
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMonolayer C60 networks: a first-principles perspective
Bo
Peng
 *a and
Michele
Pizzochero
*bc
*a and
Michele
Pizzochero
*bc
aTheory of Condensed Matter Group, Cavendish Laboratory, University of Cambridge, J. J. Thomson Avenue, Cambridge CB3 0HE, UK. E-mail: bp432@cam.ac.uk
bDepartment of Physics, University of Bath, Bath BA2 7AY, UK. E-mail: mp2834@bath.ac.uk
cSchool of Engineering and Applied Sciences, Harvard University, Cambridge, Massachusetts 02138, USA
First published on 13th June 2025
Abstract
Monolayer fullerene (C60) networks combine molecular-level rigidity with crystalline connectivity, offering a promising platform for numerous applications. In this Feature article, we review the physical and chemical properties of fullerene monolayers, focusing on first-principles studies. We first explore the structural stability of monolayer phases and investigate their thermal expansion behaviours. We then outline criteria for photocatalytic water splitting and introduce theoretical predictions which are supported by recent experimental verification. Finally, we show how interlayer stacking, molecular size, and dimensional tuning (from 2D monolayers into 3D crystals, 1D chains, or nanoribbons) offer versatile approaches to modulate their chemical functionality. Together, these insights establish fullerene networks as a novel class of carbon-based materials with tailored properties for catalysis, photovoltaics, and flexible electronics.
 Bo Peng | Bo Peng is a Junior Research Fellow at the University of Cambridge (United Kingdom), working in the Theory of Condensed Matter (TCM) group of the Cavendish Laboratory. His research focuses on designing quantum materials from first-principles calculations for a wide range of sustainable practices, bridging physics, chemistry, and materials science. He received his PhD in Physics from Cambridge (United Kingdom) and his MSc/BSc in Optics from Fudan University (China). |
 Michele Pizzochero | Michele Pizzochero is an Assistant Professor of Physics at the University of Bath (United Kingdom). His research focuses on the electronic structure of crystals, with a particular interest in low-dimensional carbon nanostructures investigated through first-principles methods and model Hamiltonians. Prior to his appointment at Bath, Michele was a Research Associate and Postdoctoral Fellow at Harvard University (United States), supported by a fellowship from the Swiss National Science Foundation. He earned his PhD in Physics from EPFL (Switzerland) and holds an MSc in Chemistry from the University of Milan (Italy). |
1 Introduction
Since the discovery of graphene in 2004,1 2D materials have attracted tremendous interest due to their extraordinary electronic,2–5 magnetic,6–10 optical,11–13 topological,14–18 and thermal19–24 properties. The basic building blocks of these 2D materials are atoms. Consider, for example, graphene: carbon atoms form a planar honeycomb lattice with sp2 hybridisation from three planar σ bonds and the out-of-plane π bonding.25 By varying constituent atoms (e.g., from C to Si or Sn), the lattice composition and crystalline symmetry can be controlled at the atomic scale,26–32 leading to tailored functionalities such as thermoelectricity33,34 and non-Abelian braiding.35,36 Additionally, structural phase transitions can be induced by external external stimuli such as temperature and light,37–40 enabling precisely tuneable physical and chemical properties. The crystal structures can be further manipulated by defect engineering,41–44 offering further degrees of freedom for applications such as single-photon sources.45–47 Despite their advantages such as atom-by-atom precision, the designed 2D crystals from atomic building blocks are constrained by their intrinsic characteristics such as limited types of chemical bonds and restricted stability as freestanding monolayers.48 Moreover, precise atomic construction remains challenging.49 As an alternative, replacing atoms with molecules as building units of crystals (Fig. 1) results in the combination of molecular rigidity and chemical tuneability, offering new opportunities in rational materials design for tailored electronic,50,51 optical,52 chemical,53 and magnetic54,55 properties. | ||
| Fig. 1 Carbon atoms and fullerene molecules as basic building units for graphene and monolayer fullerene networks respectively. | ||
The discovery of buckminsterfullerene C60 in 1985 has provided an atom-like, stable building units56–59 to form various lattice structures, as demonstrated by experimental synthesis of solid C60 in 1990–92 where fullerene molecules are bounded by van der Waals interactions with orientational ordering transition upon heating.60–64 A series of follow-up studies in 1993–96 have found that neighbouring C60 cages can be connected through covalent [2+2] cycloaddition bonds as a result of photo- or pressure-induced polymerisation,65–71 forming a rich phase diagram of fullerene polymers.72,73 These fullerene crystals exhibit distinct mechanical,74 optical,75 vibrational,76 electronic,77–79 and polaronic80,81 properties, leading to numerous applications such as photovoltaics82,83 and superconductivity.84–88 Recent breakthroughs in synthesis of monolayer fullerene networks in 2022 have demonstrated the feasibility of creating 2D materials based on C60 molecules through organic cation slicing exfoliation of Mg-intercalated bulk single crystals.89 Different from graphene with limited chemical bonding types of carbon atoms, fullerene molecules provide abundant intermolecular bonding positions with extra rotational degrees of freedom to form richer lattice geometries. As shown in Fig. 1, neighbouring carbon cages can be linked through both intermolecular [2+2] cycloaddition bonds and C–C single bonds, resulting in a nearly-triangular lattice with impossible coordination environments for individual carbon atoms. Similarly, it is possible to realise 1D, nearly-square, and nearly-hexagonal lattices in monolayer C60 networks. The diverse lattice geometries, delocalised π electrons, and large surface area in C60 monolayers endow them with promising electronic, optical, and chemical properties for energy applications.90–93 Their sizable band gaps and high charge transport are critical for use in photocatalytic water splitting, organic photovoltaics, and flexible energy storage devices. Moreover, the presence of abundant surface active sites offers opportunities for catalytic reactions such as the hydrogen evolution reaction (HER).
Despite the significant experimental progress in the synthesis and characterisation of 2D fullerene networks,89 a comprehensive understanding of their physical and chemical properties is still lacking. To this end, modern computational techniques can close this gap by providing atomistic resolution on chemical functionalities of monolayer polymeric C60. Here, we highlight three key aspects from our recent research based on first-principles calculations: (1) Are monolayer C60 networks stable? (2) Are they promising photocatalysts? (3) Are their chemical functionalities tuneable? To answer these questions, our Feature article is organised as follows: in Section 2, we summarise the structural properties of C60 monolayers, their stability and strength, as well as their thermal expansion behaviours. In Section 3, we examine the criteria for 2D C60 networks to be efficient photocatalysts and show how theoretical predictions lead to experimental verifications. In Section 4, we discuss possible strategies in tuning chemical functionalities of fullerene networks. Finally, we conclude by identifying potential avenues for future research in Section 5.
2 Are monolayer C60 networks stable?
In this Section, we address the structural stability of monolayer fullerene networks from first principles. We start with an introduction to the structural properties of the experimentally-reported crystal structures. Next, we discuss their stability and strength, including dynamic, thermodynamic, and mechanical properties to rationalise experimental observations. Lastly, we explore the dimensional stability of monolayer fullerene networks upon heating in the context of thermal expansion.2.1 Crystal structures
The crystal structures of monolayer fullerene networks reveal a remarkable diversity in intermolecular bonding environments, stemming from the intrinsic rotational degrees of freedom of C60 molecules and their multiple intermolecular bonding sites. Starting from the experimentally-reported structures,89 three distinct crystalline phases are obtained from geometry optimisation based on density functional theory (DFT),94,95 as overviewed in Fig. 2: a quasi-tetragonal phase with one-dimensional chains along b where C60 units are connected through [2+2] cycloaddition bonds (denoted as qTP1), a quasi-tetragonal phase in two-dimensional nearly-square lattices with vertical and horizontal [2+2] cycloaddition bonds along a and b respectively (denoted as qTP2), and a quasi-hexagonal phase in a nearly triangular lattice via [2+2] cycloaddition bonds along b and C–C single bonds along the diagonal of the a and b lattice vectors (denoted as qHP).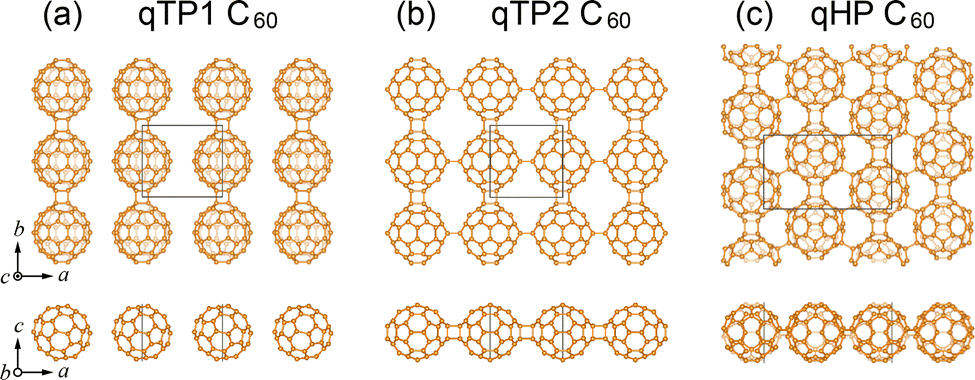 | ||
| Fig. 2 Crystal structures of (a) qTP1, (b) qTP2, and (c) qHP monolayer polymeric C60 from top and front views.90 | ||
All three networks retain the molecular curvature of individual fullerenes, but differ significantly in packing density, bond connectivity, and unit cell geometry, leading to distinct phonon spectra and elastic behaviours. Covalent polymerisation of qTP1 C60 molecules along b yields quasi-one-dimensional crystalline networks without interchain bond formation. These arrangements result in anisotropic lattice parameters and weak interchain bonding. Monolayer qTP2 C60 networks possess a more symmetric structure of space group Pmmm (no. 47) with enhanced in-plane isotropy and more robust connectivity. As the only networks with intermolecular C–C single bonds, the qHP monolayers exhibit nearly isotropic packing density but strong anisotropic intermolecular bonds, which impose anisotropic structural, elastic, and vibrational constraints on mechanical stiffness and thermal expansion, as discussed below.
2.2 Stability and strength
Comparing to the well-understood formation mechanisms and stability of fullerene molecules,96–98 when assembling C60 units into 2D configurations, it remains unclear whether ordered monolayer polymeric C60 structures are stable under ambient conditions. Experimentally, only qHP C60 has been obtained as both monolayers89 and few-layers,99–101 while all the qTP C60 flakes are few-layer.89 These results raise doubts regarding the stability of monolayer fullerene networks. Previous first-principles studies have examined a range of structural phases of monolayer C60 and confirmed the mechanical stability of several configurations.102–104 Additionally, molecular dynamics simulations have addressed their thermal stability, indicating that both qTP and qHP structures remain stable up to 800 K,105 partially corroborating experimental findings that qHP monolayers do not decompose at 600 K.89 Nevertheless, the absence of experimental exfoliation of qTP monolayers raises unresolved questions, suggesting that thermal and mechanical stability alone may be insufficient to explain their phase behaviour.To access the structural stability, a systematic investigation on dynamic, thermodynamic, and mechanical stability is required.48,106 In the following, we show how first-principles simulations can be used to address the following questions:107
(i) Are qTP and qHP C60 monolayers dynamically stable?
(ii) What is their relative thermodynamic stability?
(iii) Do their mechanical properties support phase stability?
 | ||
| Fig. 3 Low-frequency phonons of (a) qTP1, (b) qTP2, and (c) qHP polymeric C60 using the static and room-temperature lattice constants.107 | ||
For the qTP1 monolayers, small imaginary frequencies (less than 0.6i THz) are observed along the Γ–X direction when using static lattice constants, suggesting non-restorative atomic displacements110 to split the monolayers into individual 1D chains in the presence of interchain out-of-plane vibrations. The presence of a fourth, nearly zero-frequency, positive torsional mode along Γ–X further reflects the quasi-one-dimensional nature of the qTP1 structure.121,122 Incorporating finite-temperature effects, we find that at 300 K, qTP1 retains this soft mode behaviour, albeit with a reduced magnitude (<0.2i THz), thus confirming its weak dynamic stability. In contrast, the qTP2 and qHP lattices exhibit no imaginary phonon modes under either static or thermally-expanded lattice parameters, indicating that both structures reside at a local minimum on the potential energy surface and are dynamically stable over a wide temperature range.
