the anionic state, a difference that can be nicely illustrated for the three metal(I) complexes shown in Scheme 8. The cationic [Fe(IMes)2]+ readily forms Lewis-base adducts with THF ([10]+, Scheme 8A).69 In contrast, the anionic [Fe(NR2)2]− is indifferent towards any regular donor ligands (e.g. phosphines or NHCs).70 Moreover, in the interaction with the ethylene bridged diphosphine 1,2-bis(diphenylphosphine)ethylene the metal in [Fe(NR2)2]− does not bind via the P-donors but the ethylene backbone ([11]−, Scheme 8B).
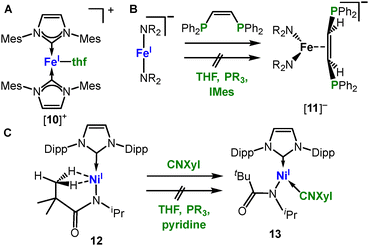 |
| | Scheme 8 Examples for the general coordination behaviour of linear open-shell metal(I) complexes towards donor ligands. | |
The neutral Ni(I) amidate complex 12 (Scheme 8C) displays bis(σ-C–H)-agostic interactions between the amidate's tert-butyl function and the nickel(I) ion due to its coordinative unsaturation. On the other hand, the complex is inert towards donor-ligands with the exception of isonitriles that are also capable of M → L backbonding (13). This observation emphasizes the role of the overall charge of the complex, irrespective of a low oxidation state of the central metal ion. The second effect of a low-coordinate state is the strong preference for higher spin-states. It was nicely demonstrated in the case of CO: its reaction with [MI]− at low temperatures yields in the isolation of monocarbonyl adducts ([13]− and [14]−, Scheme 9, top).71 Despite considerable M → ligand π-backbonding the high-spin state is maintained. The monocarbonyl complexes are however highly labile and undergo dismutation reactions (e.g. for cobalt it gives [Co(CO)4]− and [Co(NR2)3]−).71 In the case of the neutral chromium(I) complex 15 the coordination of two CO ligands are sufficient to invoke the low-spin state (16, Scheme 9, bottom).
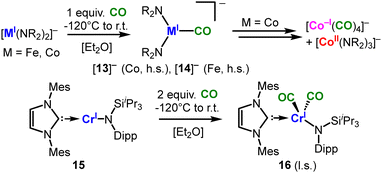 |
| | Scheme 9 Reaction of linear metal(I) complexes with CO. | |
C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e002.gif) C/C
C/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) C/CN bond activation.
Besides CO, alkenes and alkynes are known to interact strongly with low-valent 3d-metals in organometallic chemistry. The group of Tilley reported first on the heteroleptic iron(I) complex [(IDipp)Fe(N{Dipp}SiMe3)] as a catalyst for cyclotrimerisation of terminal and internal alkynes (Scheme 10).49 Detailed studies of the reaction mechanism and isolation of intermediates gave a bimetallic pathway under carbene dissociation (17).72 At 60 °C oxidative coupling takes place resulting in the dimetalla(II) butadienediyl complex 18 as evidenced by isolation of both reaction intermediates. At this point a third alkyne likely coordinates and subsequently inserts into a metal–carbon bond to generate a dimetalla cycloheptatriene that finally undergoes ring closure to produce the aromatic ring. The metal(I) silylamide fragment is either captured by the previously liberated NHC ligand or re-enters into the catalytic cycle by alkyne coordination. In the case of anionic complexes [MI]− (Cr–Co) the interaction with alkynes leads mostly only to η2-alkyne complexes ([M19R′]−, Scheme 11, top).73 For cobalt alkyne coordination is weak and reversible, yet becomes stronger when going to the lighter metals (Co → Cr). CASSCF calculations on these complex series revealed their primary electronic structure as metal(II) radical anions (Scheme 11, bottom), whereas the textbook dichotomy of such compounds as π-complexes or metallacyclopropenes are a lesser factor. One has to bear in mind that the classic description of π-complexes along the Chatt–Dewar–Duncanson concept was originally adopted for closed-shell/low-spin organometallics. Here, the π-backbonding proceeds from a doubly occupied metal centered orbital with no substantial unpaired spin transfer. The observed deviation for the [MI]− complexes shown herein is likely a result of their high-spin state in conjunction with the anionic charge that facilitates the electron transfer to the π*-orbital of the substrate. Steric factors are also in play as for the more encumbered [FeI(N{Dipp}SiMe3)2]− alkyne coordination is labile.43 A similar bond trichotomy is formulated for side-on bound organo nitrile iron complexes ([20R′]−, Scheme 11, top).74 The radical anionic nature of the bound alkyne rationalizes subsequent reduction to bismetallated alkene dianions in the case of manganese ([21]2−) and chromium.73 Moreover, for manganese a series of C
C/CN bond activation.
Besides CO, alkenes and alkynes are known to interact strongly with low-valent 3d-metals in organometallic chemistry. The group of Tilley reported first on the heteroleptic iron(I) complex [(IDipp)Fe(N{Dipp}SiMe3)] as a catalyst for cyclotrimerisation of terminal and internal alkynes (Scheme 10).49 Detailed studies of the reaction mechanism and isolation of intermediates gave a bimetallic pathway under carbene dissociation (17).72 At 60 °C oxidative coupling takes place resulting in the dimetalla(II) butadienediyl complex 18 as evidenced by isolation of both reaction intermediates. At this point a third alkyne likely coordinates and subsequently inserts into a metal–carbon bond to generate a dimetalla cycloheptatriene that finally undergoes ring closure to produce the aromatic ring. The metal(I) silylamide fragment is either captured by the previously liberated NHC ligand or re-enters into the catalytic cycle by alkyne coordination. In the case of anionic complexes [MI]− (Cr–Co) the interaction with alkynes leads mostly only to η2-alkyne complexes ([M19R′]−, Scheme 11, top).73 For cobalt alkyne coordination is weak and reversible, yet becomes stronger when going to the lighter metals (Co → Cr). CASSCF calculations on these complex series revealed their primary electronic structure as metal(II) radical anions (Scheme 11, bottom), whereas the textbook dichotomy of such compounds as π-complexes or metallacyclopropenes are a lesser factor. One has to bear in mind that the classic description of π-complexes along the Chatt–Dewar–Duncanson concept was originally adopted for closed-shell/low-spin organometallics. Here, the π-backbonding proceeds from a doubly occupied metal centered orbital with no substantial unpaired spin transfer. The observed deviation for the [MI]− complexes shown herein is likely a result of their high-spin state in conjunction with the anionic charge that facilitates the electron transfer to the π*-orbital of the substrate. Steric factors are also in play as for the more encumbered [FeI(N{Dipp}SiMe3)2]− alkyne coordination is labile.43 A similar bond trichotomy is formulated for side-on bound organo nitrile iron complexes ([20R′]−, Scheme 11, top).74 The radical anionic nature of the bound alkyne rationalizes subsequent reduction to bismetallated alkene dianions in the case of manganese ([21]2−) and chromium.73 Moreover, for manganese a series of C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bond activation processes results in alkyne cyclotrimerisation and the formation of the rare manganese(−I) arene [Mn(η6-C6Ph6)(η2-PhCCPh)].73
C bond activation processes results in alkyne cyclotrimerisation and the formation of the rare manganese(−I) arene [Mn(η6-C6Ph6)(η2-PhCCPh)].73
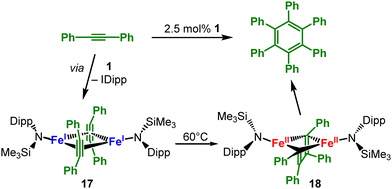 |
| | Scheme 10 [(IPr)Fe(N{SiMe3}Dipp)], 1, as a precatalyst for cyclotrimerisation of terminal and internal alkynes, and isolable reaction intermediates 17 and 18. | |
 |
| | Scheme 11 General reaction of alkynes with linear metal(I) silylamides (top) and electronic resonance structures obtained from CASSCF calculations. | |
Complex dismutation is more pronounced for the overall neutral, more encumbered [KM(N{Dipp}SiMe3)2] (M = Fe, Mn) where the alkali metal cation is not separated from the complex (Scheme 12).43 [KMn(N{Dipp}SiMe3)2] undergoes redox disproportionation, formation of a dinuclear manganese(0) complex (20) and incomplete alkyne cyclotrimerisation (21). For iron the complete cyclotrimerisation by isolation of the arene iron(−I) complex [K(Fe(C6Ph6)(PhCCPh))], 19, is observed. Together with the intermediates 17 and 18 by Tilley (Scheme 10), 20 and 21 provide snapshots into the cyclotrimerisation steps which seems dinuclear in nature for these low-coordinate systems. For the anionic metal(I) complexes only K{m}[Fe(N{Dipp}SiMe3)2]− proved competent in cyclotrimerization of diphenyl acetylene (Scheme 13A), likely by in situ complex disproportionation. The reaction of terminal alkynes with [Fe(NR2)2]− resulted not in the cyclotrimerisation, but simple side-one coordination.75 Contrastingly, the neutral [Fe(NR2)2] facilitates the selective cyclotrimerisation of terminal alkynes to the 1,2,4-substituted benzene regioisomers (Scheme 13B).76 However, the reaction presumably occurs under ligand exchange via deprotonation of the alkyne by the silylamides, and likely the formation of multinuclear iron alkinyl species. In the special case of terminal benzyl alkynes [Fe(NR2)2]− catalyses its transformation to phenyl allene within minutes (Scheme 13C)75 and to the internal alkyne within 24 hours. For the interaction of alkenes with divalent metals reports are restricted to intramolecular interactions with a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond as in [M(N{Dipp}Si{Me2}C3H5)2] (Scheme 14, top).37 The interaction is purely electrostatic with marginal CC bond elongation. It changes upon reduction as exemplified for cobalt: shortening of the Co–(CC) distance is observed (Δ = −0.4 Å) with a widening of CC-bond (Δ = 0.03 Å). Akin to the situation for metal alkynes (Scheme 11), the stronger metal–alkene interaction is likely attributed to the situation as predominantly a metal(II) alkene characteristic. It becomes more obvious for chromium where intermolecular C–C-coupling via one of the alkene functions is observed.37 In the reaction of the iron(I) silylamide [Fe(NR2)2]− with external substrates, that bear an internal CC bond, side-on complexes are formed ([25R′]− and [26R′]−, Scheme 14, bottom).75,77 Interestingly, Z-substituted substrates undergo rapid isomerisation to the thermodynamically more stable E-isomer. This observation gives evidence for the metal(I) alkenes as actual iron(II) bound radical anions (Scheme 11, bottom), which was supported by 57Fe Mössbauer spectroscopy analysis.70 The isomerisation can be performed in a catalytic fashion with either aromatic or aliphatic substituted simple alkenes such as 3-hexene. By comparison with the behaviour of the prototypical E-stilbene radical anion,77 whose first isolation was achieved by encapsulation between two K{18c6}+ moieties (27), it was concluded that the isomerisation takes place in the coordination sphere of the metal and not by intermittent decoordination of alkene substrates as “free” radical anions.
C double bond as in [M(N{Dipp}Si{Me2}C3H5)2] (Scheme 14, top).37 The interaction is purely electrostatic with marginal CC bond elongation. It changes upon reduction as exemplified for cobalt: shortening of the Co–(CC) distance is observed (Δ = −0.4 Å) with a widening of CC-bond (Δ = 0.03 Å). Akin to the situation for metal alkynes (Scheme 11), the stronger metal–alkene interaction is likely attributed to the situation as predominantly a metal(II) alkene characteristic. It becomes more obvious for chromium where intermolecular C–C-coupling via one of the alkene functions is observed.37 In the reaction of the iron(I) silylamide [Fe(NR2)2]− with external substrates, that bear an internal CC bond, side-on complexes are formed ([25R′]− and [26R′]−, Scheme 14, bottom).75,77 Interestingly, Z-substituted substrates undergo rapid isomerisation to the thermodynamically more stable E-isomer. This observation gives evidence for the metal(I) alkenes as actual iron(II) bound radical anions (Scheme 11, bottom), which was supported by 57Fe Mössbauer spectroscopy analysis.70 The isomerisation can be performed in a catalytic fashion with either aromatic or aliphatic substituted simple alkenes such as 3-hexene. By comparison with the behaviour of the prototypical E-stilbene radical anion,77 whose first isolation was achieved by encapsulation between two K{18c6}+ moieties (27), it was concluded that the isomerisation takes place in the coordination sphere of the metal and not by intermittent decoordination of alkene substrates as “free” radical anions.
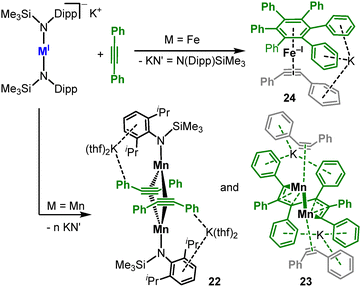 |
| | Scheme 12 Reaction of [KM(N{Dipp}SiMe3)2] (M = Fe, Mn) with PhCCPh. | |
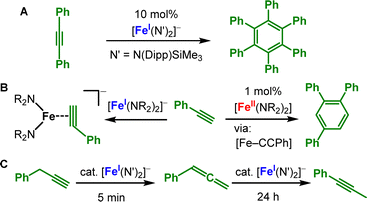 |
| | Scheme 13 Linear iron silylamides as precatalysts for alkyne trimerisation of internal alkynes (A), their oxidation state dependent coordination or cyclotrimerisation of terminal alkynes (B) and use for catalytic isomerization of terminal to internal alkynes (C) (N′ = N(Dipp)SiMe3). | |
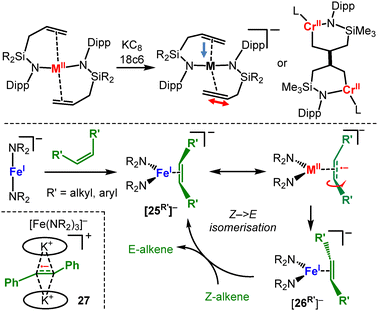 |
| | Scheme 14 Effect of oxidation state on the metal–alkene interaction in quasi-linear complexes with alkenyl substituents (top). Reaction of the linear iron(I) complex [Fe(NR2)2]− with alkenes. Inset: Isolable stilbene radical anion complex 24 (bottom). | |
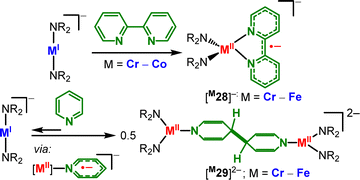 |
| | Scheme 15 Reaction of 2,2′-bipyridine with (top) and reductive coupling of pyridine by (bottom) linear metal(I) complexes. | |
The reduction is remarkable in the case of cobalt where the reduction potential of the metal(I) precursor is 1 V less negative than that of free bipyridine (Ered([Co(NR2)2]−) = −1.47 V vs. [Fc/Fc+], E1/2(bipyridine): approx. −2.67). With pyridine, the formation of binuclear metal(II) complexes [M29]2− (M = Cr–Fe, Scheme 15, bottom) occured.79 Here, pyridine underwent reversible reductive C–C coupling forming a bridging 4,4′-dihydrobipyridyl ligand via a transient monomeric metal(II) complex bearing a pyridyl radical anion. Benzophenone was also used in combination with linear anionic complexes of iron and cobalt to produce metal(II) stabilized ketyl radical anions ([M30]−, Scheme 16), supported by Mössbauer spectroscopic and computational analysis. This approach was extended to unprecedented metal bound ketiminyl ([M31]−) and aldiminyl radical anions ([32]−),80 with the reductive coordination being in part found to be reversible. Finally, [FeI]− can act as an Et2O-soluble one-electron reductant in the isolation of the first diarsene radical anion [Mes*As=AsMes*]− ([33]−).81
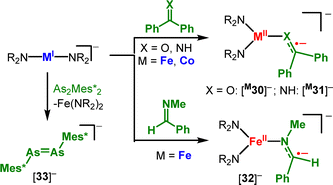 |
| | Scheme 16 Reduction of diaryl ketones, ketimines, aldimines and a diarsene with [M(NR2)2]− complexes. (Mes* = 2,6-Mes2-C6H3). | |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixtures of alkyl iron(II) and cyanido iron(II) complexes (Scheme 18).74 The capability of linear metal(I) to activate aromatic C–F bonds was further explored for fluoropyridines (Scheme 19).
:1 mixtures of alkyl iron(II) and cyanido iron(II) complexes (Scheme 18).74 The capability of linear metal(I) to activate aromatic C–F bonds was further explored for fluoropyridines (Scheme 19).
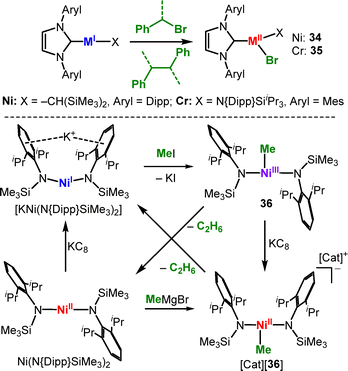 |
| | Scheme 17 Formation of methyl bis(amido) nickel complexes. | |
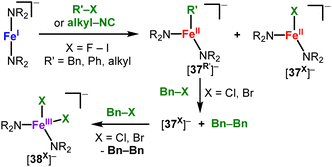 |
| | Scheme 18 Reaction of [FeI(NR2)2]− (R = SiMe3) with benzyl, phenyl and alkyl halides as well as aliphatic isonitriles. | |
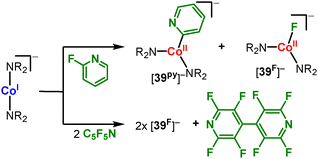 |
| | Scheme 19 Reaction of [CoI(NR2)2]− with fluorinated pyridines. | |
Depending on the degree of fluorination of the pyridine its reaction with [CoI]− yields either a 1:1 mixture of cobalt(II) pyridyl ([39py]−) and cobalt(II) fluoride ([39F]−) complexes, or homocoupling to fluorinated 4,4′-bipyridines.79 The selective formation of metal(II) fluorides is observed as well in the case of [MnI]− and [FeI]− using perfluoropyridine.79
Imido metal complexes.