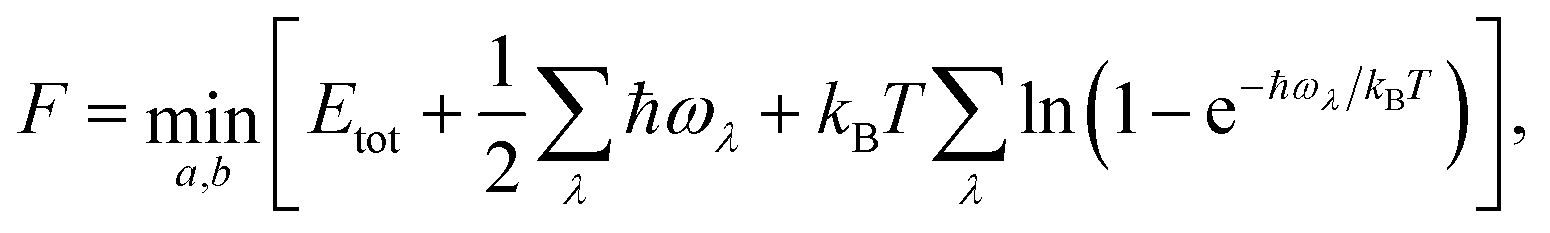 | (1) |
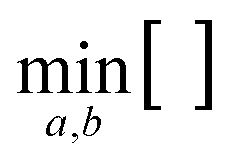 changes the lattice constants a and b to find the unique minimum value of the Helmholtz free energy, Etot is the total energy of the crystal, ħ is the reduced Planck constant, ωλ is the phonon frequency at mode λ, kB is the Boltzmann constant, and T is the temperature. As shown in Fig. 4, the temperature dependence of the Gibbs free energy reveals a crossover in phase stability. Although qTP2 is most stable at 0 K, qTP1 becomes thermodynamically preferred above 150 K due to its lower vibrational frequencies and hence greater entropy. At room temperature (300 K), the free energy of qTP1 is 47 meV f.u.−1 lower than that of qTP2, and this energetic preference increases at higher temperatures, reinforcing the relative thermodynamic stability of qTP1 at high temperatures.
changes the lattice constants a and b to find the unique minimum value of the Helmholtz free energy, Etot is the total energy of the crystal, ħ is the reduced Planck constant, ωλ is the phonon frequency at mode λ, kB is the Boltzmann constant, and T is the temperature. As shown in Fig. 4, the temperature dependence of the Gibbs free energy reveals a crossover in phase stability. Although qTP2 is most stable at 0 K, qTP1 becomes thermodynamically preferred above 150 K due to its lower vibrational frequencies and hence greater entropy. At room temperature (300 K), the free energy of qTP1 is 47 meV f.u.−1 lower than that of qTP2, and this energetic preference increases at higher temperatures, reinforcing the relative thermodynamic stability of qTP1 at high temperatures.
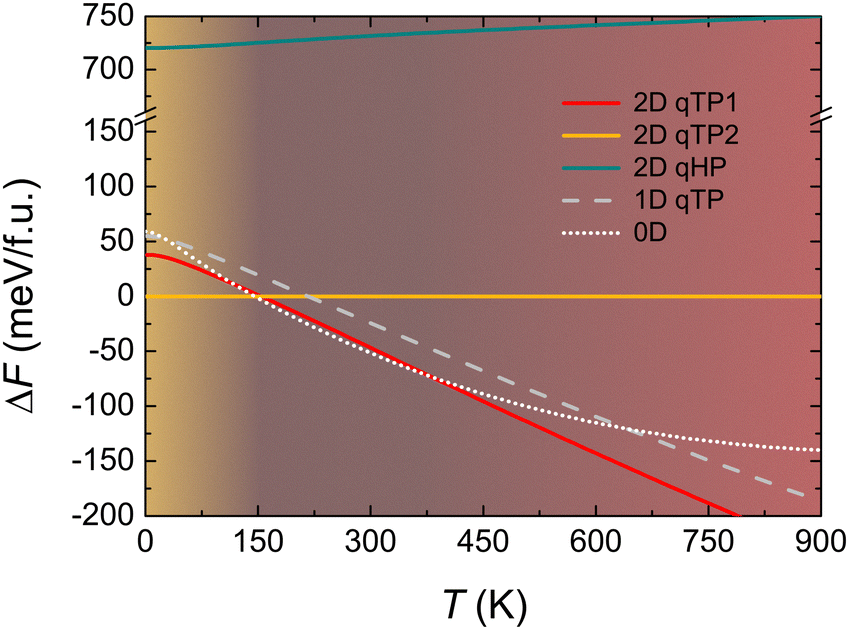 | ||
| Fig. 4 Relative thermodynamic stabilities of monolayer fullerene networks, a one-dimensional fullerene chain, and a zero-dimensional fullerene molecule, with the Gibbs free energy F of monolayer qTP2 C60 set to zero to compare the relative stabilities.107 | ||
As the qTP1 monolayer tends to split into individual chains, the relative thermodynamic stability of 1D qTP chain and 0D C60 molecule is also shown in Fig. 4. Notably, while the 2D qTP1 structure remains more stable than the 1D chain throughout the entire temperature range, the 1D chain becomes more stable than the qTP2 monolayer above 220 K. Interestingly, the 0D C60 molecule shows the steepest decrease in free energy below room temperature, becoming more stable than qTP1 and qTP2 monolayers at 120 K and 150 K, respectively. Above 380 K, however, qTP1 regains its dominance as the most stable phase. Notably, the energy difference between the 1D and 2D qTP1 lattices is comparable to the thermal fluctuation energy (kBT) at 300 K of ∼26 meV, implying the possibility for qHP1 monolayers to split into 1D chains upon thermal fluctuations. These results highlight a temperature dependence for the thermodynamically most stable phase: below 150 K, qTP2 is preferred; between 150 and 380 K, the 0D C60 molecule is energetically favoured; and above 380 K, the 2D qTP1 monolayer becomes thermodynamically dominant. It should be noticed that the quasi-harmonic approximation is appropriate at moderate temperatures <500 K, which is the temperature range of interest. Additionally, we focus on isolated, free-standing monolayers, which neglects possible interactions in experimental samples in the few-layer form or on the substrates.
| Phase | C 11 | C 22 | C 12 | C 66 |
|---|---|---|---|---|
| qTP1 | 5.4 | 123.7 | −1.2 | −0.2 |
| (2.5) | (121.3) | — | ||
| qTP2 | 149.9 | 148.7 | 22.9 | 53.4 |
| (150.5) | (141.2) | — | (54.5) | |
| qHP | 150.8 | 186.8 | 22.5 | 60.6 |
| (142.4) | (172.7) | — | (61.7) |
Notably, the elastic constant C11 in qTP1 is more than an order of magnitude smaller than C22, reflecting the pronounced anisotropy and structural softness along the a-axis. This is consistent with its dynamic instability and the weak bonding along that direction. In comparison, qTP2 monolayers exhibit nearly isotropic in-plane stiffness, with C11 ≈ C22, due to the presence of comparable [2+2] cycloaddition bonds along both a and b lattice vectors. Among the three phases, qHP C60 networks display the highest values of C11, C22, and C66, consistent with its denser intermolecular bonding networks.
2.3 Thermal expansion
Having understood the stability of various monolayer fullerene networks, the next step is to consider how these 2D frameworks respond to thermal effects. In low-dimensional materials, thermal expansion is not only a fundamental physical property but also a sensitive probe of bonding anisotropy and lattice flexibility. This motivates a detailed investigation of how temperature influences lattice constants and vibrational properties, with a particular focus on the role of intermolecular bonding geometry in driving positive or negative thermal expansion.120As shown in Fig. 5, the type of intermolecular bonds critically governs the anisotropic thermal response. In the qTP C60 networks, neighbouring cages are connected via [2+2] cycloaddition bonds along both in-plane directions, and early isotropic positive thermal expansion is observed. In contrast, the qHP monolayers incorporate C–C single bonds along one axis and [2+2] cycloaddition bonds along the other, exhibiting pronounced anisotropy with positive expansion along the cycloaddition bond direction and negative thermal expansion along the single bond direction persisting up to 500 K.
 | ||
| Fig. 5 (a) Crystal structures and (b) thermal expansion of monolayer qTP and qHP C60 networks. The schematics in (a) show the structural changes with increased temperature.120 | ||
The unique combination of rigid [2+2] and flexible single bonds in qHP fullerene networks indicates strong anisotropic thermal expansion behaviour, offering an intriguing platform for engineering thermal expansion properties through molecular-scale structural design. This contrasting behaviour arises from distinct mechanical responses of different types intermolecular bonds. The [2+2] cycloaddition bonds are structurally rigid and resist transverse deformation. On the other hand, the C–C single bonds display considerable geometric flexibility, which allows hinge-like compression in response to perpendicular strain and therefore results in thermal contraction. Additionally, vibrational characteristics suggest large negative Grüneisen parameters for low-frequency out-of-plane acoustic and optical vibrations associated with the single bonds. Conversely, vibrations associated with the cycloaddition bonds show positive or near-zero Grüneisen values, contributing to thermal expansion.
The interplay between bond rigidity and flexibility provides a general strategy for tailoring thermal expansion in molecular materials beyond conventional approaches such as crystalline networks,140,141 rigid unit modes,142–145 and transverse displacements of bridging atoms146,147 or membranes.148,149 Through varied intermolecular bonds, one can rationally modulate both the magnitude and directionality of the thermal response. These design principles extend to other systems beyond monolayer polymeric C60, where variations in molecular curvature and intermolecular bonding types further tune thermomechanical behaviour.120 Overall, these insights, in combination with known properties of monolayer fullerene networks such as superlubricity150 and high carrier mobility,89,90 offer predictive guidelines for developing 2D fullerene-based materials with controllable thermal expansion for applications in flexible electronics, precision engineering, and energy-related technologies.
3 Are 2D C60 networks promising photocatalysts?
Photocatalytic water splitting harnesses solar energy to decompose water into hydrogen and oxygen, presenting an environmentally-friendly method for green hydrogen production. Since the discovery of photocatalytic TiO2 in 1972,151 extensive research has been dedicated to developing efficient photocatalysts.152–167 To enhance photocatalytic efficiency under visible light, materials must exhibit: (i) effective light absorption to generate sufficient electron–hole pairs; (ii) efficient separation and transport of these charge carriers to reactive sites; and (iii) appropriate band-edge positions to drive the redox reactions involved in water splitting. In this Section, we present recent computational studies addressing these criteria90 and discuss the subsequent experiments that verify these predictions.1003.1 Electronic structure from first principles
Band alignment and optical absorption are central to photocatalytic processes. However, one main challenge in the theoretical description of electronic structure of monolayer C60 networks is that, conventional DFT and HSE approaches underestimate the band gaps by at least 10%102,103 and fail to predict exciton binding energy. Therefore, an accurate theoretical framework is essential for fully exploiting the potential of these fullerene-based networks in photocatalytic water splitting.| PBEsol | HSEsol | PBEsol0 | GW | Exp. | |
|---|---|---|---|---|---|
| μ | ∞ | 0.2 | 0 | — | — |
| qTP1 | 1.09 | 1.65 | 2.31 | — | — |
| qTP2 | 0.94 | 1.48 | 2.18 | — | — |
| qHP | 0.86 | 1.44 | 2.12 | 2.37 | 1.6–2.1 |
| (0.86) | (1.44) | (1.69) | (1.60) | (1.55) |
For optical band gaps, time-dependent DFT (TDDFT) or time-dependent Hartree–Fock (TDHF) calculations on top of the PBEsol or HSEsol band structures fail to capture excitonic effects. Instead, the PBEsol0 + TDHF approach accurately reproduces both the optical band gap and exciton binding energy.