Lately 3d-metal imido complexes have attracted significant interest due to their ability to catalytically transfer the metal-bound [NR] unit to organic substrates, e.g. in C–H bond amination or aziridation of alkenes.85 Despite the growing use of such reactions, the fundamental electronic and structural factors that dictate the reactivity of the [MNR] unit are still not well resolved.86 A higher spin state appears to increase the electrophilicity and reactivity of the metal–nitrogen bond by an increased population of antibonding π*-orbitals of the M–N interaction. As such, linear metal(I) complexes were employed as for the resulting imido complexes higher spin states could be expected. The neutral linear chromium(I) complex 40 was reacted with MesN3 to give the trigonal high-spin metal(III) imide 41 (Schemes 20 and 21, top), that showed however only marginal H atom abstraction capabilites.56 Trigonal high-spin chromium(IV) imides also proved to be rather stable (42, Scheme 20, top).57 Attempts for isolobal oxido complexes yielded so far only the chromium(VI) dioxide (43)57 and by that shows the range of oxidation states that can be supported by these amide ligands. In contrast, the iron silylamide [Fe(NR2)2] mediated the intramolecular C–H amination of aliphatic azides (Scheme 21, top)87 with low catalyst loading (1 mol%) yet at high reaction temperatures (120 °C). Insights into the catalytic conversion were gained by isolation and characterization of coordinatively labile aliphatic azide adducts to the two-coordinate (Fe(NR2)2) fragment (44, Scheme 21, bottom).88,89 Subsequent N2 liberation resulted in the imido complex 45, which proved however too reactive for its isolation. It immediately underwent nitrene insertion into a C–H bond of the ancillary silylamide ligand (46). In-crystallo irradiation of the azide adducts 44 effectuated N2-extrusion and gave spectroscopic and structural information about the elusive imido iron species 45 in the solid state matrix. Depending on the substituent, an intermediate88 or a high-spin89 situation with imidyl or even nitrene character was discussed. Reaction of Mes-N3 with the more encumbered Fe(N{EMe3}Dipp)2 (E = C, Si) yielded isolable high-spin aryl imido iron complexes (E47, Scheme 22).40,90 Computational analysis gave for [Fe(NMes)(N{CMe3}Dipp)2] (C47)40 an imidyl type situation. In slight contrast, for [Fe(NMes)(N{SiMe3}Dipp)2] (Si47, Scheme 22)90 spectroscopic examination in conjunction with calculations predicted a very covalent Fe–N bond with shared nitrene, imidyl and imide characteristics. For adamantyl-N3 an analogous elusive species was obtained,40 that facilitated intramolecular C–H amination as seen for the less encumbered [Fe(NR2)2]. Using the linear iron(I) precursor [Fe(N{SiMe3}Dipp)2]− resulted in isolation of [Fe(NMes)(N{SiMe3}Dipp)2]− ([Si47]−).90 Together with the neutral counterpart Si47 it gave the first pair of isostructural high-spin imido metal complexes in two oxidation states, which further could be interconverted by chemical reduction/oxidation. [Si47]− proved computationally as an iron(II) imidyl, yet reacted as an iron(III) imide with CS2 by insertion of the latter into the M–NR bond ([48]−, Scheme 22). This behaviour was not observed for the neutral Si47 which contrastingly facilitated electrophilic nitrene transfer to PEt3. Corresponding studies on [CoI(N{SiMe3}Dipp)2]− with N3Dipp yielded the first example of a high-spin (S = 2) imido cobalt species ([49]−, Scheme 23, top).91 X-ray absorption and EPR spectroscopy as well as computational studies led to the formulation of [49]− as a cobalt(II) imidyl. For the smaller substituent mesityl the analogous imidyl complex [50]− unexpectedly underwent SiMe3-group migration from the co-ligand to the imidyl nitrogen. Again, it implicated a nucleophilic character of these anionic imido complexes. Subsequent intramolecular C–H bond activation resulted in the formation of cobalt(III) alkyl species [51]−. The less encumbered [CoI]− enabled the isolation of the highly reactive alkyl imido complex [52]− for which an intermediate-spin state (S = 1) was proposed (Scheme 24, bottom).92 [52]− facilitated intermolecular C–H abstraction from external substrates – so far not seen for cobalt imidos. The resulting cobalt(II) amide [53]− also engaged unprecedentedly in H atom abstraction (HAT) via direct transfer or a stepwise deprotonation/Co–C bond cleavage mechanism (via [54]−) to regenerate [CoI]−. For the nickel(I) complex [NiI(N{SiMe3}Dipp)2]− imido complex formation was only possible for the smallest aromatic azide PhN3.93 The resulting T-shaped low-spin nickel(III) complex [55]− was found to possess a [NiNDipp] imide unit (Scheme 25) that originated from a SiMe3 shift from a co-ligand, as seen for cobalt ([50]−, Scheme 23).
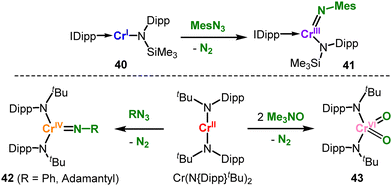 |
| | Scheme 20 Formation of trigonal chromium imides and a chromium dioxide. | |
 |
| | Scheme 21 Intramolecular C–H amination catalysed by FeII(NR2)2 (top) and detection of a trigonal imido iron complex by irradiation of an iron(II) organo azide adduct (bottom). | |
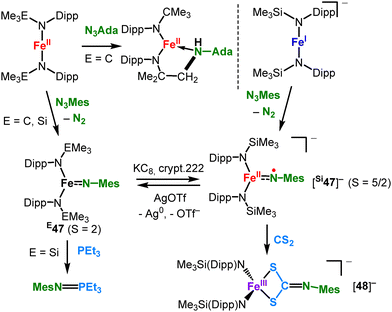 |
| | Scheme 22 Reaction of [Fe(N{EMe3}Dipp)2]0,− (E = C, Si) with RN3. | |
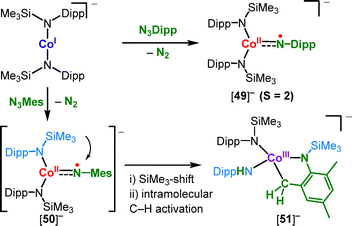 |
| | Scheme 23 Formation and behaviour of trigonal imidyl cobalt complexes. | |
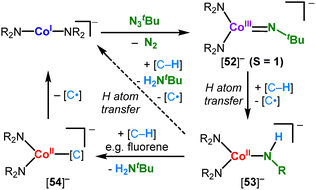 |
| | Scheme 24 Formation and reactivity of trigonal imido cobalt complexes. | |
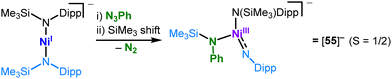 |
| | Scheme 25 Formation of a T-shaped low-spin imido nickel(III) complex. | |
Element activation and cluster formation.
Last but not least, two-coordinate complexes are predestined for the bottom-up construction of like-wise low-coordinate 3d-metal/main group units. These can give insights into the electronic situation of small metal/main group clusters motifs, and may serve as structural and/or functional models for small biological clusters (e.g. ferredoxins). The reaction of the iron(I) complex [Fe(N{Dipp}SiMe3)2]− with 0.5 equivalents of sulfur gave the diiron compound [56]2− bearing an unsupported sulfide bridge (Scheme 26A).94 [56]2− could subsequently be oxidized in a stepwise fashion to monoanionic, mixed-valent [56]− and neutral, diferric 56. The mixed valent [56]− is particularly interesting as it shows almost complete valence localisation despite identical iron sites. This unprecedented series of an unsupported, low-coordinate [Fe–S–Fe] unit in three-oxidation states resembles one of the belt sulfide functions in the iron/sulfur/molybdenum co-factor (FeMoCo) of the nitrogenase enzyme (Scheme 26A, inset),95 which is supposed to open up during substrate turn-over.96 [56]2− and [56]− could be cleaved by CS2 which yielded the mononuclear iron thiocarbonates [57]2− and [57]− under decoordination of the second [FeL2] fragment.94 The facile rupture of a [Fe–S–Fe] unit by CS2 gives insights into how CS2 might act as an inhibitor of nitrogenase FeMoco (and other iron–sulfur clusters) – a behaviour that is not yet fully understood.97 An analogous study with red selenium gave the dianionic complex [59]2− (Scheme 26B).98 Variation of the Fe:Se ratio resulted in the mononuclear iron(III) di- (582−) and triselenides (602−). It is contemplated that elemental selenium is initially reduced by [Fe(N{Dipp}SiMe3)2]− to anionic polyselenides. Subsequently, the chalcogenide anions recombine with the now Lewis-acidic iron(II) complexes – with or without further oxidation to iron(III) – to yield monomeric iron polyselenides or dimeric diiron monoselenides depending on the Fe:Se ratio. Accordingly, pre-reduced polychalcogenides can be used like-wise in conjunction with divalent complexes (Scheme 26B).98 If the steric demand of the silylamide ligands of the employed iron(I) complex is lowered, a bis-μ-S bridged complex with an [Fe2S2] diamond core was obtained (61, Scheme 26C).99 It corresponds to the central [Fe2S2] motif of well-known Rieske-type clusters. 61 could be deconstructed using CS2 to yield the mononuclear iron(III) thiocarbonate 62. Reaction of 61 with additional [Fe(NR2)2]− lead to reduction of the iron sulfur complex and was accompanied by decoordination of one silyl amide ligand per iron (63). Two other synthetic super-reduced [Fe2S2]0 clusters were so far known, which however retained the four-coordinate coordination environment due to the use of chelating ligands.10063 thus represents the first example of a super-reduced [Fe2S2]0 complex with only three-coordinate iron. It is important to note that formation of 63 does not occur using the K{crypt.222} salt of [FeI]− or KC8 in the presence of 18-crown-6. It reveals a dual role of the employed [Fe(NR2)2]0,−: it acts first as a 1e−–reductant in the oxidation state +I and then as a strong Lewis acid in the divalent state. The approach of combining a linear metal(II) fragment with unusual polyanions is also applicable for Zintl anions (Scheme 27).101 Reaction of Mn(NR2)2 with K{crypt.222}[TlBi3], which is only soluble in very polar solvents such as ethylene diamine or NH3(l), allowed for the unprecedented reactive dissolution of Zintl ions in regular solvents such as THF. The integrity of the Zintl ion remains in formed [64]2−, which is also rather uncommon. This was emphasized for [TrBi3]2− (Tr = Ga, In), where a [Bi2]2− bridged dinuclear complex ([65]2−) is observed with concomittant101 elimination of the triel metal. Moreover, the reaction with [TrBi3]2− also yielded additional ligand exchange and redox state changes of the Mn(NR2)2 fragment, as shown for the tetranuclear cluster [66]2−. [66]2− is a mixed [MnI2MnII2] complex, thus underscoring the ease of redox state changes in these low-coordinate high-spin metal ions.