Notably, a more recent study has applied the most accurate many-body perturbation theory GW calculations for electronic band gaps, as well as the Bethe–Salpeter equation (BSE) for optical band gaps and exciton binding energy, which yield agreeable results with PBEsol0 + TDHF.180 Therefore, the PBEsol0 + TDHF approach accurately describes low dielectric screening in fullerene networks and is essential to obtain physically meaningful band alignments for evaluating photocatalytic performance and to predict the excitonic absorption spectrum.
 | ||
| Fig. 6 Band alignment of qTP1, qTP2, and qHP monolayers calculated with PBEsol, HSEsol, and PBEsol0.90 | ||
3.2 Carrier dynamics
We next discuss the carrier dynamics in photocatalysis. Specially, the photocatalysts need to: (i) generate sufficient electron–hole pairs upon photoexcitation; (ii) separate the photoexcited electrons and holes effectively; and (iii) transport carriers efficiently on the monolayer surface. | ||
| Fig. 7 (a) Crystal structures and (b) PBEsol electronic structures of qTP2-PbTe heterostructures.90 | ||
 | ||
| Fig. 8 Mobility of monolayer qHP C60 networks at 300 K as a function of carrier concentration, as well as the corresponding partial charge density of the VBM and CBM states.90 | ||
3.3 Reaction pathways
Thermodynamic analysis reveals favourable adsorption energies for water on the surface of monolayer fullerene networks,90 which is the initial step of photocatalytic reaction. To thermodynamically drive the redox reactions, the next step is for the free-energy diagram to exhibit downhill reaction pathway. As shown in Fig. 9(a), the hydrogen evolution reaction has two steps. In the first step, monolayer fullerene networks (denoted as *) combine with a proton (H+) and an electron (e−) to form H* species. In the second step, H2 molecules are formed from the H* species. | ||
| Fig. 9 (a) Gibbs free energy changes associated with the HER in qTP2 C60 networks at a pH of 0 and room temperature. (b) Adsorption sites for qTP2 C60 networks, with lower to higher numbers corresponding to lower to higher free energies of the intermediates.91 | ||
Without photoexcitation, there is always energy barrier posed by the intermediate adsorbate. Upon photoexcitation, the external potential generated by the photoexcited electrons in the CBM exceeds the energy barriers for hydrogen evolution for all three phases at the level of unscreened hybrid functional PBEsol0.90–93 The presence of photoexcitation also creates a more favorable Gibbs free energy between the intermediates and the products, enabling spontaneous HER under acidic conditions at room temperature. The photocatalytic activity is further enhanced by the high surface area and abundant active sites provided by the spherical C60 units, as shown in Fig. 9(b). In addition, the hydrogen storage capacity of various fullerene structures194–202 offers a unique dual function in facilitating hydrogen evolution and serving as hydrogen storage materials.
3.4 Experimental verification
Conventional fullerene-based heterostructures typically involve sparse and non-periodically distributed C60 molecules.203 In comparison, monolayer polymeric C60 networks offer atomically smooth surfaces with periodic C60 arrangements. This enhanced structural order leads to improved crystallinity and higher C60 content, thereby increasing the density of active sites and overall photocatalytic activity. Recent experimental studies100 have supported the theoretical predictions90 regarding the photocatalytic capabilities of monolayer fullerene networks. In particular, the photocatalytic efficiency of 2D fullerene networks is much higher than those of 0D C60 molecules or 3D C60 crystals.100 These findings not only validate the previous theoretical predictions but also pave the way for further research into the optimisation and integration of 2D fullerene networks in photocatalytic systems.From a materials design perspective, monolayer fullerene networks represent a promising class of photocatalysts owing to their unique combination of molecular features and crystalline characteristics. The C60 building blocks retain high surface area with delocalised π electrons for abundant active sites, while their covalent 2D connectivity enables efficient charge transport across the monolayers. These structural features allow for strong optical absorption, high carrier mobility, and enhanced chemical reactivity. Compared to traditional metal oxides,204–211 monolayer fullerene networks offer a chemically pure, carbon-based alternative that combines structural flexibility and high efficiency. Compared to covalent organic frameworks212–215 or molecular heterojunctions,216–219 monolayer polymeric C60 has higher densities of π electrons on larger surface area of nearly-spherical units in highly crystalline frameworks. These properties make monolayer fullerene networks appealing candidates for next-generation photocatalysts.
4 Are functionalities of fullerene networks tuneable?
The chemical functionalities of monolayer fullerene networks can be further modulated through structural design at multiple levels. Three key tuning strategies have emerged: variation in stacking configurations, control of molecular size, and dimensionality engineering. Vertical stacking of monolayers introduces interlayer degrees of freedom that alter optical absorption and exciton dynamics, offering a pathway to enhance light harvesting.93 At the molecular scale, substituting C60 with smaller cage units such as C24 modifies the electronic structure and increases the density of active sites, improving catalytic performance.92 Beyond monolayers, extending or reducing the lattice dimensionality yields rich behaviour: three-dimensional van der Waals crystals exhibit diverse structural phases,220 one-dimensional chains enhance reactivity via increased surface active sites,91 and quasi-one-dimensional nanoribbons display strongly geometry- and edge-dependent band structures.221 Among these, fullerene nanoribbons are especially interesting due to their tuneable edge shapes, quantum confinement effects, and potential for directional charge transport, positioning them as promising candidates for next-generation molecular optoelectronics and catalysis.4.1 Stacking degree of freedom
While monolayer polymeric qHP C60 networks possess promising optoelectronic and photocatalytic properties, they suffer from limited thermodynamic stability, making experimental isolation challenging. As a result, most experimentally-realised qHP structures exist in the few-layer form,99–101 which benefit from enhanced structural stability due to interlayer van der Waals interactions. These bilayer systems not only retain the desirable properties of monolayers such as appropriate band alignment for water splitting but also introduce new stacking degrees of freedom such as sliding, orientation, and twisting angles, which can be exploited to tune their functionalities. Taking AB-stacked bilayer fullerene networks as an example, it can be viewed as two van der Waals layers in a closely-packed stacking pattern for C60 molecules with space group P2/c (no. 13), as shown in Fig. 10. | ||
| Fig. 10 3D and top views of the crystal structures of (a) monolayer and (b) bilayer fullerene networks.93 | ||
From an electronic structure perspective, the bilayer exhibits a slightly reduced direct band gap at Γ (2.05 eV) compared to the monolayer (2.08 eV). Importantly, the band-edge positions of the bilayer still straddle the redox potentials of water at pH from 0 to 7, indicating that the bilayer retains its suitability for overall water splitting.93 The presence of two layers gives rise to nearly degenerate states at the VBM and CBM, with minor energy splittings between in-phase and out-of-phase combinations of the top and bottom layer states. The interlayer interaction slightly modifies the anisotropic effective mass, particularly in the conduction band, allowing for directional tuning of charge transport.93
In terms of optical properties, the bilayer C60 network demonstrates enhanced absorbance across the entire visible spectrum compared to its monolayer counterpart (Fig. 11). This enhancement arises from stronger excitonic transitions involving multiple degenerate states and is accompanied by increased anisotropy in polarisation-dependent absorbance. Notably, while the monolayer shows relatively isotropic exciton absorption, the bilayer exhibits more pronounced absorbance differences for polarisation along the a and b axes. Such anisotropy can be exploited in polarised light detectors and direction-sensitive photonic devices.93
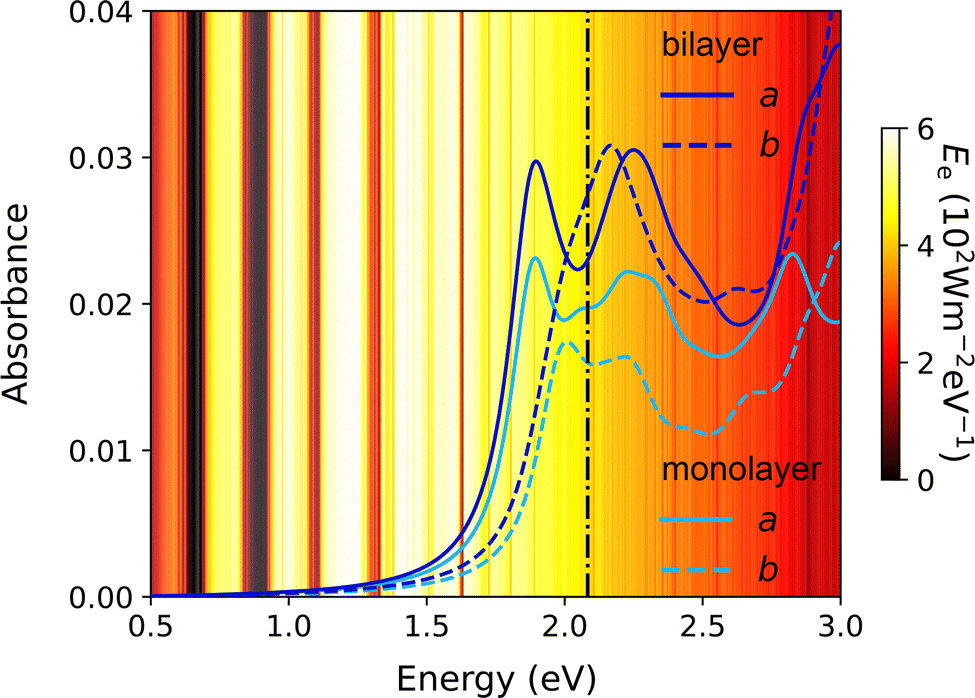 | ||
| Fig. 11 Optical absorbance of monolayer and bilayer fullerene networks for both a and b polarisations. The black vertical dash-dot lines represent the direct band gaps from independent-particle approximations in the absence of excitons. The background shows the global total spectral irradiance Ee from the Sun.93 | ||
Overall, stacking monolayers into bilayer fullerene networks preserves photocatalytic performance while enhancing structural stability and optoelectronic tuneability. These stacking degrees of freedom also open avenues for emerging applications such as flexible displays, memory storage, and ferroelectric devices, where interlayer sliding and twist engineering may give rise to novel phenomena such as sliding ferroelectricity222 and moiré patterns.99
4.2 Molecular size
The choice of molecular building block provides further tuneablity in the stability and functionality of 2D fullerene networks. While C60-based monolayers have attracted significant attention for their photocatalytic90,100 and optoelectronic101 properties, their relatively large molecular size limit the structural stability and density of accessible active sites. Recent studies have demonstrated that reducing the molecular size to the smallest stable conventional [5,6]fullerene unit, C24, yields 2D networks with superior stability and enhanced photocatalytic performance.92 | ||
| Fig. 12 Top and side views of crystal structures of (a) qTP and (b) qHP C24 monolayers.92 | ||
Compared to monolayer polymeric C60, C24 monolayers exhibit significantly improved dynamic, thermodynamic, and mechanical stability.92 Cohesive energy calculations demonstrate that the formation of polymeric C24 networks is energetically favourable, owing to the release of stereochemical strain through the formation of noncoplanar sp3-like bonds. These networks are also dynamically stable, as confirmed by phonon dispersion calculations showing no imaginary modes. Mechanically, the C24 lattices exhibit larger elastic and shear moduli than their C60 counterparts, because of the smaller cage size and therefore higher density of covalent interfullerene bonds.