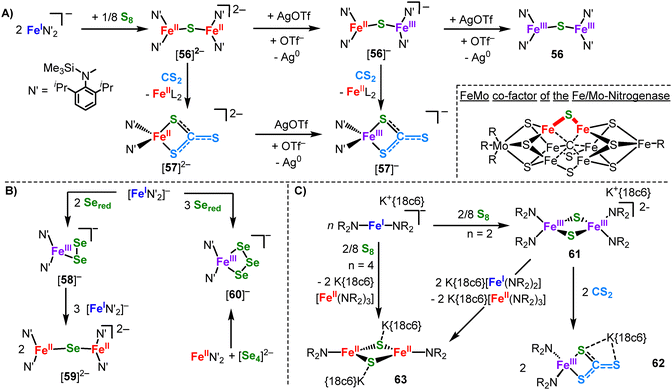 |
| | Scheme 26 (A) Reaction of a bulky linear iron(I) silylamide with elemental sulfur and subsequent oxidation and Fe–S bond cleavage with CS2. Inset: Iron/sulfur/molybdenum co-factor (FeMoCo) of the nitrogenase enzyme. (B) Reaction of bulky linear iron(I) and iron(II) silylamides with selenide sources. (C) Reaction of the less encumbered K{18c6}[Fe{NR2}2] (R = SiMe3) with elemental sulfur and subsequent cleavage of a [Fe2S2]2+ cluster with CS2. | |
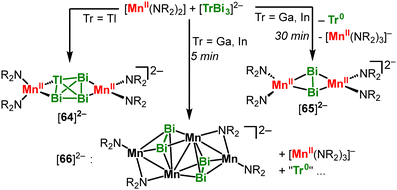 |
| | Scheme 27 Reaction of Mn(NR2)2 with tetraatomic trielbismuthates [TrBi3]2− (Tr = In, Ga, Ta). K{crypt.222} cations are omitted. | |
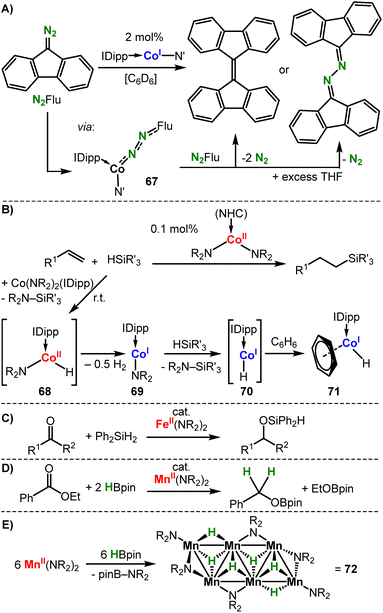 |
| | Scheme 28 (A) Cobalt mediated coupling of diazoalkanes to either alkenes or hydrazine derivatives. (B) Hydrosilylation of a variety of terminal alkenes with tertiary silanes catalysed by [(NHC)Co(N{SiMe3}2)2] via linear cobalt(I) intermediates. (C) Iron-mediated hydrosilylation of ketones. (D) Manganese mediated hydroboration of esters. (E) Isolation of a polymeric manganese hydride cluster intermediate. | |
In the presence of an excess of the diazoalkane the catalytic homocoupling to an alkene under N2 liberation is observed in non-polar solvents. The reactivity switches to hydrazine formation when donor solvents are employed. This is attributed to blocking of the metal site by the solvent and by that prevents complete metal-mediated N2 liberation.50 Metal(II) hexamethyldisilazanides have also been used in a number of hydro-elementation reactions using silanes or boranes (Scheme 28B–D). These transformations rely on the loss of complex integrity via amide/hydride exchange and redox-processes: the cobalt(II) amide Co(NR2)2 or their NHC adduct (Scheme 28B) catalyses the hydrosilylation of a variety of terminal alkenes with tertiary silanes.102 Stoichiometric studies on NHC adducts revealed reduction to the linear cobalt(I) amide 69via presumed hydride formation (68) and subsequent reductive H2 liberation. The linear hydride 70, as inferred from the isolable arene complex 71 (or the silyl derivative), are likely the active alkene hydrosilylation agents. The neutral iron and manganese complexes MII(NR2)2 were found to be active in the hydrosilylation or hydroboration of carbonyl compounds at room temperature (Scheme 28C and D).103 Under catalytic conditions MII(NR2)2 serves as a precatalyst and initially forms mono- or polymeric metal hydrides.104,105 The latter was evidenced exemplarily for Mn(NR2)2, that reacted with HAliPr2 to form the hexameric, sheet-like [MnII6H6(NR2)6] (72, Scheme 28E).105
Beyond linear metal amides: notable examples of other linear 3d-metal complexes.
As mentioned in the introduction, some linear complexes bearing other ligands were successfully employed in chemical transformations, mostly in catalysis. For example, linear FeII and MnII terphenyls served as precatalysts in hydroamination (Scheme 29A)106 and hydrophosphination (B)107 of isocyanates as well as cyclotrimerisation of isocyanates (C)108 and alkynes.109 However, the reaction conditions and substrate types implicate in situ transformation of the central two-coordinate metal fragment into higher-coordinate metal species. For manganese(II) aryl compounds the amineborane dehydrogenation was also reported (Scheme 29D), which proceeds by either a homogenous or heterogeneous (via in situ nanoparticle formation) mechanism.110 A two-coordinate iron alkoxide served as a precatalyst for stereospecific ring-opening metathesis polymerization of norbornene (Scheme 29E).111 An increase in the polymerization rate was observed by in situ addition of one equivalent of a more acidic fluorinated alcohol, by presumed intermittent formation of a linear heteroleptic alkoxide complex. Purely NHC based linear complexes should also be mentioned. Mn(cAAC)2 is able to cleave H2 under ambient conditions, yielding the Mn(II) bisalkyl complex 73 (Scheme 30).27 For Fe(cAAC)2 reversible coordination of N2 was reported.112 The labile N2-adduct 74 can be stabilized by further reduction with KC8 and 18c6 to the anionic 75, which allows for subsequent (catalytic) N2 silylation. Co(cAAC)2 serves amongst others as a coordination site for diazoalkanes (76) or alkynes (77).113 Computational analysis of the alkyne complex 77 revealed only marginal π-backdonation to the alkyne (or even its reduction). It contrasts the calculated situation for the anionic [CoI(NR2)2(η2-PhCCPh)]− ([Co19Ph]−, Scheme 11), despite Co(cAAC)2 having a lower oxidation state.
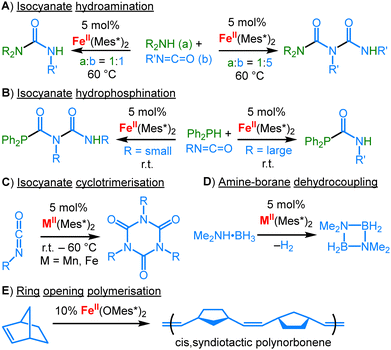 |
| | Scheme 29 Application of linear aryl and alkoxide metal complexes as precatalysts for isocyananate hydroamination (A), hydrophosphination (B) as well as cyclotrimerisation (C), for amine-borane dehydrocoupling (D) and for ring opening polymerisation (E). | |
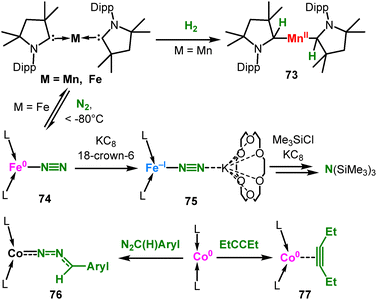 |
| | Scheme 30 Reactivity of linear, cAAC based low-valent 3d-metal complexes. | |
Conclusion
For decades, linear open-shell 3d-metal complexes have been a curiosity and synthetic challenge in coordination chemistry. However, in recent years their potential as single ion magnets, as reaction partners and even as (pre)catalysts, has been developed. Of these, silylamide based complexes have gained prominence, either in the divalent or monovalent state. Divalent metal silylamides can serve most notably as platforms for imido metal species. They exhibit intriguing electronic structures of mixed metal(III) imidyl and metal(II) nitrene characteristics, which were further exploited for catalytic C–H amination catalysis. In this context, the pronounced Lewis acidity of M(NR2)2 helped in the isolation of weakly bound organoazide adducts as the initial species in the catalytic cycle. These adducts can be irradiated in-crystallo under N2 liberation and metal imido formation, which were otherwise too reactive in solution chemistry. Looking also to other linear, mainly aryl-based metal(II) complexes, they served as precatalysts for a variety of transformations. However, the employed compounds are subject to intricate transformation reactions that can involve substrate insertion into the original metal–ligand bonds as well as substantial redox state changes.
The chemistry of the monovalent siblings, either anionic and homoleptic or neutral and heteroleptic with NHC ligation, exhibits a larger reactivity variety due to a more electron-rich metal centre. The most salient features of linear metal(I) complexes are a pronounced loss of Lewis acidity despite the coordinatively unsaturated state. Furthermore, the interaction of these compounds with substrates is characterized by one-electron transfer processes. This property is most pronounced for anionic complexes ([M(NR2)2]−) and was exploited for the generation of a number of metal(II) stabilized radical anions. It was exemplified for carbonyl or iminyl based substrates and could also be extended to alkynes and alkenes. Complexes with the latter could undergo radical-anion based bond transformations, such as cis → trans isomerisation. Here, certain limits of linear metal(I) based chemistry with π-accepting ligands also became evident as the interaction with CC triple bonds can lead to complex dismutation driven by the formation of comparably stable 18 VE organometal complexes. Like for the neutral metal(II) species, anionic and neutral linear metal(I) complexes are suitable platforms for imido metal complexes in higher spin states using organo azides as transfer agents. This allows for catalytic applications, with evidence that the employed metal silylamide fragments remains intact. For similar diazo methane derivatives, they can be catalytically coupled by a linear cobalt(I) complex under N2 extrusion to give either alkenes or hydrazine derivatives.
A very different area is the bottom-up construction of small metal/main-group clusters, in which the dual role of metal silylamides is emphasized. Anionic iron(I) silylamides can activate elemental chalcogenes, that likely proceeds via initial single-electron-transfer from the metal to yield chalcogenide (poly)anions. These immediately recombine with the now Lewis acidic iron(II) silylamides to unusual metal sulfide or selenide motifs. Accordingly, neutral metal(II) silylamides can directly be combined with main-group polyanions (e.g. zintl ions). In all cases, further redox state changes can be observed, yielding highly unusual clusters with low-coordinate metal ions.
Outlook
Despite the presented knowledge for linear metal silylamides, it is evident that for this compound class alone a multitude of combinations with already examined substrates are still open for discovery. Possible variations concern for example other main-group element/metal combinations or advancing the field of catalytically relevant imido metal species. Furthermore, the flurry of intriguing organic transfer-agents in recent years, such as diazoolefins (NHCCN2)114 or the carbon atom transfer agent Ph2SCN2,115 might pose unchartered territory with regards to metal-main group multiple bonds in higher spin state.
Looking beyond, the introduction of NHC ligation further opened up the variability in terms of charge and gave access to formally zero- or even subvalent compounds. Here, the non-innocence of alkyl amino carbene ligands is prospective to facilitate multi-electron bond activation processes. These NHC complexes were so far only little examined in a broader fashion, which would be needed to address the integrity and stability of the employed two-coordinate [M(NHC)2]n fragments during bond activation processes. In this context, the NHC based metallates, namely [Fe(cAAC)2(N2)]− or the higher coordinate [Co(IMes)2(N2)]−116 with their labile N2 ligation, are possible candidates. These can serve as M−I synthons, similar to arene metallates such as [Fe(anthracen)2]−,117 yet with more persistent ancillary ligands. The anionic charge might prevent unwanted decomposition or aggregation reactions, enhance the likelihood of higher spin states and provide beneficial crystallisation properties as seen for anionic metal(I) silylamides. In conclusion, the chemistry of linear open shell complexes still leaves a wide open chemical space. Its further exploration is promising for uncovering new and unusual reactivity patterns, which we and others will continue to address.
Author contributions
A. C. and C. G. W. conceptionalised and wrote the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
C. G. W. acknowledges the Deutsche Forschungsgemeinschaft (grants WE 5627/7-1 and WE 5627/8-1).
Notes and references
-
(a)
M. F. Lappert, Metal amide chemistry, Wiley, Chichester, UK, 2009 Search PubMed;
(b)
Metal and metalloid amides: syntheses, structures, and physical and chemical properties, Chichester, 1979 Search PubMed;
(c) P. P. Power, Comment. Inorg. Chem., 1989, 8, 177–202 CrossRef CAS;
(d) P. P. Power, Chemtracts, 1994, 6, 181–194 CAS;
(e) D. L. Kays, Dalton Trans., 2011, 40, 769–778 RSC;
(f) A. Noor, Coord. Chem. Rev., 2023, 476, 214941 CrossRef CAS;
(g) L. J. Taylor and D. L. Kays, Dalton Trans., 2019, 48, 12365–12381 RSC.
- P. P. Power, Chem. Rev., 2012, 112, 3482–3507 CrossRef CAS PubMed.
- H. Bürger and U. Wannagat, Monatsh. Chem., 1963, 94, 1007–1012 CrossRef.
- H. Bürger and U. Wannagat, Monatsh. Chem., 1964, 95, 1099–1102 CrossRef.
-
(a) D. C. Bradley, M. B. Hursthouse, K. M. Abdul Malik and R. Möseler, Transition Met. Chem., 1978, 3, 253–254 CrossRef CAS;
(b) B. D. Murray and P. P. Power, Inorg. Chem., 1984, 23, 4584–4588 CrossRef CAS;
(c) R. A. Andersen, K. Faegri, J. C. Green, A. Haaland, M. F. Lappert, W. P. Leung and K. Rypdal, Inorg. Chem., 1988, 27, 1782–1786 CrossRef CAS.
- M. M. Olmstead, P. P. Power and S. C. Shoner, Inorg. Chem., 1991, 30, 2547–2551 CrossRef CAS.
- A. M. Bryan, G. J. Long, F. Grandjean and P. P. Power, Inorg. Chem., 2013, 52, 12152–12160 CrossRef CAS PubMed.
- D. C. Bradley, M. B. Hursthouse, C. W. Newing and A. J. Welch, Chem. Commun., 1972, 567 RSC.
- D. C. Bradley, M. B. Hursthouse, A. A. Ibrahim, K. Malik, M. Motevalli, R. Möseler, H. Powell, J. D. Runnacles and A. C. Sullivan, Polyhedron, 1990, 9, 2959–2964 CrossRef CAS.
- M. Faust, A. M. Bryan, A. Mansikkamäki, P. Vasko, M. M. Olmstead, H. M. Tuononen, F. Grandjean, G. J. Long and P. P. Power, Angew. Chem., Int. Ed., 2015, 54, 12914–12917 CrossRef CAS PubMed.
-
(a) D. L. J. Broere, I. Čorić, A. Brosnahan and P. L. Holland, Inorg. Chem., 2017, 56, 3140–3143 CrossRef CAS PubMed;
(b) R. Weller, A. Gonzalez, H. Gottschling, C. von Hänisch and C. G. Werncke, Z. Anorg. Allg. Chem., 2022, 648, e202100338 CrossRef CAS.
- A. M. Borys and E. Hevia, Organometallics, 2021, 40, 442–447 CrossRef CAS.
- A. Reckziegel, B. Battistella, A. Schmidt and C. G. Werncke, Inorg. Chem., 2022, 61, 7794–7803 CrossRef CAS PubMed.
-
P. P. Power, Inorganic Syntheses, ed. P. P. Power, John Wiley & Sons, 2018, vol. 37, pp. 1–14 Search PubMed.
- R. A. Andersen, A. Haaland, K. Rypdal and H. V. Volden, J. Chem. Soc., Chem. Commun., 1985, 1807 RSC.
- N. H. Buttrus, C. Eaborn, P. B. Hitchcock, J. D. Smith and A. C. Sullivan, J. Chem. Soc., Chem. Commun., 1985, 1380 RSC.
-
(a) W. Seidel, H. Müller and H. Görls, Angew. Chem., Int. Ed. Engl., 1995, 34, 325–327 CrossRef;
(b) R. J. Wehmschulte and P. P. Power, Organometallics, 1995, 14, 3264–3267 CrossRef CAS;
(c) C. Ni, T. A. Stich, G. J. Long and P. P. Power, Chem. Commun., 2010, 46, 4466 RSC.