The reduction in molecular size also leads to decreased screening and hence increased band gaps. Unscreened hybrid functional calculations show that monolayer C24 networks have wide band gaps (3.10–3.74 eV depending on phase), which are comparable to those of TiO2,204–211 allowing for visible-to-UV light harvesting. Both qTP and qHP C24 structures have band-edge positions that straddle the redox potentials of water across a broad pH range, satisfying the thermodynamic requirements for overall water splitting. Moreover, strongly bound bright excitons contribute to strong optical absorbance, particularly in the UV regimes, facilitating efficient generation of photoexcited carriers.
 | ||
| Fig. 13 Free energy profiles of hydrogen evolution at different adsorption sites through the Volmer–Tafel mechanism for (a) qTP and (b) qHP C24 and the Volmer–Heyrovsky mechanism for (c) qTP and (d) qHP C24 at pH = 0, with grey and blue lines representing the absence and presence of photoexcitation respectively. Symmetry-irreducible adsorption sites for (e) qTP and (f) qHP C24. (g) Volmer–Tafel and (h) Volmer–Heyrovsky reaction mechanisms. (i) Correlation between adsorption free energy and average bond angle strain. (j) Area density σa of active sites for various phases of fullerene monolayers at changing pH.92 | ||
To further understand adsorption behaviour, we assess the correlation between hydrogen adsorption free energy and local bond angle strain. The bond angle strain is defined as the deviation of the average bond angle at a given site from the ideal sp2 value of 120°, providing a geometric descriptor of local carbon environments. As depicted in Fig. 13(i), adsorption free energy exhibits a strong correlation with the bond angle strain, indicating that more curved C24 cages enhance hydrogen adsorption. This suggests that the higher curvature of monolayer C24 networks plays a significant role in increasing photocatalytic efficiency.
Finally, the area density σa of thermodynamically active sites at different pH values is summarised in Fig. 13(j). While qTP and qHP C24 monolayers exhibit similar active site densities under acidic conditions (pH = 0) to their C60 counterparts, increasing pH to 3 significantly reduces the number of active sites on C60 networks. On the other hand, C24 monolayers retain full catalytic reactivity. As a result, the active site density in C24 monolayers becomes approximately three times that of C60 at moderate acidity, with this ratio increasing further under near-neutral conditions. This resilience under varying pH highlights the superior catalytic performance of C24 networks for hydrogen evolution across a broader operational range.
4.3 Lattice dimensionality
Changes in lattice dimensionality offer a useful strategy to modulate the physical and chemical behaviour of fullerene networks. When increasing lattice dimension to 3D van der Waals crystals of C60, molecular orientations and their corresponding crystalline symmetry govern electronic band gaps, excitonic effects, and phase stability, which are crucial for applications in optoelectronics and energy harvesting devices.220 Conversely, reducing lattice dimension to 1D polymeric chains increases the band gap and the number of active sites, which are advantageous for photocatalytic HER.91 Quasi-1D C60 nanoribbons, split from 2D monolayers, represent an intermediate geometry where quantum confinement and edge states give rise to tuneable band gaps and effective masses, with electronic properties highly sensitive to nanoribbon width and edge configuration.221 Together, these dimensional variants unlock even richer physical and chemical properties, offering design flexibility for targeted functionalities (Fig. 14). | ||
Fig. 14 Crystal structures of solid C60 in conventional unit cells with space group (a) Immm, (b) R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, (c) Fm, and (d) Pa.220 m, (c) Fm, and (d) Pa.220 | ||
m) phases, C60 molecules are covalently bonded in quasi-2D sheets via [2+2] cycloaddition bonds, with adjacent layers stacked through van der Waals interactions. These van der Waals layered structures exhibit dynamic stability220 and are viable for experimental synthesis under moderate pressure and temperature conditions.65–73
Instead of layered crystals, the cubic Fm and Paphases remove all covalent intermolecular bonds in favour of purely non-covalent van der Waals interactions. In these structures, the C60 units have extra rotational degrees of freedom with varied molecular orientations. The Fm phase exhibits higher symmetry with a single C60 molecule per primitive unit cell (i.e., all molecules have the same orientation), while the Pa structure contains four inequivalent molecules with distinct orientations, leading to reduced symmetry but enhanced packing diversity. This rotationally-disordered phase can be realised at elevated temperatures as a result of the phase transitions observed near 255 K.64 These 3D van der Waals phases display large band gaps (2.293–2.461 eV) and strong exciton binding energies (>200 meV),220 which agree well with previous GW calculations224,225 and (inverse-)photoemission spectra.78,226–229
The transition from 2D to 3D provides more than structural diversity. It introduces additional avenues for tuneable functionalities through control of crystalline symmetry and intermolecular spacing.220 While the layered Immm and Rm phases offer anisotropic optical absorption and exciton confinement suitable for photonic devices, the cubic Fm and Pa phases exhibit more isotropic absorption profiles, with the absorption peak (>105 cm−1) comparable to halide perovskites in the green-blue light region.230,231 These distinctions highlight the rich structural tuneability in fullerene-based networks and provide a versatile platform for energy harvesting and storage. Recent synthesis of a thin layer of an ionic salt of C60 significantly increases mechanical stability of the electron transport layer in inverted perovskite solar cells with a roughly threefold increase in interfacial toughness.232 Notably, fullerene molecules are also highly stable in outer space and contribute to 1% of the cosmic carbon in the interstellar medium,233–235 which further extends their applications to space photovoltaics.
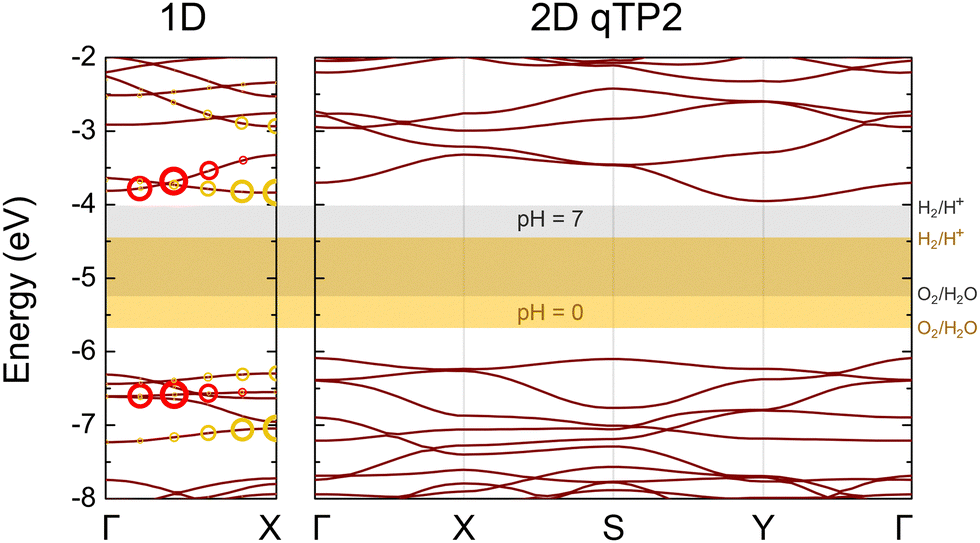 | ||
| Fig. 15 PBEsol0 band structures of the 1D C60 chain and 2D C60 networks. The red and yellow circles drawn on the 1D band structures indicate the contributions of the corresponding electron–hole pairs to the two brightest excitons near the band edges. The radii of these circles represent their corresponding oscillator strength.91 | ||
To understand carrier separation in 1D chains, the electron–hole pairs for the two brightest excitons in 1D are indicated by red and yellow circles in Fig. 15, with larger radius representing higher oscillator strength. Because the holes are in much lower valence states than the VBM, their thermalisation to the VBM tends to dissociate the excitons, giving rise to effective electron–hole separation to their respective redox half reactions.
Benefiting from the favourable band-edge alignments for HER, 1D C60 chains demonstrate higher catalytic potential than their 2D counterparts under a wider pH range. These chains also possess twice the number of surface active sites per C60 unit compared to monolayer networks, leading to higher photocatalytic efficiency. Furthermore, the 1D chain is thermodynamically more stable than monolayer fullerene networks at room temperature, making it a more promising photocatalyst with higher efficiency and superior stability than the monolayers.
In Fig. 16(a), we show a representative qHP nanoribbon. Both edges terminate at the lattice site corresponding to intermolecular single bonds. This nanoribbon can be formed upon cleavage along the armchair crystallographic orientation of the monolayer. Therefore, we denote it as qHP-AC nanoribbons.221 The width of the nanoribbon W is defined as the number of C60 units spanning the non-periodic (transverse) direction. The qHP-AC nanoribbons crystallise in the P2221 space group (no. 17), with a lattice constant of 9.15 Å and a width of 30.75 Å for W = 4.
 | ||
| Fig. 16 (a) Crystal structures of a representative qHP-AC nanoribbon, and (b) their corresponding band structures as a function of W.221 | ||
The electronic band structure of qHP-AC nanoribbons shows additional in-gap states absent in the monolayer [Fig. 16(b)]. These states are localised on the ribbon edges and remain fixed in number (two nearly-degenerate conduction bands and two nearly-degenerate valence bands) regardless of width W, whereas the bulk-like valence and conduction bands display replicas proportional to W. For narrow ribbons (W = 2), the two in-gap valence states and the two in-gap conduction states appear within ± 0.5 eV. As W increases, interactions between the two edges diminish, leading to complete degeneracy of these in-gap states. As the CBMs at both Γ and Y originate from the edge states, their effective masses quickly converge once W > 2. The same holds for the VBM at Y. On the other hand, the VBM at Γ transits from edge-localised states to monolayer-like characters as W increases, causing a sign reversal in the effective mass between between W = 3 and W = 4. Notably, the CBM at Γ has negative effective mass, while the VBM at Γ has positive effective mass with larger absolute value than that of the CBM. Therefore, the electron and hole at Γ have positive total mass [m(e) + m(h)] but negative reduced mass [1/m(e) + 1/m(h)]. In a classical picture, the electron–hole pairs at Γ are expected to form excitons that behave differently from gravitational objects such as a binary star system.255
In terms of band gaps, while the monolayer exhibits a direct band gap at Γ, the nanoribbons display their smallest direct band gap at Y. The presence of edge states leads to a 420 meV reduction in the band gap relative to the monolayer. The band-gap reduction from edge states may account for the experimentally-observed discrepancy in electronic band gaps (1.60–2.05 eV)89,99,100 and in optical band gaps (1.10–1.55 eV),89,101 due to the finite size of the measured samples.
Besides qHP-AC nanoribbons, a variety of other nanoribbons can be realised, depending on the crystalline direction and the resulting edge geometry, as shown in Fig. 17. For the qHP monolayers, nanoribbons cleaved along a1 adopt a zigzag-like edge geometry, which is denoted as qHP-ZZ. The electronic structures of qHP-ZZ nanoribbons also exhibit in-gap edge states. Different from the dispersive in-gap states in the qHP-AC nanoribbons, the edge states in qHP-ZZ nanoribbons show flat-band features.221
 | ||
| Fig. 17 Crystal structures of monolayer (a) qTP and (b) qHP fullerene networks, as well as their corresponding nanoribbons.221 | ||
For the qTP monolayers, the nanoribbons can be formed along either the vertical or the horizontal [2+2] cycloaddition bonding directions, which are denoted as qTP-V and qTP-H respectively. The two qTP nanoribbons also show a rich variety of electronic properties, including crossover from positive to negative effective mass with increased W, transitions from indirect to direct band gaps, and coexistence of both flat and dispersive bands.221
Overall, fullerene nanoribbons, derived from experimentally-observed monolayer networks, offer a promising platform with tuneable electronic properties governed by edge geometry and width.221 First-principles calculations reveal diverse behaviours: depending on their crystallographic orientation, qTP nanoribbons exhibit either direct or indirect band gaps, while qHP nanoribbons host prominent in-gap states localised on the edges. These edge states lead to flat bands and unconventional effective masses, distinct from those of their parent monolayers. The evolution of band gaps, effective masses, and band widths with increasing nanoribbon width W highlights the importance of finite-size effects. These findings position fullerene nanoribbons as versatile nanostructures for future nanoscale devices.