-
(a) C. Ni and P. P. Power, Chem. Commun., 2009, 5543–5545 RSC;
(b) J. J. Ellison, K. Ruhlandt-Senge and P. P. Power, Angew. Chem., Int. Ed. Engl., 1994, 33, 1178–1180 CrossRef;
(c) T. Nguyen, A. D. Sutton, M. Brynda, J. C. Fettinger, G. J. Long and P. P. Power, Science, 2005, 310, 844–847 CrossRef CAS PubMed;
(d) A. M. Bryan, G. J. Long, F. Grandjean and P. P. Power, Inorg. Chem., 2014, 53, 2692–2698 CrossRef CAS PubMed.
-
(a) J. N. Boynton, J.-D. Guo, J. C. Fettinger, C. E. Melton, S. Nagase and P. P. Power, J. Am. Chem. Soc., 2013, 135, 10720–10728 CrossRef CAS PubMed;
(b) J. Li, H. Song, C. Cui and J.-P. Cheng, Inorg. Chem., 2008, 47, 3468–3470 CrossRef CAS PubMed;
(c) A. M. Bryan, W. A. Merrill, W. M. Reiff, J. C. Fettinger and P. P. Power, Inorg. Chem., 2012, 51, 3366–3373 CrossRef CAS PubMed.
- W. Alexander Merrill, T. A. Stich, M. Brynda, G. J. Yeagle, J. C. Fettinger, R. D. Hont, W. M. Reiff, C. E. Schulz, R. D. Britt and P. P. Power, J. Am. Chem. Soc., 2009, 131, 12693–12702 CrossRef CAS PubMed.
- J. N. Boynton, W. A. Merrill, W. M. Reiff, J. C. Fettinger and P. P. Power, Inorg. Chem., 2012, 51, 3212–3219 CrossRef CAS PubMed.
-
(a) H. Chen, R. A. Bartlett, M. M. Olmstead, P. P. Power and S. C. Shoner, J. Am. Chem. Soc., 1990, 112, 1048–1055 CrossRef CAS;
(b) R. A. Bartlett and P. P. Power, J. Am. Chem. Soc., 1987, 109, 7563–7564 CrossRef CAS;
(c) S. N. Konig, C. Schadle, C. Maichle-Mossmer and R. Anwander, Inorg. Chem., 2014, 53, 4585–4597 CrossRef PubMed;
(d) K. Freitag, C. R. Stennett, A. Mansikkamäki, R. A. Fischer and P. P. Power, Inorg. Chem., 2021, 60, 4108–4115 CrossRef CAS PubMed.
- D. J. Liptrot and P. P. Power, Nat. Rev. Chem., 2017, 1, 0004 CrossRef.
-
(a) Y. Liu, N. T. Coles, N. Cajiao, L. J. Taylor, E. S. Davies, A. Barbour, P. J. Morgan, K. Butler, B. Pointer-Gleadhill, S. P. Argent, J. McMaster, M. L. Neidig, D. Robinson and D. L. Kays, Chem. Sci., 2024, 15, 9599–9611 RSC;
(b) H. R. Sharpe, A. M. Geer, L. J. Taylor, B. M. Gridley, T. J. Blundell, A. J. Blake, E. S. Davies, W. Lewis, J. McMaster, D. Robinson and D. L. Kays, Nat. Commun., 2018, 9, 3757 CrossRef PubMed.
- S. Roy, K. C. Mondal and H. W. Roesky, Acc. Chem. Res., 2016, 49, 357–369 CrossRef CAS PubMed.
- S. Kumar Kushvaha, A. Mishra, H. W. Roesky and K. Chandra Mondal, Chem. – Asian J., 2022, 17, e202101301 CrossRef CAS PubMed.
- P. P. Samuel, K. C. Mondal, H. W. Roesky, M. Hermann, G. Frenking, S. Demeshko, F. Meyer, A. C. Stückl, J. H. Christian, N. S. Dalal, L. Ungur, L. F. Chibotaru, K. Pröpper, A. Meents and B. Dittrich, Angew. Chem., Int. Ed., 2013, 52, 11817–11821 CrossRef CAS PubMed.
- G. Ung, J. Rittle, M. Soleilhavoup, G. Bertrand and J. C. Peters, Angew. Chem., Int. Ed., 2014, 53, 8427–8431 CrossRef CAS PubMed.
- K. C. Mondal, S. Roy, S. De, P. Parameswaran, B. Dittrich, F. Ehret, W. Kaim and H. W. Roesky, Chem. – Eur. J., 2014, 20, 11646–11649 CrossRef CAS PubMed.
- K. C. Mondal, P. P. Samuel, Y. Li, H. W. Roesky, S. Roy, L. Ackermann, N. S. Sidhu, G. M. Sheldrick, E. Carl, S. Demeshko, S. De, P. Parameswaran, L. Ungur, L. F. Chibotaru and D. M. Andrada, Eur. J. Inorg. Chem., 2014, 818–823 CrossRef CAS.
-
(a) P. P. Samuel, R. Neufeld, K. Chandra Mondal, H. W. Roesky, R. Herbst-Irmer, D. Stalke, S. Demeshko, F. Meyer, V. C. Rojisha, S. De, P. Parameswaran, A. C. Stückl, W. Kaim, J. H. Christian, J. K. Bindra and N. S. Dalal, Chem. Sci., 2015, 6, 3148–3153 RSC;
(b) R. C. Poulten, M. J. Page, A. G. Algarra, J. J. Le Roy, I. López, E. Carter, A. Llobet, S. A. Macgregor, M. F. Mahon, D. M. Murphy, M. Murugesu and M. K. Whittlesey, J. Am. Chem. Soc., 2013, 135, 13640–13643 CrossRef CAS PubMed.
- C. G. Werncke, E. Suturina, P. C. Bunting, L. Vendier, J. R. Long, M. Atanasov, F. Neese, S. Sabo-Etienne and S. Bontemps, Chem. – Eur. J., 2016, 22, 1668–1674 CrossRef CAS PubMed.
- I. C. Cai, M. I. Lipschutz and T. D. Tilley, Chem. Commun., 2014, 50, 13062–13065 RSC.
- J. M. Zadrozny, M. Atanasov, A. M. Bryan, C.-Y. Lin, B. D. Rekken, P. P. Power, F. Neese and J. R. Long, Chem. Sci., 2013, 4, 125–138 RSC.
- C.-Y. Lin, J.-D. Guo, J. C. Fettinger, S. Nagase, F. Grandjean, G. J. Long, N. F. Chilton and P. P. Power, Inorg. Chem., 2013, 52, 13584–13593 Search PubMed.
-
(a) C. L. Wagner, L. Tao, J. C. Fettinger, R. D. Britt and P. P. Power, Inorg. Chem., 2019, 58, 8793–8799 CrossRef CAS PubMed;
(b) M. I. Lipschutz and T. D. Tilley, Chem. Commun., 2012, 48, 7146–7148 RSC.
- R. Weller, L. Ruppach, A. Shlyaykher, F. Tambornino and C. G. Werncke, Dalton Trans., 2021, 50, 10947–10963 RSC.
- M. I. Lipschutz, X. Yang, R. Chatterjee and T. D. Tilley, J. Am. Chem. Soc., 2013, 135, 15298–15301 CrossRef CAS PubMed.
- I. C. Cai, M. S. Ziegler, P. C. Bunting, A. Nicolay, D. S. Levine, V. Kalendra, P. W. Smith, K. V. Lakshmi and T. D. Tilley, Organometallics, 2019, 38, 1648–1663 CrossRef CAS.
- P.-C. Yang, K.-P. Yu, C.-T. Hsieh, J. Zou, C.-T. Fang, H.-K. Liu, C.-W. Pao, L. Deng, M.-J. Cheng and C.-Y. Lin, Chem. Sci., 2022, 13, 9637–9643 RSC.
-
(a) Y. W. Chao, P. A. Wexler and D. E. Wigley, Inorg. Chem., 1989, 28, 3860–3868 CrossRef CAS;
(b) D. K. Kennepohl, S. Brooker, G. M. Sheldrick and H. W. Roesky, Chem. Ber., 1991, 124, 2223–2225 CrossRef CAS;
(c) R. J. Schwamm, C. M. Fitchett and M. P. Coles, Chem. – Asian J., 2019, 14, 1204–1211 CrossRef CAS PubMed.
- C. L. Wagner, L. Tao, E. J. Thompson, T. A. Stich, J. Guo, J. C. Fettinger, L. A. Berben, R. D. Britt, S. Nagase and P. P. Power, Angew. Chem., Int. Ed., 2016, 55, 10444–10447 CrossRef CAS PubMed.
- R. Weller, I. Müller, C. Duhayon, S. Sabo-Etienne, S. Bontemps and C. G. Werncke, Dalton Trans., 2021, 50, 4890–4903 RSC.
- R. Weller, M. Atanasov, S. Demeshko, T.-Y. Chen, I. Mohelsky, E. Bill, M. Orlita, F. Meyer, F. Neese and C. G. Werncke, Inorg. Chem., 2023, 62, 3153–3161 CrossRef CAS PubMed.
-
(a) N. L. Bell, M. Gladkikh, C. Fraser, M. Elsayed, E. Richards and R. D. Turnbull, Angew. Chem., Int. Ed., 2025, e202505408 CAS;
(b) A. Eichhöfer, Y. Lan, V. Mereacre, T. Bodenstein and F. Weigend, Inorg. Chem., 2014, 53, 1962–1974 CrossRef PubMed.
- C. G. Werncke, P. C. Bunting, C. Duhayon, J. R. Long, S. Bontemps and S. Sabo-Etienne, Angew. Chem., Int. Ed., 2015, 54, 245–248 CrossRef CAS PubMed.
-
(a) D.-Y. Lu, J.-S. K. Yu, T.-S. Kuo, G.-H. Lee, Y. Wang and Y.-C. Tsai, Angew. Chem., Int. Ed., 2011, 50, 7611–7615 CrossRef CAS PubMed;
(b) J. Chai, H. Zhu, A. C. Stückl, H. W. Roesky, J. Magull, A. Bencini, A. Caneschi and D. Gatteschi, J. Am. Chem. Soc., 2005, 127, 9201–9206 CrossRef CAS PubMed.
- A. A. Danopoulos, P. Braunstein, K. Y. Monakhov, J. van Leusen, P. Kögerler, M. Clémancey, J.-M. Latour, A. Benayad, M. Tromp, E. Rezabal and G. Frison, Dalton Trans., 2017, 46, 1163–1171 RSC.
- M. I. Lipschutz, T. Chantarojsiri, Y. Dong and T. D. Tilley, J. Am. Chem. Soc., 2015, 137, 6366–6372 CrossRef CAS PubMed.
- Y. Dong, M. I. Lipschutz, R. J. Witzke, J. A. Panetier and T. D. Tilley, ACS Catal., 2021, 11, 11160–11170 CrossRef CAS.
- R. J. Witzke, D. Hait, M. Head-Gordon and T. D. Tilley, Organometallics, 2021, 40, 1758–1764 CrossRef CAS.
- C. A. Laskowski and G. L. Hillhouse, J. Am. Chem. Soc., 2008, 130, 13846–13847 CrossRef CAS PubMed.
- C. A. Laskowski, G. R. Morello, C. T. Saouma, T. R. Cundari and G. L. Hillhouse, Chem. Sci., 2013, 4, 170–174 RSC.
- T. Inatomi, Y. Fukahori, Y. Yamada, R. Ishikawa, S. Kanegawa, Y. Koga and K. Matsubara, Catal. Sci. Technol., 2019, 9, 1784–1793 RSC.
- M. I. Lipschutz and T. D. Tilley, Organometallics, 2014, 33, 5566–5570 CrossRef CAS PubMed.
- A. Gonzalez, T.-Y. Chen, S. Demeshko, F. Meyer and C. G. Werncke, Organometallics, 2023, 42, 1198–1204 CrossRef CAS.
- K.-C. Hsiao, P.-C. Yang, C.-T. Fang, H.-K. Liu and C.-Y. Lin, Chem. – Asian J., 2024, 19, e202300924 CrossRef CAS PubMed.
- J. M. Zadrozny, D. J. Xiao, M. Atanasov, G. J. Long, F. Grandjean, F. Neese and J. R. Long, Nat. Chem., 2013, 5, 577–581 CrossRef CAS PubMed.
- P. C. Bunting, M. Atanasov, E. Damgaard-Møller, M. Perfetti, I. Crassee, M. Orlita, J. Overgaard, J. van Slageren, F. Neese and J. R. Long, Science, 2018, 362, 7319 CrossRef PubMed.
- M. K. Thomsen, A. Nyvang, J. P. S. Walsh, P. C. Bunting, J. R. Long, F. Neese, M. Atanasov, A. Genoni and J. Overgaard, Inorg. Chem., 2019, 58, 3211–3218 CrossRef CAS PubMed.
- M. Atanasov, J. M. Zadrozny, J. R. Long and F. Neese, Chem. Sci., 2013, 4, 139–156 RSC.
- W. M. Reiff, A. M. LaPointe and E. H. Witten, J. Am. Chem. Soc., 2004, 126, 10206–10207 CrossRef CAS PubMed.
- D. Errulat, K. L. M. Harriman, D. A. Gálico, J. S. Ovens, A. Mansikkamäki and M. Murugesu, Inorg. Chem. Front., 2021, 8, 5076–5085 RSC.
-
(a) A. M. Bryan, C.-Y. Lin, M. Sorai, Y. Miyazaki, H. M. Hoyt, A. Hablutzel, A. LaPointe, W. M. Reiff, P. P. Power and C. E. Schulz, Inorg. Chem., 2014, 53, 12100–12107 CrossRef CAS PubMed;
(b) X.-N. Yao, J.-Z. Du, Y.-Q. Zhang, X.-B. Leng, M.-W. Yang, S.-D. Jiang, Z.-X. Wang, Z.-W. Ouyang, L. Deng, B.-W. Wang and S. Gao, J. Am. Chem. Soc., 2017, 139, 373–380 CrossRef CAS PubMed.
- J. M. Frost, K. L. M. Harriman and M. Murugesu, Chem. Sci., 2016, 7, 2470–2491 RSC.
- J. M. Zadrozny, D. J. Xiao, J. R. Long, M. Atanasov, F. Neese, F. Grandjean and G. J. Long, Inorg. Chem., 2013, 52, 13123–13131 Search PubMed.
- P. P. Samuel, K. C. Mondal, N. Amin Sk, H. W. Roesky, E. Carl, R. Neufeld, D. Stalke, S. Demeshko, F. Meyer, L. Ungur, L. F. Chibotaru, J. Christian, V. Ramachandran, J. van Tol and N. S. Dalal, J. Am. Chem. Soc., 2014, 136, 11964–11971 CrossRef CAS PubMed.
- Y.-S. Meng, Z. Mo, B.-W. Wang, Y.-Q. Zhang, L. Deng and S. Gao, Chem. Sci., 2015, 6, 7156–7162 Search PubMed.
- Z. Ouyang, J. Du, L. Wang, J. L. Kneebone, M. L. Neidig and L. Deng, Inorg. Chem., 2015, 54, 8808–8816 CrossRef CAS PubMed.
- C. G. Werncke and I. Müller, Chem. Commun., 2020, 56, 2268–2271 RSC.
- C. Schneider, L. Guggolz and C. G. Werncke, Dalton Trans., 2021, 51, 179–184 RSC.
- R. J. Witzke, D. Hait, K. Chakarawet, M. Head-Gordon and T. D. Tilley, ACS Catal., 2020, 10, 7800–7807 CrossRef CAS.
- I. Müller, D. Munz and C. G. Werncke, Inorg. Chem., 2020, 59, 9521–9537 CrossRef PubMed.
- C. G. Werncke, I. Müller, K. Weißer and C. Limberg, Inorg. Chem., 2024, 63, 15236–15246 CrossRef CAS PubMed.
- R. Weller, I. Müller and G. Werncke, Eur. J. Inorg. Chem., 2022, e202100955 CrossRef CAS.
- D. Brenna, M. Villa, T. N. Gieshoff, F. Fischer, M. Hapke and A. Jacobi von Wangelin, Angew. Chem., Int. Ed., 2017, 56, 8451–8454 CrossRef CAS PubMed.
- G. Sieg, I. Müller, K. Weißer and G. Werncke, Chem. Sci., 2022, 13, 13872 RSC.
- I. Müller, C. Schneider, C. Pietzonka, F. Kraus and C. G. Werncke, Inorganics, 2019, 7, 117 CrossRef.
- I. Müller and C. G. Werncke, Chem. – Eur. J., 2021, 27, 4932–4938 CrossRef PubMed.
- G. Sieg, Q. Pessemesse, S. Reith, S. Yelin, C. Limberg, D. Munz and C. G. Werncke, Chem. – Eur. J., 2021, 27, 16760–16767 CrossRef CAS PubMed.
- G. Sieg, M. Fischer, F. Dankert, J.-E. Siewert, C. Hering-Junghans and C. G. Werncke, Chem. Commun., 2022, 58, 9786–9789 RSC.
-
(a) S. Sandl and A. Jacobi von Wangelin, Angew. Chem., Int. Ed., 2020, 59, 5434–5437 CrossRef CAS PubMed;
(b) J. D. Sears, P. G. N. Neate and M. L. Neidig, J. Am. Chem. Soc., 2018, 140, 11872–11883 CrossRef CAS PubMed;
(c) C.-Y. Lin and P. P. Power, Chem. Soc. Rev., 2017, 46, 5347–5399 RSC.
- M. I. Lipschutz and T. D. Tilley, Angew. Chem., Int. Ed., 2014, 53, 7290–7294 CrossRef CAS PubMed.
- C. G. Werncke, J. Pfeiffer, I. Müller, L. Vendier, S. Sabo-Etienne and S. Bontemps, Dalton Trans., 2019, 48, 1757–1765 RSC.
-
(a) P. Müller and C. Fruit, Chem. Rev., 2003, 103, 2905–2920 CrossRef PubMed;
(b) L. Zhang and L. Deng, Chin. Sci. Bull., 2012, 57, 2352–2360 CrossRef CAS;
(c) Y. Park, Y. Kim and S. Chang, Chem. Rev., 2017, 117, 9247–9301 CrossRef CAS PubMed;
(d) Y. Liu, T. You, H.-X. Wang, Z. Tang, C.-Y. Zhou and C.-M. Che, Chem. Soc. Rev., 2020, 49, 5310–5358 RSC;
(e)
K. Kawakita, Y. Kakiuchi, H. Tsurugi, K. Mashima, B. F. Parker, J. Arnold and I. A. Tonks, Coord. Chem. Rev., 2020, 407, 213118 Search PubMed.