5 Conclusion and outlook
In conclusion, monolayer fullerene networks represent an emerging family of molecular 2D materials with unique structural, electronic, and chemical properties distinct from atomically constructed monolayers. Their diversity in intermolecular bonding motifs enables the formation of rich lattice geometries with tuneable functionalities. Through first-principles calculations, we have provided a thorough understanding of the phase stability of the experimentally-observed structures and clarified the impact of intermolecular bonds on thermal expansion. Beyond their stability, monolayer polymeric C60 fulfills all key criteria for photocatalysts: they possess suitable band-edge alignments, demonstrate strong optical absorption, and support efficient carrier separation and transport. Theoretical predictions of photocatalytic water splitting have been subsequently corroborated by experimental observations. Furthermore, the molecular nature of fullerene building blocks allows for functional tuning via interlayer stacking, molecular size, and dimensionality from 3D crystals to 1D chains and nanoribbons. These findings highlight a robust platform for the rational design of carbon-based nanomaterials.Looking ahead, the development of controlled synthesis techniques and device integration strategies will be essential to fully harness the potential of 2D fullerene networks, while future theoretical studies will prove invaluable in complementing, guiding, and driving the experimental work to address potential challenges in practical applications of monolayer fullerene networks, such as structural stability under varied operational conditions (light, moisture, and temperature) for long-term performance, scalable synthesis of defect-free monolayers over large areas, and surface functionalisation control for tuning band alignment and catalytic activity. Opportunities also lie in engineering moiré superlattices, heterostructures, and strain-tuneable systems for emergent quantum phenomena such as ferroelectricity and superconductivity. The intrinsic structural resilience as atom-like, stable building units, combined with the richness of their chemical degrees of freedom, positions monolayer fullerene networks as a platform to realise next-generation materials and devices for a wide range of applications at the nanoscale.
Conflicts of interest
There are no conflicts to declare.Data availability
The code VASP can be found at [https://www.vasp.at/]. The version of the code employed for this study is version vasp.6.3.1.Acknowledgements
B. P. acknowledges support from Magdalene College Cambridge for a Nevile Research Fellowship. The calculations were performed using resources provided by the Cambridge Service for Data Driven Discovery (CSD3) operated by the University of Cambridge Research Computing Service (https://www.csd3.cam.ac.uk), provided by Dell EMC and Intel using Tier-2 funding from the Engineering and Physical Sciences Research Council (capital grant EP/T022159/1), and DiRAC funding from the Science and Technology Facilities Council (https://www.dirac.ac.uk), as well as with computational support from the UK Materials and Molecular Modelling Hub, which is partially funded by EPSRC (EP/T022213/1, EP/W032260/1 and EP/P020194/1), for which access was obtained via the UKCP consortium and funded by EPSRC grant ref EP/P022561/1.References
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666 CrossRef CAS PubMed.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, M. I. Katsnelson, I. V. Grigorieva, S. V. Dubonos and A. A. Firsov, Nature, 2005, 438, 197–200 CrossRef CAS PubMed.
- Y. Zhang, Y.-W. Tan, H. L. Stormer and P. Kim, Nature, 2005, 438, 201–204 CrossRef CAS PubMed.
- A. K. Geim, Science, 2009, 324, 1530–1534 CrossRef CAS PubMed.
- K. S. Novoselov, V. I. Fal'ko, L. Colombo, P. R. Gellert, M. G. Schwab and K. Kim, Nature, 2012, 490, 192–200 CrossRef CAS PubMed.
- C. Gong, L. Li, Z. Li, H. Ji, A. Stern, Y. Xia, T. Cao, W. Bao, C. Wang, Y. Wang, Z. Q. Qiu, R. J. Cava, S. G. Louie, J. Xia and X. Zhang, Nature, 2017, 546, 265 CrossRef CAS PubMed.
- B. Huang, G. Clark, E. Navarro-Moratalla, D. R. Klein, R. Cheng, K. L. Seyler, D. Zhong, E. Schmidgall, M. A. McGuire, D. H. Cobden, W. Yao, D. Xiao, P. Jarillo-Herrero and X. Xu, Nature, 2017, 546, 270 CrossRef CAS PubMed.
- Y. Deng, Y. Yu, Y. Song, J. Zhang, N. Z. Wang, Z. Sun, Y. Yi, Y. Z. Wu, S. Wu, J. Zhu, J. Wang, X. H. Chen and Y. Zhang, Nature, 2018, 563, 94–99 CrossRef CAS PubMed.
- M. Pizzochero, R. Yadav and O. V. Yazyev, 2D Mater., 2020, 7, 035005 CrossRef CAS.
- M. Pizzochero and O. V. Yazyev, J. Phys. Chem. C, 2020, 124, 7585–7590 CrossRef CAS.
- S. Zhang, Z. Yan, Y. Li, Z. Chen and H. Zeng, Angew. Chem., Int. Ed., 2015, 54, 3112–3115 CrossRef CAS PubMed.
- B. Peng, H. Zhang, H. Shao, Y. Xu, R. Zhang and H. Zhu, J. Mater. Chem. C, 2016, 4, 3592 RSC.
- B. Peng, H. Zhang, H. Shao, K. Xu, G. Ni, L. Wu, J. Li, H. Lu, Q. Jin and H. Zhu, ACS Photonics, 2018, 5, 4081–4088 CrossRef CAS.
- C. L. Kane and E. J. Mele, Phys. Rev. Lett., 2005, 95, 226801 CrossRef CAS PubMed.
- M. Wada, S. Murakami, F. Freimuth and G. Bihlmayer, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 121310 CrossRef.
- C.-C. Liu, H. Jiang and Y. Yao, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 195430 CrossRef.
- B. Cai, S. Zhang, Z. Hu, Y. Hu, Y. Zou and H. Zeng, Phys. Chem. Chem. Phys., 2015, 17, 12634–12638 RSC.
- F.-F. Zhu, W.-J. Chen, Y. Xu, C.-L. Gao, D.-D. Guan, C.-H. Liu, D. Qian, S.-C. Zhang and J.-F. Jia, Nat. Mater., 2015, 14, 1020–1025 CrossRef CAS PubMed.
- A. A. Balandin, S. Ghosh, W. Bao, I. Calizo, D. Teweldebrhan, F. Miao and C. N. Lau, Nano Lett., 2008, 8, 902–907 CrossRef CAS PubMed.
- J. H. Seol, I. Jo, A. L. Moore, L. Lindsay, Z. H. Aitken, M. T. Pettes, X. Li, Z. Yao, R. Huang, D. Broido, N. Mingo, R. S. Ruoff and L. Shi, Science, 2010, 328, 213–216 CrossRef CAS PubMed.
- B. Peng, H. Zhang, H. Shao, Y. Xu, X. Zhang and H. Zhu, Sci. Rep., 2016, 6, 20225 CrossRef CAS PubMed.
- B. Peng, H. Zhang, H. Shao, Y. Xu, X. Zhang and H. Zhu, Ann. Phys., 2016, 528, 504–511 CrossRef CAS.
- B. Peng, H. Zhang, H. Shao, Y. Xu, R. Zhang, H. Lu, D. W. Zhang and H. Zhu, ACS Appl. Mater. Interfaces, 2016, 8, 20977–20985 CrossRef CAS PubMed.
- Y. Chen, B. Peng, C. Cong, J. Shang, L. Wu, W. Yang, J. Zhou, P. Yu, H. Zhang, Y. Wang, C. Zou, J. Zhang, S. Liu, Q. Xiong, H. Shao, Z. Liu, H. Zhang, W. Huang and T. Yu, Adv. Mater., 2019, 31, 1804979 CrossRef PubMed.
- H. Şahin, S. Cahangirov, M. Topsakal, E. Bekaroglu, E. Akturk, R. T. Senger and S. Ciraci, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 155453 CrossRef.
- S. Cahangirov, M. Topsakal, E. Aktürk, H. Şahin and S. Ciraci, Phys. Rev. Lett., 2009, 102, 236804 CrossRef CAS PubMed.
- A. O'Hare, F. V. Kusmartsev and K. I. Kugel, Nano Lett., 2012, 12, 1045–1052 CrossRef PubMed.
- N. J. Roome and J. D. Carey, ACS Appl. Mater. Interfaces, 2014, 6, 7743–7750 CrossRef CAS PubMed.
- B. Peng, H. Zhang, H. Shao, Y. Xu, X. Zhang and H. Zhu, RSC Adv., 2016, 6, 5767–5773 RSC.
- B. Peng, H. Zhang, H. Shao, Y. Xu, G. Ni, R. Zhang and H. Zhu, Phys. Rev. B, 2016, 94, 245420 CrossRef.
- B. Peng, D. Zhang, H. Zhang, H. Shao, G. Ni, Y. Zhu and H. Zhu, Nanoscale, 2017, 9, 7397 RSC.
- B. Peng, B. Mortazavi, H. Zhang, H. Shao, K. Xu, J. Li, G. Ni, T. Rabczuk and H. Zhu, Phys. Rev. Appl., 2018, 10, 034046 CrossRef CAS.
- B. Peng, H. Zhang, H. Shao, K. Xu, G. Ni, J. Li, H. Zhu and C. M. Soukoulis, J. Mater. Chem. A, 2018, 6, 2018–2033 RSC.
- B. Peng, H. Mei, H. Zhang, H. Shao, K. Xu, G. Ni, Q. Jin, C. M. Soukoulis and H. Zhu, Inorg. Chem. Front., 2019, 6, 920–928 RSC.
- B. Peng, A. Bouhon, B. Monserrat and R.-J. Slager, Nat. Commun., 2022, 13, 423 CrossRef CAS PubMed.
- B. Peng, A. Bouhon, R.-J. Slager and B. Monserrat, Phys. Rev. B, 2022, 105, 085115 CrossRef CAS.
- S. Cho, S. Kim, J. H. Kim, J. Zhao, J. Seok, D. H. Keum, J. Baik, D.-H. Choe, K. J. Chang, K. Suenaga, S. W. Kim, Y. H. Lee and H. Yang, Science, 2015, 349, 625 CrossRef CAS PubMed.
- B. Peng, H. Zhang, W. Chen, B. Hou, Z.-J. Qiu, H. Shao, H. Zhu, B. Monserrat, D. Fu, H. Weng and C. M. Soukoulis, npj 2D Mater. Appl., 2020, 4, 14 CrossRef CAS.
- J. Gou, H. Bai, X. Zhang, Y. L. Huang, S. Duan, A. Ariando, S. A. Yang, L. Chen, Y. Lu and A. T. S. Wee, Nature, 2023, 617, 67–72 CrossRef CAS PubMed.
- B. Peng, G. F. Lange, D. Bennett, K. Wang, R.-J. Slager and B. Monserrat, Phys. Rev. Lett., 2024, 132, 116601 CrossRef CAS PubMed.
- H. Qiu, T. Xu, Z. Wang, W. Ren, H. Nan, Z. Ni, Q. Chen, S. Yuan, F. Miao, F. Song, G. Long, Y. Shi, L. Sun, J. Wang and X. Wang, Nat. Commun., 2013, 4, 2642 CrossRef PubMed.
- B. Peng, Z. Ning, H. Zhang, H. Shao, Y. Xu, G. Ni and H. Zhu, J. Phys. Chem. C, 2016, 120, 29324–29331 CrossRef CAS.
- M. Pizzochero and O. V. Yazyev, 2D Mater., 2018, 5, 025022 CrossRef.
- M. Pizzochero, J. Phys. D: Appl. Phys., 2020, 53, 244003 CrossRef CAS.
- S. Li and A. Gali, J. Phys. Chem. Lett., 2022, 13, 9544–9551 CrossRef CAS PubMed.
- D. J. Trainer, J. Nieminen, F. Bobba, B. Wang, X. Xi, A. Bansil and M. Iavarone, npj 2D Mater. Appl., 2022, 6, 13 CrossRef CAS.
- S. Li, P. Li and A. Gali, Appl. Phys. Lett., 2025, 126, 062104 CrossRef CAS.
- B. Peng, H. Zhang, H. Shao, Z. Ning, Y. Xu, G. Ni, H. Lu, D. W. Zhang and H. Zhu, Mater. Res. Lett., 2017, 5, 399–407 CrossRef CAS.
- A. J. Mannix, B. Kiraly, M. C. Hersam and N. P. Guisinger, Nat. Rev. Chem., 2017, 1, 0014 CrossRef CAS.
- W. Niu, S. Sopp, A. Lodi, A. Gee, F. Kong, T. Pei, P. Gehring, J. Nägele, C. S. Lau, J. Ma, J. Liu, A. Narita, J. Mol, M. Burghard, K. Müllen, Y. Mai, X. Feng and L. Bogani, Nat. Mater., 2023, 22, 180–185 CrossRef CAS PubMed.
- B. Qie, Z. Wang, J. Jiang, Z. Zhang, P. H. Jacobse, J. Lu, X. Li, F. Liu, A. N. Alexandrova, S. G. Louie, M. F. Crommie and F. R. Fischer, Science, 2024, 384, 895–901 CrossRef CAS PubMed.
- R. K. Dubey, M. Melle-Franco and A. Mateo-Alonso, J. Am. Chem. Soc., 2021, 143, 6593–6600 CrossRef CAS PubMed.
- W. Niu, Y. Fu, G. Serra, K. Liu, J. Droste, Y. Lee, Z. Ling, F. Xu, J. D. Cojal González, A. Lucotti, J. P. Rabe, M. Ryan Hansen, W. Pisula, P. W. M. Blom, C.-A. Palma, M. Tommasini, Y. Mai, J. Ma and X. Feng, Angew. Chem., Int. Ed., 2023, 62, e202305737 CrossRef CAS PubMed.
- X. Su, Z. Ding, Y. Hong, N. Ke, K. Yan, C. Li, Y.-F. Jiang and P. Yu, Nat. Synth., 2025, 4, 694–701 CrossRef.
- X. Fu, L. Huang, K. Liu, J. C. G. Henriques, Y. Gao, X. Han, H. Chen, Y. Wang, C.-A. Palma, Z. Cheng, X. Lin, S. Du, J. Ma, J. Fernández-Rossier, X. Feng and H.-J. Gao, Nat. Synth., 2025, 4, 684–693 CrossRef.
- H. W. Kroto, J. R. Heath, S. C. O'Brien, R. F. Curl and R. E. Smalley, Nature, 1985, 318, 162–163 CrossRef CAS.
- H. Kroto, Science, 1988, 242, 1139–1145 CrossRef CAS PubMed.
- H. Kroto, Pure Appl. Chem., 1990, 62, 407–415 CrossRef CAS.
- L. D. Lamb and D. R. Huffman, J. Phys. Chem. Solids, 1993, 54, 1635–1643 CrossRef CAS.
- W. Krätschmer, L. D. Lamb, K. Fostiropoulos and D. R. Huffman, Nature, 1990, 347, 354–358 CrossRef.
- P. A. Heiney, J. E. Fischer, A. R. McGhie, W. J. Romanow, A. M. Denenstein, J. P. McCauley Jr., A. B. Smith and D. E. Cox, Phys. Rev. Lett., 1991, 66, 2911–2914 CrossRef CAS PubMed.
- J. E. Fischer, P. A. Heiney, A. R. McGhie, W. J. Romanow, A. M. Denenstein, J. P. McCauley and A. B. Smith, Science, 1991, 252, 1288–1290 CrossRef CAS PubMed.
- W. I. F. David, R. M. Ibberson, J. C. Matthewman, K. Prassides, T. J. S. Dennis, J. P. Hare, H. W. Kroto, R. Taylor and D. R. M. Walton, Nature, 1991, 353, 147–149 CrossRef CAS.
- P. A. Heiney, J. Phys. Chem. Solids, 1992, 53, 1333–1352 CrossRef CAS.
- A. M. Rao, P. Zhou, K.-A. Wang, G. T. Hager, J. M. Holden, Y. Wang, W.-T. Lee, X.-X. Bi, P. C. Eklund, D. S. Cornett, M. A. Duncan and I. J. Amster, Science, 1993, 259, 955–957 CrossRef CAS.
- Y. Iwasa, T. Arima, R. M. Fleming, T. Siegrist, O. Zhou, R. C. Haddon, L. J. Rothberg, K. B. Lyons, H. L. Carter, A. F. Hebard, R. Tycko, G. Dabbagh, J. J. Krajewski, G. A. Thomas and T. Yagi, Science, 1994, 264, 1570–1572 CrossRef CAS PubMed.
- M. Núñez-Regueiro, L. Marques, J. L. Hodeau, O. Béthoux and M. Perroux, Phys. Rev. Lett., 1995, 74, 278–281 CrossRef PubMed.
- P. Eklund, A. Rao, P. Zhou, Y. Wang and J. Holden, Thin Solid Films, 1995, 257, 185–203 CrossRef CAS.
- C. H. Xu and G. E. Scuseria, Phys. Rev. Lett., 1995, 74, 274–277 CrossRef CAS PubMed.
- M. Springborg, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 52, 2935–2940 CrossRef CAS PubMed.
- L. Marques, J. L. Hodeau, M. Núñez Regueiro and M. Perroux, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, R12633 CrossRef CAS PubMed.
- F. Giacalone and N. Martín, Chem. Rev., 2006, 106, 5136–5190 CrossRef CAS PubMed.
- M. Álvarez Murga and J. Hodeau, Carbon, 2015, 82, 381–407 CrossRef.
- R. S. Ruoff and A. L. Ruoff, Nature, 1991, 350, 663–664 CrossRef.
- U. D. Venkateswaran, D. Sanzi, J. Krishnappa, L. Marques, J.-L. Hodeau, M. Núñez-Regueiro, A. M. Rao and P. C. Eklund, Phys. Status Solidi B, 1996, 198, 545–552 CrossRef CAS.
- A. M. Rao, P. C. Eklund, J.-L. Hodeau, L. Marques and M. Nunez-Regueiro, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 55, 4766–4773 CrossRef CAS.
- L. Forró and L. Mihály, Rep. Prog. Phys., 2001, 64, 649 CrossRef.
- T. L. Makarova, Semiconductors, 2001, 35, 243–278 CrossRef CAS.
- J. Sun, W. Liang, J. Yang and J. Gao, J. Mol. Struct. THEOCHEM, 2005, 755, 105–111 CrossRef CAS.
- V. R. Belosludov, T. M. Inerbaev, R. V. Belosludov and Y. Kawazoe, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67, 155410 CrossRef.
- V. Belosludov, T. Inerbaev, R. Belosludov, Y. Kawazoe and J. Kudoh, Comput. Mater. Sci., 2006, 36, 17–25 CrossRef CAS.
- R. D. Kennedy, A. L. Ayzner, D. D. Wanger, C. T. Day, M. Halim, S. I. Khan, S. H. Tolbert, B. J. Schwartz and Y. Rubin, J. Am. Chem. Soc., 2008, 130, 17290–17292 CrossRef CAS PubMed.
- G. Dennler, M. C. Scharber and C. J. Brabec, Adv. Mater., 2009, 21, 1323–1338 CrossRef CAS.
- P. W. Stephens, L. Mihaly, P. L. Lee, R. L. Whetten, S.-M. Huang, R. Kaner, F. Deiderich and K. Holczer, Nature, 1991, 351, 632–634 CrossRef CAS.
- O. Chauvet, G. Oszlànyi, L. Forro, P. W. Stephens, M. Tegze, G. Faigel and A. Jànossy, Phys. Rev. Lett., 1994, 72, 2721–2724 CrossRef CAS PubMed.
- P. W. Stephens, G. Bortel, G. Faigel, M. Tegze, A. Jánossy, S. Pekker, G. Oszlanyi and L. Forró, Nature, 1994, 370, 636–639 CrossRef CAS.
- O. Gunnarsson, Rev. Mod. Phys., 1997, 69, 575–606 CrossRef CAS.
- A. Huq, P. Stephens, G. M. Bendele and R. Ibberson, Chem. Phys. Lett., 2001, 347, 13–22 CrossRef CAS.
- L. Hou, X. Cui, B. Guan, S. Wang, R. Li, Y. Liu, D. Zhu and J. Zheng, Nature, 2022, 606, 507–510 CrossRef CAS PubMed.
- B. Peng, J. Am. Chem. Soc., 2022, 144, 19921–19931 CrossRef CAS PubMed.
- C. Jones and B. Peng, J. Phys. Chem. Lett., 2023, 14, 11768–11773 CrossRef CAS PubMed.
- J. Wu and B. Peng, J. Am. Chem. Soc., 2025, 147, 1749–1757 CrossRef CAS PubMed.
- D. Shearsby, J. Wu, D. Yang and B. Peng, Nanoscale, 2025, 17, 2616–2620 RSC.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864–B871 CrossRef.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- H. W. Kroto, Nature, 1987, 329, 529–531 CrossRef CAS.
- N. S. Goroff, Acc. Chem. Res., 1996, 29, 77–83 CrossRef CAS.
- J. J. Bernal, P. Haenecour, J. Howe, T. J. Zega, S. Amari and L. M. Ziurys, Astrophys. J., Lett., 2019, 883, L43 CrossRef CAS.
- E. Meirzadeh, A. M. Evans, M. Rezaee, M. Milich, C. J. Dionne, T. P. Darlington, S. T. Bao, A. K. Bartholomew, T. Handa, D. J. Rizzo, R. A. Wiscons, M. Reza, A. Zangiabadi, N. Fardian-Melamed, A. C. Crowther, P. J. Schuck, D. N. Basov, X. Zhu, A. Giri, P. E. Hopkins, P. Kim, M. L. Steigerwald, J. Yang, C. Nuckolls and X. Roy, Nature, 2023, 613, 71–76 CrossRef CAS PubMed.
- T. Wang, L. Zhang, J. Wu, M. Chen, S. Yang, Y. Lu and P. Du, Angew. Chem., Int. Ed., 2023, 62, e202311352 CrossRef CAS PubMed.
- Y. Zhang, Y. Xie, H. Mei, H. Yu, M. Li, Z. He, W. Fan, P. Zhang, A. G. Ricciardulli, P. Samorì, M. Li and S. Yang, Adv. Mater., 2025, 37, 2416741 CrossRef CAS PubMed.
- L. Yu, J. Xu, B. Peng, G. Qin and G. Su, J. Phys. Chem. Lett., 2022, 13, 11622–11629 CrossRef CAS PubMed.
- R. M. Tromer, L. A. Ribeiro and D. S. Galvão, Chem. Phys. Lett., 2022, 804, 139925 CrossRef CAS.
- P. Ying, H. Dong, T. Liang, Z. Fan, Z. Zhong and J. Zhang, Extreme Mech. Lett., 2023, 58, 101929 CrossRef.
- L. Ribeiro, M. Pereira, W. Giozza, R. Tromer and D. S. Galvão, Chem. Phys. Lett., 2022, 807, 140075 CrossRef CAS.
- R. F. Zhang, D. Legut, Z. J. Lin, Y. S. Zhao, H. K. Mao and S. Veprek, Phys. Rev. Lett., 2012, 108, 255502 CrossRef CAS PubMed.
- B. Peng, Nano Lett., 2023, 23, 652–658 CrossRef CAS PubMed.
- O. I. Malyi, K. V. Sopiha and C. Persson, ACS Appl. Mater. Interfaces, 2019, 11, 24876–24884 CrossRef CAS PubMed.
- D. Luo, K. Yin and R. Dronskowski, J. Am. Chem. Soc., 2022, 144, 5155–5162 CrossRef CAS PubMed.
- I. Pallikara, P. Kayastha, J. M. Skelton and L. D. Whalley, Electron. Struct., 2022, 4, 033002 CrossRef CAS.