-
(a) P. F. Kuijpers, J. I. van der Vlugt, S. Schneider and B. de Bruin, Chem. – Eur. J., 2017, 23, 13819–13829 CrossRef CAS PubMed;
(b) K. Ray, F. Heims and F. F. Pfaff, Eur. J. Inorg. Chem., 2013, 3784–3807 CrossRef CAS;
(c) A. Grünwald, S. S. Anjana and D. Munz, Eur. J. Inorg. Chem., 2021, 4147–4166 CrossRef.
- W. Stroek and M. Albrecht, Chem. Sci., 2023, 14, 2849–2859 RSC.
- W. Stroek, M. Keilwerth, L. A. Malaspina, S. Grabowsky, K. Meyer and M. Albrecht, Chem. – Eur. J., 2024, 30, e202303410 CrossRef CAS PubMed.
- A. Gonzalez, S. Demeshko, F. Meyer and C. G. Werncke, Chem. Commun., 2023, 59, 11532–11535 RSC.
- S. Reith, S. Demeshko, B. Battistella, A. Reckziegel, C. Schneider, A. Stoy, C. Lichtenberg, F. Meyer, D. Munz and C. G. Werncke, Chem. Sci., 2022, 13, 7907–7913 RSC.
- A. Reckziegel, M. Kour, B. Battistella, S. Mebs, K. Beuthert, R. Berger and C. G. Werncke, Angew. Chem., Int. Ed., 2021, 60, 15376–15380 CrossRef CAS PubMed.
- A. Reckziegel, C. Pietzonka, F. Kraus and C. G. Werncke, Angew. Chem., Int. Ed., 2020, 59, 8527–8531 CrossRef CAS PubMed.
- A. Reckziegel, B. Battistella and C. G. Werncke, Eur. J. Inorg. Chem., 2022, e202101102 CrossRef CAS.
- C. Schneider, S. Demeshko, F. Meyer and C. G. Werncke, Chem. – Eur. J., 2021, 27, 6348–6353 CrossRef CAS PubMed.
-
(a) K. M. Lancaster, M. Roemelt, P. Ettenhuber, Y. Hu, M. W. Ribbe, F. Neese, U. Bergmann and S. DeBeer, Science, 2011, 334, 974–977 CrossRef CAS PubMed;
(b) T. Spatzal, M. Aksoyoglu, L. Zhang, S. L. A. Andrade, E. Schleicher, S. Weber, D. C. Rees and O. Einsle, Science, 2011, 334, 940 CrossRef CAS PubMed;
(c) O. Einsle, F. A. Tezcan, S. L. A. Andrade, B. Schmid, M. Yoshida, J. B. Howard and D. C. Rees, Science, 2002, 297, 1696–1700 CrossRef CAS PubMed;
(d) D. C. Rees, F. Akif Tezcan, C. A. Haynes, M. Y. Walton, S. Andrade, O. Einsle and J. B. Howard, Philos. Trans. R. Soc., A, 2005, 363, 971 CrossRef CAS PubMed.
-
(a) D. Sippel, M. Rohde, J. Netzer, C. Trncik, J. Gies, K. Grunau, I. Djurdjevic, L. Decamps, S. L. A. Andrade and O. Einsle, Science, 2018, 359, 1484–1489 CrossRef CAS PubMed;
(b) W. Kang, C. C. Lee, A. J. Jasniewski, M. W. Ribbe and Y. Hu, Science, 2020, 368, 1381–1385 CrossRef CAS PubMed.
-
(a) M. J. Ryle, H. I. Lee, L. C. Seefeldt and B. M. Hoffman, Biochemistry, 2000, 39, 1114–1119 CrossRef CAS PubMed;
(b) M. Kumar, W. P. Lu and S. W. Ragsdale, Biochemistry, 1994, 33, 9769–9777 CrossRef CAS PubMed;
(c) L. C. Seefeldt, M. E. Rasche and S. A. Ensign, Biochemistry, 1995, 34, 5382–5389 CrossRef CAS PubMed.
- C. Schneider, S. Ivlev and C. G. Werncke, Eur. J. Inorg. Chem., 2022, e202200706 Search PubMed.
- C. Schneider, S. J. Groß, S. Demeshko, S. Bontemps, F. Meyer and C. G. Werncke, Chem. Commun., 2021, 57, 10751–10754 RSC.
-
(a) A. Albers, S. Demeshko, K. Pröpper, S. Dechert, E. Bill and F. Meyer, J. Am. Chem. Soc., 2013, 135, 1704–1707 CrossRef CAS PubMed;
(b) S. Yao, F. Meier, N. Lindenmaier, R. Rudolph, B. Blom, M. Adelhardt, J. Sutter, S. Mebs, M. Haumann, K. Meyer, M. Kaupp and M. Driess, Angew. Chem., Int. Ed., 2015, 54, 12506–12510 CrossRef CAS PubMed.
- J. Rienmüller, A. Schmidt, N. J. Yutronkie, R. Clérac, C. G. Werncke, F. Weigend and S. Dehnen, Angew. Chem., Int. Ed., 2022, 61, e202210683 CrossRef PubMed.
- Y. Liu and L. Deng, J. Am. Chem. Soc., 2017, 139, 1798–1801 CrossRef CAS PubMed.
- J. Yang and T. D. Tilley, Angew. Chem., Int. Ed., 2010, 49, 10186–10188 CrossRef CAS PubMed.
-
(a) Y. Ohki, Y. Shimizu, R. Araake, M. Tada, W. M. C. Sameera, J.-I. Ito and H. Nishiyama, Angew. Chem., Int. Ed., 2016, 55, 15821–15825 CrossRef CAS PubMed;
(b) T. Higaki, K. Tanaka, H. Izu, S. Oishi, K. Kawamoto, M. Tada, W. M. C. Sameera, R. Takahata, T. Teranishi, S. Kikkawa, S. Yamazoe, T. Shiga, M. Nihei, T. Kato, R. E. Cramer, Z. Zhang, K. Meyer and Y. Ohki, J. Am. Chem. Soc., 2025, 147, 3215–3222 CrossRef CAS PubMed;
(c) T. N. Gieshoff, U. Chakraborty, M. Villa and A. Jacobi von Wangelin, Angew. Chem., Int. Ed., 2017, 56, 3585–3589 CrossRef CAS PubMed.
- U. Chakraborty, E. Reyes-Rodriguez, S. Demeshko, F. Meyer and A. Jacobi von Wangelin, Angew. Chem., Int. Ed., 2018, 57, 4970–4975 CrossRef CAS PubMed.
- A. J. South, A. M. Geer, L. J. Taylor, H. R. Sharpe, W. Lewis, A. J. Blake and D. L. Kays, Organometallics, 2019, 38, 4115–4120 CrossRef CAS.
- H. R. Sharpe, A. M. Geer, W. Lewis, A. J. Blake and D. L. Kays, Angew. Chem., Int. Ed., 2017, 56, 4845–4848 CrossRef CAS PubMed.
- H. R. Sharpe, A. M. Geer, H. E. L. Williams, T. J. Blundell, W. Lewis, A. J. Blake and D. L. Kays, Chem. Commun., 2017, 53, 937–940 RSC.
- A. M. Geer, J. Navarro, P. Alamán-Valtierra, N. T. Coles, D. L. Kays and C. Tejel, ACS Catal., 2023, 13, 6610–6618 CrossRef CAS PubMed.
- H. R. Sharpe, A. M. Geer, T. J. Blundell, F. R. Hastings, M. W. Fay, G. A. Rance, W. Lewis, A. J. Blake and D. L. Kays, Catal. Sci. Technol., 2018, 8, 229–235 RSC.
- D. S. Belov, L. Mathivathanan, M. J. Beazley, W. B. Martin and K. V. Bukhryakov, Angew. Chem., Int. Ed., 2021, 60, 2934–2938 CrossRef CAS PubMed.
- G. Ung and J. C. Peters, Angew. Chem., Int. Ed., 2015, 54, 532–535 CrossRef CAS PubMed.
- J. Du, W. Chen, Q. Chen, X. Leng, Y.-S. Meng, S. Gao and L. Deng, Organometallics, 2020, 39, 729–739 CrossRef CAS.
- P. Varava, Z. Dong, R. Scopelliti, F. Fadaei-Tirani and K. Severin, Nat. Chem., 2021, 13, 1055–1060 CrossRef CAS PubMed.
- Q. Sun, J.-N. Belting, J. Hauda, D. Tymann, P. W. Antoni, R. Goddard and M. M. Hansmann, Science, 2025, 387, 885–892 CrossRef CAS PubMed.
- Y. Gao, G. Li and L. Deng, J. Am. Chem. Soc., 2018, 140, 2239–2250 CrossRef CAS PubMed.
-
(a) W. W. Brennessel, R. E. Jilek and J. E. Ellis, Angew. Chem., Int. Ed., 2007, 46, 6132–6136 CrossRef CAS PubMed;
(b) W. W. Brennessel, V. G. Young and J. E. Ellis, Angew. Chem., Int. Ed., 2006, 45, 7268–7271 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ab
*ab
