- S. Baroni, S. de Gironcoli, A. Dal Corso and P. Giannozzi, Rev. Mod. Phys., 2001, 73, 515–562 CrossRef CAS.
- X. Gonze, Phys. Rev. A: At., Mol., Opt. Phys., 1995, 52, 1086–1095 CrossRef PubMed.
- X. Gonze, Phys. Rev. A: At., Mol., Opt. Phys., 1995, 52, 1096–1114 CrossRef CAS PubMed.
- M. T. Dove, Introduction to Lattice Dynamics, Cambridge University Press, 1993 Search PubMed.
- A. Togo, F. Oba and I. Tanaka, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 134106 CrossRef.
- A. Togo and I. Tanaka, Scr. Mater., 2015, 108, 1–5 CrossRef CAS.
- L.-F. Huang, P.-L. Gong and Z. Zeng, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 205433 CrossRef.
- L.-F. Huang, X.-Z. Lu, E. Tennessen and J. M. Rondinelli, Comput. Mater. Sci., 2016, 120, 84–93 CrossRef CAS.
- B. Peng, I. Bravić, J. L. MacManus-Driscoll and B. Monserrat, Phys. Rev. B, 2019, 100, 161101 CrossRef CAS.
- A. Shaikh and B. Peng, arXiv, 2025, preprint, arXiv:2504.02037 DOI:10.48550/arXiv.2504.02037.
- B. Peng, K. Xu, H. Zhang, Z. Ning, H. Shao, G. Ni, J. Li, Y. Zhu, H. Zhu and C. M. Soukoulis, Adv. Theory Simul., 2018, 1, 1700005 CrossRef.
- G. F. Lange, A. Bouhon, B. Monserrat and R.-J. Slager, Phys. Rev. B, 2022, 105, 064301 CrossRef CAS.
- P. Pavone, S. Baroni and S. de Gironcoli, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 10421–10423 CrossRef CAS.
- P. Pavone, J. Phys.: Condens. Matter, 2001, 13, 7593–7610 CrossRef CAS.
- A. Masago, K. Shirai and H. Katayama-Yoshida, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 104102 CrossRef.
- M. J. van Setten, M. A. Uijttewaal, G. A. de Wijs and R. A. de Groot, J. Am. Chem. Soc., 2007, 129, 2458–2465 CrossRef CAS PubMed.
- R. P. Stoffel, C. Wessel, M.-W. Lumey and R. Dronskowski, Angew. Chem., Int. Ed., 2010, 49, 5242–5266 CrossRef CAS PubMed.
- R. F. Zhang, Z. J. Lin, H.-K. Mao and Y. Zhao, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 060101 CrossRef.
- V. L. Deringer, R. P. Stoffel and R. Dronskowski, Cryst. Growth Des., 2014, 14, 871–878 CrossRef CAS.
- V. L. Deringer, R. P. Stoffel and R. Dronskowski, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 094303 CrossRef.
- M. A. White, A. B. Cerqueira, C. A. Whitman, M. B. Johnson and T. Ogitsu, Angew. Chem., Int. Ed., 2015, 54, 3626–3629 CrossRef CAS PubMed.
- J. Nyman, O. S. Pundyke and G. M. Day, Phys. Chem. Chem. Phys., 2016, 18, 15828–15837 RSC.
- J. M. Skelton, L. A. Burton, F. Oba and A. Walsh, J. Phys. Chem. C, 2017, 121, 6446–6454 CrossRef CAS PubMed.
- I. Pallikara and J. M. Skelton, Phys. Chem. Chem. Phys., 2021, 23, 19219–19236 RSC.
- C. J. Bartel, J. Mater. Sci., 2022, 57, 10475–10498 CrossRef CAS.
- Y. Le Page and P. Saxe, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 104104 CrossRef.
- X. Wu, D. Vanderbilt and D. R. Hamann, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 035105 CrossRef.
- M. Born and K. Huang, Dynamical theory of crystal lattices, Clarendon Press, Oxford, 1954 Search PubMed.
- Z.-J. Wu, E.-J. Zhao, H.-P. Xiang, X.-F. Hao, X.-J. Liu and J. Meng, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 054115 CrossRef.
- A. L. Goodwin, M. Calleja, M. J. Conterio, M. T. Dove, J. S. O. Evans, D. A. Keen, L. Peters and M. G. Tucker, Science, 2008, 319, 794–797 CrossRef CAS PubMed.
- M. T. Dove and H. Fang, Rep. Prog. Phys., 2016, 79, 066503 CrossRef PubMed.
- A. K. A. Pryde, K. D. Hammonds, M. T. Dove, V. Heine, J. D. Gale and M. C. Warren, J. Phys.: Condens. Matter, 1996, 8, 10973 CrossRef CAS.
- A. W. Sleight, Inorg. Chem., 1998, 37, 2854–2860 CrossRef CAS.
- M. G. Tucker, A. L. Goodwin, M. T. Dove, D. A. Keen, S. A. Wells and J. S. O. Evans, Phys. Rev. Lett., 2005, 95, 255501 CrossRef PubMed.
- L. Tan, V. Heine, G. Li and M. T. Dove, Rep. Prog. Phys., 2024, 87, 126501 CrossRef PubMed.
- J. N. Hancock, C. Turpen, Z. Schlesinger, G. R. Kowach and A. P. Ramirez, Phys. Rev. Lett., 2004, 93, 225501 CrossRef PubMed.
- A. L. Goodwin and C. J. Kepert, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 140301 CrossRef.
- L.-F. Huang, X.-Z. Lu and J. M. Rondinelli, Phys. Rev. Lett., 2016, 117, 115901 CrossRef PubMed.
- N. Z. Koocher, L.-F. Huang and J. M. Rondinelli, Phys. Rev. Mater., 2021, 5, 053601 CrossRef CAS.
- P. Ying, O. Hod and M. Urbakh, Nano Lett., 2024, 24, 10599–10604 CrossRef CAS PubMed.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef PubMed.
- J. Rossmeisl, Z.-W. Qu, H. Zhu, G.-J. Kroes and J. Nørskov, J. Electroanal. Chem., 2007, 607, 83–89 CrossRef CAS.
- L. Zhang, W. Wang, L. Zhou and H. Xu, Small, 2007, 3, 1618–1625 CrossRef CAS PubMed.
- Y. Zhang, J. Yu, D. Yu, X. Zhou and W. Lu, Rare Met., 2011, 30, 192–198 CrossRef CAS.
- T. Suzuki, T. Hisatomi, K. Teramura, Y. Shimodaira, H. Kobayashi and K. Domen, Phys. Chem. Chem. Phys., 2012, 14, 15475–15481 RSC.
- H.-Y. Jiang, J. Liu, K. Cheng, W. Sun and J. Lin, J. Phys. Chem. C, 2013, 117, 20029–20036 CrossRef CAS.
- J. Xu, L. Li, C. Guo, Y. Zhang and W. Meng, Appl. Catal., B, 2013, 130–131, 285–292 CrossRef CAS.
- J. Zhu, Z. Yin, D. Yang, T. Sun, H. Yu, H. E. Hoster, H. H. Hng, H. Zhang and Q. Yan, Energy Environ. Sci., 2013, 6, 987–993 RSC.
- T. Le Bahers, M. Rérat and P. Sautet, J. Phys. Chem. C, 2014, 118, 5997–6008 CrossRef.
- Y. Zheng, Y. Jiao, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2015, 54, 52–65 CrossRef CAS PubMed.
- M. Qiao, J. Liu, Y. Wang, Y. Li and Z. Chen, J. Am. Chem. Soc., 2018, 140, 12256–12262 CrossRef CAS PubMed.
- H. Yang, Y. Ma, S. Zhang, H. Jin, B. Huang and Y. Dai, J. Mater. Chem. A, 2019, 7, 12060–12067 RSC.
- L. Ju, J. Shang, X. Tang and L. Kou, J. Am. Chem. Soc., 2020, 142, 1492–1500 CrossRef CAS PubMed.
- A. Nakada, D. Kato, R. Nelson, H. Takahira, M. Yabuuchi, M. Higashi, H. Suzuki, M. Kirsanova, N. Kakudou, C. Tassel, T. Yamamoto, C. M. Brown, R. Dronskowski, A. Saeki, A. Abakumov, H. Kageyama and R. Abe, J. Am. Chem. Soc., 2021, 143, 2491–2499 CrossRef CAS PubMed.
- G. Wang, J. Chang, W. Tang, W. Xie and Y. S. Ang, J. Phys. D: Appl. Phys., 2022, 55, 293002 CrossRef CAS.
- C. Fu, G. Wang, Y. Huang, Y. Chen, H. Yuan, Y. S. Ang and H. Chen, Phys. Chem. Chem. Phys., 2022, 24, 3826–3833 RSC.
- J. P. Perdew, Int. J. Quantum Chem., 1985, 28, 497–523 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. P. Perdew, A. Ruzsinszky, G. I. Csonka, O. A. Vydrov, G. E. Scuseria, L. A. Constantin, X. Zhou and K. Burke, Phys. Rev. Lett., 2008, 100, 136406 CrossRef PubMed.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207 CrossRef CAS.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2006, 124, 219906 CrossRef.
- J. E. Peralta, J. Heyd, G. E. Scuseria and R. L. Martin, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 073101 CrossRef.
- L. Schimka, J. Harl and G. Kresse, J. Chem. Phys., 2011, 134, 024116 CrossRef PubMed.
- J. P. Perdew, M. Ernzerhof and K. Burke, J. Chem. Phys., 1996, 105, 9982–9985 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1996, 104, 1040–1046 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- M. Ernzerhof and G. E. Scuseria, J. Chem. Phys., 1999, 110, 5029–5036 CrossRef CAS.
- M. Bernardi, J. Phys.: Condens. Matter, 2020, 32, 385501 CrossRef CAS PubMed.
- A. Champagne, M. Camarasa-Gómez, F. Ricci, L. Kronik and J. B. Neaton, Nano Lett., 2024, 24, 7033–7039 CrossRef CAS PubMed.
- V. Chakrapani, J. C. Angus, A. B. Anderson, S. D. Wolter, B. R. Stoner and G. U. Sumanasekera, Science, 2007, 318, 1424–1430 CrossRef CAS PubMed.
- X. Zhang, A. Chen, Z. Zhang, M. Jiao and Z. Zhou, Nanoscale Adv., 2019, 1, 154–161 RSC.
- H. Chen, J. Zhao, X. Wang, X. Chen, Z. Zhang and M. Hua, Nanoscale, 2022, 14, 5551–5560 RSC.
- M. Bernardi, C. Ataca, M. Palummo and J. C. Grossman, Nanophotonics, 2017, 6, 479–493 CrossRef.
- Y. Li, Y.-L. Li, C. M. Araujo, W. Luo and R. Ahuja, Catal. Sci. Technol., 2013, 3, 2214–2220 RSC.
- E. Parzinger, B. Miller, B. Blaschke, J. A. Garrido, J. W. Ager, A. Holleitner and U. Wurstbauer, ACS Nano, 2015, 9, 11302–11309 CrossRef CAS PubMed.
- D. O. Scanlon, C. W. Dunnill, J. Buckeridge, S. A. Shevlin, A. J. Logsdail, S. M. Woodley, C. R. A. Catlow, M. J. Powell, R. G. Palgrave, I. P. Parkin, G. W. Watson, T. W. Keal, P. Sherwood, A. Walsh and A. A. Sokol, Nat. Mater., 2013, 12, 798–801 CrossRef CAS PubMed.
- V. O. Özçelik, J. G. Azadani, C. Yang, S. J. Koester and T. Low, Phys. Rev. B, 2016, 94, 035125 CrossRef.
- W. Hu and J. Yang, J. Mater. Chem. C, 2017, 5, 12289–12297 RSC.
- X. Yang, D. Singh and R. Ahuja, Catalysts, 2020, 10, 1111 CrossRef CAS.
- D. Rode, in Semiconductors and Semimetals, ed. R. Willardson and A. C. Beer, Elsevier, 1975, vol. 10, pp. 1–89 Search PubMed.
- A. Faghaninia, J. W. Ager and C. S. Lo, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 235123 CrossRef.
- A. M. Ganose, J. Park, A. Faghaninia, R. Woods-Robinson, K. A. Persson and A. Jain, Nat. Commun., 2021, 12, 2222 CrossRef CAS PubMed.
- Y. Zhao, Y.-H. Kim, A. C. Dillon, M. J. Heben and S. B. Zhang, Phys. Rev. Lett., 2005, 94, 155504 CrossRef PubMed.
- Y.-H. Kim, Y. Zhao, A. Williamson, M. J. Heben and S. B. Zhang, Phys. Rev. Lett., 2006, 96, 016102 CrossRef PubMed.
- M. Yoon, S. Yang, C. Hicke, E. Wang, D. Geohegan and Z. Zhang, Phys. Rev. Lett., 2008, 100, 206806 CrossRef PubMed.
- O. V. Pupysheva, A. A. Farajian and B. I. Yakobson, Nano Lett., 2008, 8, 767–774 CrossRef CAS PubMed.
- Q. Wang, Q. Sun, P. Jena and Y. Kawazoe, J. Chem. Theory Comput., 2009, 5, 374–379 CrossRef CAS PubMed.
- Q. Sun, Q. Wang and P. Jena, Appl. Phys. Lett., 2009, 94, 013111 CrossRef.
- D. Durbin, N. Allan and C. Malardier-Jugroot, Int. J. Hydrogen Energy, 2016, 41, 13116–13130 CrossRef CAS.
- D. Sankar De, J. A. Flores-Livas, S. Saha, L. Genovese and S. Goedecker, Carbon, 2018, 129, 847–853 CrossRef.
- Y. Ren, Y. Lu and D. Zhang, J. Phys. Chem. Lett., 2023, 14, 11051–11057 CrossRef CAS PubMed.
- J. Guan, J. Wu, D. Jiang, X. Zhu, R. Guan, X. Lei, P. Du, H. Zeng and S. Yang, Int. J. Hydrogen Energy, 2018, 43, 8698–8706 CrossRef CAS.
- P. Deák, B. Aradi and T. Frauenheim, J. Phys. Chem. C, 2011, 115, 3443–3446 CrossRef.
- V. Pfeifer, P. Erhart, S. Li, K. Rachut, J. Morasch, J. Brötz, P. Reckers, T. Mayer, S. Rühle, A. Zaban, I. Mora Seró, J. Bisquert, W. Jaegermann and A. Klein, J. Phys. Chem. Lett., 2013, 4, 4182–4187 CrossRef CAS.
- M.-G. Ju, G. Sun, J. Wang, Q. Meng and W. Liang, ACS Appl. Mater. Interfaces, 2014, 6, 12885–12892 CrossRef CAS PubMed.
- Y. Mi and Y. Weng, Sci. Rep., 2015, 5, 11482 CrossRef CAS PubMed.
- D. Zhang, M. Yang and S. Dong, Phys. Chem. Chem. Phys., 2015, 17, 29079–29084 RSC.
- P. Deák, J. Kullgren, B. Aradi, T. Frauenheim and L. Kavan, Electrochim. Acta, 2016, 199, 27–34 CrossRef.
- L. Chiodo, J. M. García-Lastra, A. Iacomino, S. Ossicini, J. Zhao, H. Petek and A. Rubio, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 045207 CrossRef.
- B. Li, S. Wu and X. Gao, Nanotechnol. Rev., 2020, 9, 1080–1103 CrossRef CAS.
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef PubMed.
- P. J. Waller, F. Gándara and O. M. Yaghi, Acc. Chem. Res., 2015, 48, 3053–3063 CrossRef CAS PubMed.
- N. Huang, P. Wang and D. Jiang, Nat. Rev. Mater., 2016, 1, 16068 CrossRef CAS.
- C. S. Diercks and O. M. Yaghi, Science, 2017, 355, eaal1585 CrossRef PubMed.
- J. Roncali, Acc. Chem. Res., 2009, 42, 1719–1730 CrossRef CAS PubMed.
- J. Shin, S. Yang, Y. Jang, J. S. Eo, T.-W. Kim, T. Lee, C.-H. Lee and G. Wang, Nat. Commun., 2020, 11, 1412 CrossRef CAS PubMed.
- L. Zhang, J. Liu and Y.-Q. Lan, Acc. Chem. Res., 2024, 57, 870–883 CrossRef CAS PubMed.
- J. Royakkers, H. Yang, A. J. Gillett, F. Eisner, P. Ghosh, D. G. Congrave, M. Azzouzi, Z. Andaji-Garmaroudi, A. Leventis, A. Rao, J. M. Frost, J. Nelson and H. Bronstein, Nat. Chem., 2024, 16, 1453–1461 CrossRef CAS PubMed.
- D. Kayley and B. Peng, Comput. Mater. Today, 2025, 6, 100030 CrossRef.
- B. Peng and M. Pizzochero, arXiv, 2025, preprint, arXiv:2504.07790 DOI:10.48550/arXiv.2504.07790.
- X. Wang, Y. Ren, S. Qiu, F. Zhang, X. Li, J. Gao, W. Gao and J. Zhao, npj Comput. Mater., 2025, 11, 5 CrossRef CAS.
- N. Danilovic, R. Subbaraman, D. Strmcnik, K.-C. Chang, A. P. Paulikas, V. R. Stamenkovic and N. M. Markovic, Angew. Chem., Int. Ed., 2012, 51, 12495–12498 CrossRef CAS PubMed.
- E. L. Shirley and S. G. Louie, Phys. Rev. Lett., 1993, 71, 133–136 CrossRef CAS PubMed.
- E. L. Shirley, L. X. Benedict and S. G. Louie, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 10970–10977 CrossRef CAS PubMed.
- J. H. Weaver, J. L. Martins, T. Komeda, Y. Chen, T. R. Ohno, G. H. Kroll, N. Troullier, R. E. Haufler and R. E. Smalley, Phys. Rev. Lett., 1991, 66, 1741–1744 CrossRef CAS PubMed.
- R. W. Lof, M. A. van Veenendaal, B. Koopmans, H. T. Jonkman and G. A. Sawatzky, Phys. Rev. Lett., 1992, 68, 3924–3927 CrossRef CAS PubMed.
- R. Lof, M. van Veenendaal, H. Jonkman and G. Sawatzky, J. Electron Spectrosc. Relat. Phenom., 1995, 72, 83–87 CrossRef CAS.
- R. Schwedhelm, L. Kipp, A. Dallmeyer and M. Skibowski, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 13176–13180 CrossRef CAS.
- Y. Xie, B. Peng, I. Bravić, Y. Yu, Y. Dong, R. Liang, Q. Ou, B. Monserrat and S. Zhang, Adv. Sci., 2020, 7, 2001698 CrossRef CAS PubMed.
- B. Peng, D. Bennett, I. Bravić and B. Monserrat, Phys. Rev. Mater., 2022, 6, L082401 CrossRef CAS.
- S. You, H. Zhu, Z. Shen, X. Wang, B. Shao, Q. Wang, J. Lu, Y. Yuan, B. D. Dou, E. M. Sanehira, T. Russell, A. Lorenz, Y. Dong, L. Chen, M. Casareto, N. Rolston, M. C. Beard, J. J. Berry, M. Freitag, Y. Yan, O. M. Bakr and K. Zhu, Science, 2025, 388, 964–968 CrossRef CAS PubMed.
- P. Ehrenfreund and B. Foing, Adv. Space Res., 1997, 19, 1033–1042 CrossRef CAS PubMed.
- J. Cami, J. Bernard-Salas, E. Peeters and S. E. Malek, Science, 2010, 329, 1180–1182 CrossRef CAS PubMed.
- P. Woods, Nat. Astron., 2020, 4, 299–305 CrossRef.
- K. Nakada, M. Fujita, G. Dresselhaus and M. S. Dresselhaus, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 17954–17961 CrossRef CAS PubMed.
- O. V. Yazyev, Acc. Chem. Res., 2013, 46, 2319–2328 CrossRef CAS PubMed.
- Z. Chen, A. Narita and K. Müllen, Adv. Mater., 2020, 32, 2001893 CrossRef CAS PubMed.
- H. Wang, H. S. Wang, C. Ma, L. Chen, C. Jiang, C. Chen, X. Xie, A.-P. Li and X. Wang, Nat. Rev. Phys., 2021, 3, 791–802 CrossRef CAS.
- Y.-W. Son, M. L. Cohen and S. G. Louie, Phys. Rev. Lett., 2006, 97, 216803 CrossRef PubMed.
- Y.-C. Chen, T. Cao, C. Chen, Z. Pedramrazi, D. Haberer, D. G. de Oteyza, F. R. Fischer, S. G. Louie and M. F. Crommie, Nat. Nanotechnol., 2015, 10, 156–160 CrossRef CAS PubMed.
- G. D. Nguyen, H.-Z. Tsai, A. A. Omrani, T. Marangoni, M. Wu, D. J. Rizzo, G. F. Rodgers, R. R. Cloke, R. A. Durr, Y. Sakai, F. Liou, A. S. Aikawa, J. R. Chelikowsky, S. G. Louie, F. R. Fischer and M. F. Crommie, Nat. Nanotechnol., 2017, 12, 1077–1082 CrossRef CAS PubMed.
- K. Černevičs, O. V. Yazyev and M. Pizzochero, Phys. Rev. B, 2020, 102, 201406 CrossRef.
- H. Raza and E. C. Kan, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 245434 CrossRef.
- M. Pizzochero, N. V. Tepliakov, A. A. Mostofi and E. Kaxiras, Nano Lett., 2021, 21, 9332–9338 CrossRef CAS PubMed.
- Y.-W. Son, M. L. Cohen and S. G. Louie, Nature, 2006, 444, 347–349 CrossRef CAS PubMed.
- M. Pizzochero and E. Kaxiras, Nano Lett., 2022, 22, 1922–1928 CrossRef CAS PubMed.
- O. V. Yazyev, Rep. Prog. Phys., 2010, 73, 056501 CrossRef.
- R. Ma, N. V. Tepliakov, A. A. Mostofi and M. Pizzochero, J. Phys. Chem. Lett., 2025, 16, 1680–1685 CrossRef CAS PubMed.
- O. Gröning, S. Wang, X. Yao, C. A. Pignedoli, G. Borin Barin, C. Daniels, A. Cupo, V. Meunier, X. Feng, A. Narita, K. Müllen, P. Ruffieux and R. Fasel, Nature, 2018, 560, 209–213 CrossRef PubMed.
- D. J. Rizzo, G. Veber, T. Cao, C. Bronner, T. Chen, F. Zhao, H. Rodriguez, S. G. Louie, M. F. Crommie and F. R. Fischer, Nature, 2018, 560, 204–208 CrossRef CAS PubMed.
- N. V. Tepliakov, J. Lischner, E. Kaxiras, A. A. Mostofi and M. Pizzochero, Phys. Rev. Lett., 2023, 130, 026401 CrossRef CAS PubMed.
- Y. Tong, H. Liu, S. Dai and D.-E. Jiang, Nano Lett., 2023, 23, 7470–7476 CrossRef CAS PubMed.
- Y. Tong, H. Liu, S. M. Mahurin, S. Dai and D.-E. Jiang, Comput. Mater. Today, 2024, 4, 100013 CrossRef.
- K.-Q. Lin, C. S. Ong, S. Bange, P. E. Faria Junior, B. Peng, J. D. Ziegler, J. Zipfel, C. Bäuml, N. Paradiso, K. Watanabe, T. Taniguchi, C. Strunk, B. Monserrat, J. Fabian, A. Chernikov, D. Y. Qiu, S. G. Louie and J. M. Lupton, Nat. Commun., 2021, 12, 5500 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |